Abstract
Thirty-four years ago, the groundbreaking work of John McCafferty and Sir Gregory Winter in developing phage display technology revolutionized the discovery of human antibodies, paving the way for diverse applications. Since then, numerous phage-derived antibodies have been successfully developed and advanced into clinical studies, resulting in the approval of more than a dozen therapeutic antibodies. These antibodies have demonstrated efficacy across a spectrum of medical conditions, ranging from autoimmune diseases to various cancers. In this article, we provide an in-depth review of the development of phage display libraries as powerful platforms for therapeutic antibody discovery, elucidating the intricate procedures involved in antibody development. Additionally, we conduct a review of the current ntibody drugs for cancer treatment that have been developed using the phage display platform. Furthermore, we discuss the challenges inherent in this technology, offering insights into potential solutions to enhance crucial steps and facilitate more efficient drug discovery in the field of phage display technology.
1. Introduction
Therapeutic monoclonal antibodies have emerged as transformative agents in the realm of cancer treatment, fundamentally altering the landscape of oncology. These antibodies, designed to target specific proteins associated with cancer cells, exemplify precision medicine. By leveraging the body’s immune system to identify and attack cancer cells, monoclonal antibodies have proven instrumental in the treatment of various cancers [1,2,3].
In 1985, George P. Smith pioneered the concept of phage display, unveiling a groundbreaking technique for exhibiting peptides on filamentous phages. By fusing the gene encoding the desired protein to the 5′ region of a filamentous phage’s coat protein, such as protein III or protein VIII, Smith showcased an innovative method that exhibited distinct peptides on the outer surfaces of viral clones [4]. The subsequent screening step in this process isolated peptides with the highest binding affinity, marking a pivotal advancement in molecular selection methods. The refinement of this technique continued as Stephen Parmley and George Smith introduced biopanning in 1988, demonstrating that recursive rounds of selection could effectively enrich for clones present at remarkably low frequencies, even as rare as 1 in a billion or less. In 1990, McCafferty et al. achieved a groundbreaking milestone by demonstrating the display of complete antibody variable domains on the surface of phages. This revolutionary technique enabled the selection of phages capable of binding to specific antigens [5]. Moreover, Jamie Scott and George Smith extended these capabilities by describing the creation of extensive random peptide libraries displayed on filamentous phages [6]. This methodological evolution laid the foundation for the subsequent refinement and enhancement of phage display technology. The significant contributions of George P. Smith and Greg Winter culminated in the recognition of a half share of the 2018 Nobel Prize in Chemistry for their seminal work in developing phage display.
Up to now, the impact of phage display in generating antibodies extends across diverse applications. In the field of therapeutics, it has played a pivotal role in the development of monoclonal antibodies for various diseases, including cancer, autoimmune disorders, infectious diseases, and neurological conditions. Furthermore, phage display has significantly contributed to the discovery of antibodies targeting specific viral pathogens, such as influenza and HIV, providing valuable tools for both diagnosis and treatment [7,8]. Beyond therapeutics, phage display has been instrumental in antibody engineering, epitope mapping, and the study of protein interactions. Additionally, it has facilitated the identification of novel peptide ligands for drug targeting, the development of biosensors for disease detection, and the creation of protein libraries for enzyme optimization and protein–protein interaction studies [9]. Overall, phage display continues to revolutionize biotechnology and biomedical research by enabling the rapid and efficient generation of high-affinity antibodies and protein-based therapeutics for a wide range of applications.
2. Overview of Procedures of Phage Display Technology in Monoclonal Antibody Production
2.1. Library Construction
The journey of generating antibodies through phage display technology commences with the construction of a diverse library. This process entails integrating genetic material encoding antibody fragments, such as antibodies’ heavy and light chains, single-chain variable fragments (scFvs), Fabs, or peptides, into the genome of bacteriophages, notably filamentous ones like M13 [10]. This fusion results in the expression of a multitude of unique antibody candidates on the surface of phages, forming the basis of the library. The diversity of this library, ranging from millions to billions of variants, lays the groundwork for the subsequent exploration of potential binding partners (Figure 1) [11].
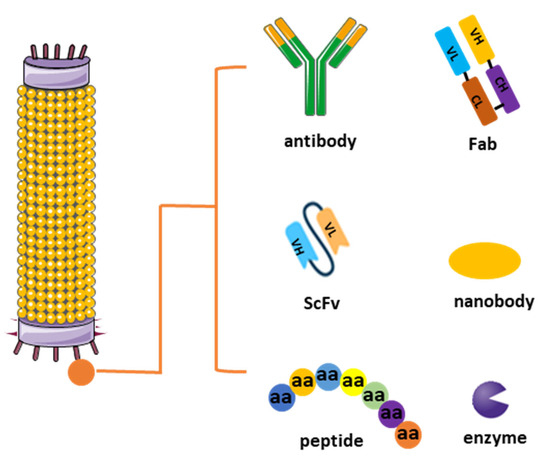
Figure 1.
Different types of phage display libraries. Phage display technology enables the selection and identification of peptides, antibodies, or enzymes that bind to specific target molecules. This approach entails generating filamentous fusion phages that showcase random foreign peptides or antibodies linked to a phage coat protein. Subsequently, the filamentous phages are assembled into a phage display library comprising random peptide sequences or antibodies.
Constructing a phage display library involves several key steps aimed at generating a diverse repertoire of displayed peptides or proteins for subsequent screening against target molecules. Initially, a source of genetic material encoding the desired peptides or proteins is obtained, which can include synthetic DNA sequences, cDNA libraries, or genomic DNA. Next, this genetic material is ligated into a phagemid vector, which contains the genetic elements necessary for phage display, including a phage coat protein gene fused to the peptide or protein of interest. The ligated DNA is then introduced into competent Escherichia coli cells via transformation, where it undergoes replication and amplification. Following transformation, the bacterial cells are infected with helper phage, which provides the necessary components for packaging the phagemid DNA into phage particles. The infected cells are then cultured to allow for phage production, and the resulting phage display library is harvested by collecting the culture supernatant or lysing the bacterial cells to release the phage particles. The library can be further characterized by sequencing a subset of clones to assess diversity and by performing binding assays to confirm the display of the desired peptides or proteins (Figure 2) [12].
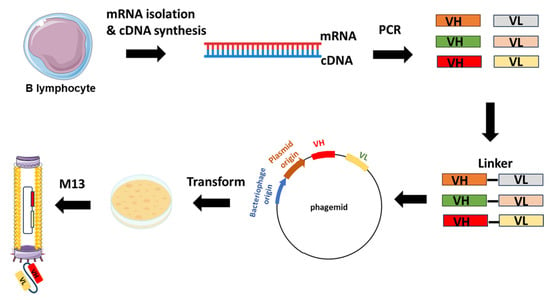
Figure 2.
Example of scFv-phage display library construction. The procedure starts with the extraction of total RNA from B-lymphocytes acquired from either immunized or non-immunized healthy donors. Subsequent to RNA isolation, cDNA is synthesized through reverse transcription. The repertoire of VH and VL genes is amplified from the cDNA using forward and reverse primers designed to target the variable domains. The VH and VL genes are ligated with linker sequences. Subsequently, scFvs sequences are cloned into the phagemid vectors, leading to the generation of a phage library.
Phage construction involves the genetic modification of bacteriophages like those in the M13 group to display specific peptides or proteins on their surface. Typically, this modification targets either the tail or the main coat protein of the phage, depending on the desired outcome and the characteristics of the phage being used. Tail proteins are often chosen for peptide insertion when the aim is to display peptides for binding to specific receptors or targets on host cells. This approach allows for the display of larger peptides or proteins without compromising the structural integrity of the main coat protein. Consequently, modifying the tail protein can enhance the specificity and efficiency of phage-mediated targeting, particularly in applications such as targeted drug delivery or tissue-specific imaging.
On the other hand, main coat proteins, like the major coat protein (pVIII) in M13 phages, are commonly targeted for peptide insertion due to their abundance and accessibility on the phage surface. Genetic fusion techniques are typically employed for peptide insertion into the main coat protein, where the peptide sequence is fused to the coding sequence of the coat protein. This strategy enables the display of peptides on every copy of the coat protein, potentially leading to a higher display density compared to tail protein modifications. However, modifications to the main coat protein may impact phage assembly and stability, necessitating careful design and optimization [13].
Comparing tail and main coat protein modification reveals differences in specificity, efficiency, and structural integrity. Tail protein modification may offer higher specificity in targeting host cells or specific receptors due to the potential for more diverse peptide sequences and a larger display capacity. In contrast, main coat protein modification often results in higher display densities but may lack specificity compared to tail modifications. Furthermore, tail protein modification typically preserves the structural integrity of the main coat protein, crucial for phage assembly and stability, while main coat protein modification may disrupt these properties, potentially affecting phage viability and infectivity. Ultimately, the choice between tail and main coat protein modification depends on the specific requirements of the intended application, balancing factors such as the targeting specificity, display density, and phage stability [14].
2.2. Biopanning, Elution and Amplification
The heart of the procedure lies in the targeted selection of antibodies through a process known as panning. The phage display library is exposed to the specific target of interest, such as a protein or cell type [10,15]. Those phages displaying antibodies that bind to the target are selectively enriched through successive rounds of panning. This iterative process involves incubating the library with the target, followed by stringent washing to remove weakly bound or non-specific phages. The remaining phages, carrying antibodies with higher affinity for the target, are recovered and subjected to additional rounds of panning, amplifying the specificity of the antibody repertoire (Figure 3) [16].
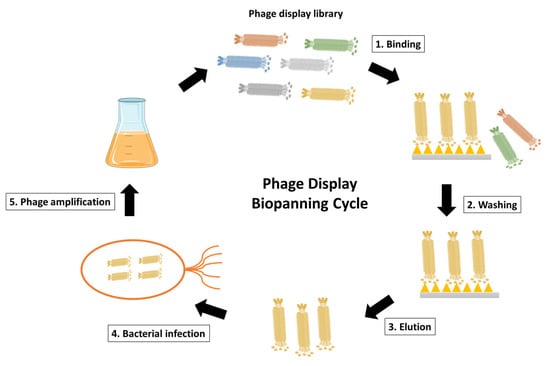
Figure 3.
Schematic representation of phage display biopanning cycle. The phage display library after construction is incubated with the desired antigen, such as the surface-immobilized antigen or the cell surface antigen. Unspecific phages that fail to bind are then eliminated through washing steps. The antigen-specific phages are subsequently eluted and used to infect E. coli for phage amplification. Coinfection with a helper phage enables the production and amplification of the desired phages. The phage display biopanning cycle is typically repeated for 2–3 cycles to achieve phage enrichment.
Once the panning process is complete, the phages displaying antibodies of interest are eluted from the target, disrupting the interaction between the displayed molecules and the immobilized target. These eluted phages represent a pool enriched with potential antibody hits. Subsequently, the selected phages are amplified by infecting bacterial hosts, ensuring the preservation and proliferation of the enriched antibody candidates. This step provides a sufficient quantity of the selected phages for downstream analysis and characterization [17].
In the process of panning and elution in phage display libraries, the choice of buffers with varying pH levels can significantly influence the characteristics of the displayed peptides and the efficiency of the selection process. Buffers with high pH, such as carbonate-bicarbonate buffer (pH 9.6), are commonly employed during the panning step of phage display selections. The high pH environment helps to ensure the stability and solubility of target proteins or ligands by minimizing their aggregation or precipitation. Additionally, at higher pH levels, the surface charge of both the target molecule and the phage-displayed peptides may be altered, which can influence the strength and specificity of their interactions. Peptides displayed on the phage surface may adopt different conformations or exhibit altered electrostatic properties under high pH conditions, potentially enhancing their affinity for specific targets or enabling the recognition of unique epitopes. Conversely, buffers with low pH, such as glycine-HCl buffer (pH 2.2), are often utilized during the elution step to dissociate phage–target complexes and recover specifically bound phages. The acidic environment disrupts electrostatic interactions and weakens the binding affinity between the displayed peptides and their target molecules, thereby facilitating the release of bound phages. Low-pH elution buffers are particularly effective for eluting phages that bind tightly to their targets, as they provide a robust means of dissociating these interactions without affecting phage viability or infectivity. However, it is important to note that excessively low pH conditions can also potentially damage the phage particles or denature the displayed peptides, leading to the reduced efficacy of the selection process. By optimizing the pH conditions to suit the specific characteristics of the target molecule and the desired selection outcome, researchers can enhance the success of phage display experiments and expedite the identification of novel peptide ligands or protein–protein interactions [18,19].
2.3. DNA Sequencing and Antibody Production
Individual phage clones from the enriched pool are isolated, and the genetic sequences of the displayed peptides or antibodies are determined through DNA sequencing. This information is crucial for the subsequent production of the identified antibodies. Once the genetic sequence is known, the corresponding antibodies can be expressed and produced in various formats, such as scFv or full-length antibodies, for further analysis and characterization [20].
2.4. Characterization and Optimization
The generated antibodies undergo rigorous characterization to evaluate their binding specificity, affinity, and potential therapeutic applications. This may involve assays such as ELISA or surface plasmon resonance. Further optimization steps can be employed to enhance the desired properties of the antibodies, including improvements in the binding affinity and stability, or reduced immunogenicity. Iterative rounds of optimization may be conducted to refine the antibodies for specific therapeutic or diagnostic purposes [21]. In essence, the procedures used in phage display technology to generate antibodies encompass a strategic and iterative process, from the construction of diverse libraries to the targeted selection of high-affinity binders and the subsequent characterization and optimization of the identified antibodies. This robust methodology has been pivotal in advancing the field of antibody discovery and engineering.
3. Categories of Phage Display Libraries for Antibody Development
Phage libraries derived from rearranged V-gene repertoires are created using mRNA or RNA isolated from B cells obtained from either immunized or naïve donors. The construction process entails initially preparing the cDNA template through reverse transcription polymerase chain reaction (RT-PCR). Subsequently, the repertoire of variable heavy (VH) and variable light (VL) chain genes is amplified via PCR before being cloned into the phagemid vector [22]. While phage display technology has been widely used for novel antibody development, it is noteworthy that there are different types of phage display libraries after many years of development.
As one of the first libraries constructed, scFv libraries are commonly used for antibody selection. They consist of antibody fragments that include the VH and VL chains linked by a flexible peptide linker. This format allows the expression of functional antigen-binding sites in a single polypeptide chain. For instance, the Tomlinson I and J libraries are well-known scFv libraries derived from human germline sequences. They offer a diverse source of antibody fragments for various applications. Another well-known application is the selection of scFv antibodies against specific tumor antigens using phage display, as demonstrated by Lee et al. [23]. In contrast, Fab libraries display antibody fragments that consist of the variable regions of both the heavy and light chains, along with the constant regions of the heavy chain. Fabs maintain the antigen-binding capacity of antibodies while providing additional structural stability. The HuCAL (Human Combinatorial Antibody Library) Fab library is a widely used human-derived library for the generation of Fab fragments with therapeutic potential [24]. In addition, a study by Gram et al. utilized a Fab phage display library to isolate antibodies specific to the human epidermal growth factor receptor 2 (HER2) [25].
In addition to the libraries from human and mice, single-domain antibody (nanobody) libraries have been derived from the variable domains of the heavy-chain-only antibodies found in camelids, such as llamas and camels. Renowned for their unique structural and functional attributes, nanobodies are single polypeptide chains that retain the antigen-binding capabilities of conventional antibodies [26,27]. One remarkable feature of nanobodies is their compact size, comprising only the variable domain without the additional heavy or light chains. This simplicity grants nanobodies several advantages, including enhanced tissue penetration, increased stability, and the ability to recognize cryptic epitopes that might be challenging for larger antibodies. The absence of a light chain also simplifies production and engineering processes. The selection process from nanobody libraries often involves immunizing animals with the target antigen of interest, followed by the generation of the library and subsequent panning against the antigen. Additionally, synthetic or naïve nanobody libraries can be created without prior immunization, providing a valuable resource for discovering binders against a wide range of targets [28].
In addition, chimeric phage display libraries are a hybrid class of libraries that incorporate genetic material from multiple sources, combining elements to enhance the diversity and functionality of displayed peptides or proteins. The construction of chimeric libraries involves the fusion of genetic sequences derived from different antibody fragments or protein domains, resulting in a mosaic-like collection of variants presented on the surface of bacteriophages [29]. These libraries aim to harness the strengths of different antibody formats or functional domains to create molecules with optimized properties. One common strategy for constructing chimeric libraries is the fusion of variable regions from human antibodies with constant regions from non-human sources, such as mice. This chimerization process seeks to retain the specificity and affinity of human antibodies while benefiting from the stability and expression advantages offered by non-human constant regions. Chimeric libraries can also be generated by combining variable domains from different antibodies, allowing for the exploration of diverse binding specificities within a single library [30].
In another way, phage display antibody libraries can be classified into four distinct types based on the origin of their sequences: naive, immune, semi-synthetic, and synthetic libraries [31,32]. Naive libraries, sourced from natural entities such as primary B-cells of non-immunized donors, harness naturally rearranged variable region genes. With extensive repertoires reaching up to 1011 members, naive libraries have the capacity to generate antibodies targeting a diverse array of antigens. In contrast, immune libraries derive from B-cell antibody repertoires of immunized or immune donors, exhibiting a predisposition to a limited panel of antigens and generally being smaller in size. While adept at addressing specific antigens, immune libraries are less suited for identifying antibody fragments against a broad spectrum of antigens, particularly self-antigens [30]. Synthetic libraries leverage computational design and gene synthesis, affording precise control over the composition of complementarity-determining regions (CDRs) [28]. Designed in silico, synthetic antibody libraries enable the creation of highly diverse libraries with predetermined structural features, incorporating non-natural amino acids. For example, MorphoSys’s HuCAL PLATINUM library is a synthetic antibody library meticulously designed for optimal diversity and human-like antibody properties. Similarly, semi-synthetic libraries amalgamate CDRs from natural sources with in silico-designed elements. Early iterations of semi-synthetic libraries maintained diversity through various framework genes. For example, in 1992, Hoogenboom and Winter introduced semi-synthetic scFv-antibody phage display libraries, integrating 49 germline VH sequences and a single V_lambda 3 light chain sequence. These libraries, with a size of 1 × 107, employed the PCR-based randomization of five or eight residues in the CDR-H3 to enhance diversity [33].
4. Phage Display Libraries for Antibody Development for Cancer Treatment
4.1. The Advantages of Using Phage Display Libraries for Antibody Development
Nowadays, there are many technologies for monoclonoal antibody discovery, such as phage display, hybridoma or single-cell sorting [34,35]. Hybridoma technology and phage display libraries represent two major approaches in antibody development, each with their unique strengths and considerations. In hybridoma technology, monoclonal antibodies are generated by fusing immunized animal B cells with myeloma cells, yielding antibodies with fixed specificity from the natural immune response. This method is effective for well-defined antigens and stable targets, but it is limited in diversity and speed due to the necessity of immunization and subsequent cell fusion. In contrast, phage display libraries provide a versatile and accelerated platform for antibody development. Phage display libraries are particularly advantageous for challenging targets, such as complex antigens or poorly immunogenic molecules. The diversity of antibodies generated by phage display libraries surpasses that of hybridoma technology, allowing for the screening of large repertoires against a variety of antigens. Moreover, the in vitro nature of phage display libraries enables rapid development without the need for immunization. Synthetic libraries, in particular, provide precise control over antibody composition, introducing non-natural amino acids and allowing for the creation of fully synthetic antibodies. This flexibility extends to humanization, as phage display libraries can be designed to produce fully human antibodies or undergo in vitro evolution for humanization. While hybridoma technology remains relevant for specific applications, the advanced capabilities of phage display libraries make them a preferred choice for modern antibody development needs. Their ability to rapidly generate diverse antibodies against a range of targets, including complex or challenging antigens, positions phage display libraries as a powerful and adaptable tool in the field of therapeutic antibody discovery [36].
4.2. Phage Display-Derived mAbs for Cancer Treatment
As previously mentioned, phage display technology has underscored its strength and consistency as a platform for discovering human antibodies. Currently, 14 monoclonal antibodies developed through this technology have gained approval for diverse diseases, including cancer. Furthermore, numerous others derived from phage display are undergoing pre-clinical development or progressing through clinical trials. This section delves into a comprehensive discussion of the five phage display-derived antibodies that have received approval for cancer treatment, emphasizing the valuable role of antibody phage display technology in shaping the landscape of the biopharmaceutical industry (Table 1). It is noteworthy that nine other approved phage display-derived antibodies have been successfully used for non-cancer treatments. For instance, Abicipar pegol, an anti-vascular endothelial growth factor (VEGF) antibody fragment, has been approved for the treatment of neovascular (wet) age-related macular degeneration. Belimumab (Benlysta), targeting the soluble B lymphocyte stimulator, which is involved in the survival and activation of B cells, is employed to reduce disease activity in systemic lupus erythematosus patients [37].

Table 1.
Phage display-derived antibodies in clinical trials or approved for cancer treatment.
The process of constructing these engineered antibodies follows a similar procedure. Initially, a phage library is generated by cloning a diverse array of antibody variable region sequences into a phage display vector. This library encompasses a broad spectrum of antibody fragments, each showcased on the surface of individual phage particles. Through multiple rounds of biopanning, phages displaying antibodies that specifically bind to the target antigen are selected from the library. These chosen phages are subsequently amplified and propagated to enrich for high-affinity binders. Finally, the DNA sequences encoding the variable regions of the selected antibodies are isolated and inserted into expression vectors for the production of full-length antibodies. Atezolizumab, for instance, was discovered through screening a human phage display library against a recombinant extracellular domain-Fc fusion of human PD-L1. From this screening, a high-affinity antibody was isolated from a single phage clone, utilizing a human IgG1 backbone. Due to PD-L1 expression on activated T cells, the Fc region of atezolizumab underwent engineering to abolish the antibody-dependent cytotoxicity (ADCC) effect. This engineering involved introducing an Asn to Ala substitution at position 298 within the CH2 domain of each heavy chain. As a result, atezolizumab was rendered incapable of binding to human Fcγ receptors.
Comparing the use of a phage-derived monoclonal antibody (mAb) to the original mAb in cancer treatment reveals several advantages of phage display-derived antibodies. Firstly, phage display allows for the generation of antibodies with enhanced specificity and affinity through iterative rounds of selection and optimization. This enables the identification of antibodies with superior binding properties compared to those obtained through traditional hybridoma-based methods [36]. Additionally, phage display offers greater versatility in antibody engineering, allowing for the incorporation of specific modifications, such as humanization or affinity maturation, to improve therapeutic efficacy and reduce immunogenicity. Moreover, phage display-derived antibodies offer potential benefits in terms of manufacturing scalability and cost effectiveness. Unlike traditional mAbs produced from hybridoma cell lines, phage-derived antibodies can be generated entirely in vitro, eliminating the need for animal immunization and cell culture-based production systems. This streamlined production process enables rapid and cost-effective antibody development, making phage display an attractive platform for the generation of therapeutic antibodies [22].
4.2.1. Atezolizumab
Atezolizumab is a monoclonal antibody (humanized IgG1κ) developed by Genentech, a subsidiary of Roche [38,39]. It is part of a class of immunotherapy drugs known as immune checkpoint inhibitors and specifically targets the programmed death-ligand 1 (PD-L1) protein. Numerous immune and tumor-infiltrating cells exhibit the expression of PD-L1, which plays a negative regulatory role in the activation of cytotoxic T-lymphocytes by engaging with programmed death-1 (PD-1) and B7.1 (CD80) receptors [63]. This interaction results in the suppression of T-cell migration, proliferation, and the secretion of cytotoxic mediators, ultimately hindering the effective killing of tumor cells. By binding to PD-L1, atezolizumab disrupts its interaction with PD-1 on T cells, which cancer cells often exploit to evade the immune system, therefore unleashing the immune system to recognize and attack cancer cells.
Atezolizumab has received approval for various indications, reflecting its efficacy across different cancer types. It is utilized with approval in the treatment of metastatic non-small cell lung cancer (NSCLC), triple-negative breast cancer, urothelial carcinoma, and bladder cancer. The approval status and specific indications may vary by region and are often in the context of advanced or metastatic stages. Currently, atezolizumab is also involved in many clinical trials for solid tumors and hematologic disorders. As of 2023, there are more than 400 ongoing trials using atezolizumab as a monotherapy or in combination with other therapies for cancer treatment. The ongoing trials have extended to include colon cancers, melanoma, glioblastoma, osteosarcoma, NHL, and DLBCL, etc. Its remarkable efficacy regarding overall survival, in comparison to some traditional chemotherapies such as docetaxel, has been consistently observed across various studies [64]. Atezolizumab has demonstrated promising results in combination therapies. For instance, the triple therapy combining the PD-L1 inhibitor atezolizumab with the BRAF inhibitor vemurafenib and MEK inhibitor cobimetinib has gained approval as a first-line treatment for advanced melanoma patients with BRAFV600 mutations [65]. Building on the success of the IMbrave150 trial, the combination of atezolizumab (an anti-PD-L1 antibody) and bevacizumab (an anti-VEGF antibody) has become the standard frontline treatment for patients with advanced-stage hepatocellular carcinoma (HCC) [66]. In the IMpassion031 trial, the combination of atezolizumab and nab-paclitaxel exhibited increased overall survival compared to placebo plus nab-paclitaxel in early triple-negative breast cancer (TNBC) patients, irrespective of the PD-L1 status [67]. These findings underscore the versatility and effectiveness of atezolizumab across a spectrum of cancers and treatment contexts.
4.2.2. Avelumab
Similar to atezolizumab, avelumab is another fully human IgG1 monoclonal antibody developed by Merck KGaA and Pfizer and designed to target PD-L1 protein [47]. By targeting PD-L1, avelumab disrupts the interaction between PD-L1 and PD-1, thereby promoting an anti-tumor immune response. Avelumab has been approved for the treatment of various cancers, including metastatic Merkel cell carcinoma, renal cell carcinoma (RCC), and advanced urothelial carcinoma, and as a maintenance treatment for patients with locally advanced or metastatic urothelial carcinoma that has not progressed following first-line platinum-containing chemotherapy. As of 2023, there are nearly 100 ongoing clinical trials employing avelumab in monotherapy and combination therapies. Besides the ones mentioned above, several other cancer types have been investigated, such as TNBC, thymic carcinoma, Hodgkin lymphoma, small cell lung cancer, and colon cancers. In the realm of combination therapy, it has demonstrated remarkable effectiveness by addressing various aspects of cancer biology and fostering a synergistic anti-cancer impact. The approval of avelumab in conjunction with axitinib marks a significant advancement for the first-line treatment of advanced RCC [68]. In a single-arm multicenter phase 2 trial (CAVEATT) comprising 32 enrolled patients, 27 with thymic carcinoma, 3 with type B3 thymoma, and 2 with a mixed-type B3 thymoma and thymic carcinoma, noteworthy outcomes were observed. The overall response rate stood at 34% (90% CI 21–50), with 11 patients (34%) achieving a partial response, 18 (56%) demonstrating stable disease, and 2 patients (6%) experiencing progressive disease as the best response. The combination of avelumab and axitinib exhibited promising anti-tumor activity, with acceptable toxicity in patients with advanced type B3 thymoma and thymic carcinoma, particularly for those progressing after chemotherapy. This combination holds the potential to emerge as a new standard treatment option in this challenging clinical setting [69].
4.2.3. Moxetumomab Pasudotox-tdfk
Marketed under the brand name LUMOXITI, Moxetumomab pasudotox-tdfk is a monoclonal antibody developed for the treatment of certain types of blood cancers [40,41,42]. It represents a pioneering recombinant immunotoxin, comprising a genetically fused recombinant murine scFv with a truncated form of pseudomonas exotoxin A (PE38). This medication is a CD22-directed cytotoxin and is indicated for the treatment of adult patients with relapsed or refractory hairy cell leukemia (HCL) who have received at least two prior systemic therapies, including treatment with a purine nucleoside analog [70]. The unique construct allows for the targeted delivery of the toxin to CD22-positive cells, specifically hairy cells in the case of HCL. By binding to CD22 on the surface of cancer cells, moxetumomab pasudotox-tdfk enters the cell, disrupting protein synthesis and leading to cell death.
Until now, moxetumomab pasudotox has shown tremendous efficacy in patients with relapsed/refractory (R/R) HCL. For instance, in a pivotal open-label trial with a multicenter design (NCT01829711), a total of eighty adult patients were enrolled. These individuals had undergone a minimum of two prior systemic therapies, including at least two purine nucleoside analogs (PNAs) or one PNA followed by rituximab or a BRAF inhibitor. With a median follow-up period of 24.6 months, the trial demonstrated a durable complete response (CR) rate of 36%, with a CR lasting at least 360 days observed in 33% of cases and an overall CR of 41%. Notably, 82% of complete responders were negative for minimal residual disease (MRD), accounting for 34% of all patients. The rate of CR lasting at least 60 months was 61%, and the median progression-free survival rate without the loss of CR was 71.7 months. Moxetumomab pasudotox demonstrated a notably high rate of durable responses and MRD negativity, presenting a manageable safety profile for heavily pre-treated patients with HCL who had inadequate responses to previous therapies [71]. Beyond its application in treating HCL, moxetumomab pasudotox has undergone evaluation for the treatment of other hematologic disorders, including acute lymphoblastic leukemia (ALL). A phase 2 study conducted internationally across multiple centers involved the enrollment of 32 patients with relapsed/refractory B-cell precursor ALL. Of the 28 patients evaluated for response, the objective response rate reached 28.6%. Notably, three patients (10.7%) achieved morphologic CR, and five patients (17.9%) achieved a partial response. While these findings hint at the heightened efficacy of moxetumomab pasudotox, particularly through continuous infusion or combination regimens, ongoing studies are underway to refine and optimize both the efficacy and safety aspects of moxetumomab pasudotox in the treatment of ALL [72].
4.2.4. Necitumumab
Commercially known as Portrazza, necitumumab is a fully human IgG1 kappa monoclonal antibody developed as an epidermal growth factor receptor (EGFR) antagonist [43,44]. By targeting EGFR, it interferes with the signaling pathways involved in cancer cell growth and survival. To be precise, it hampers the activation of downstream signaling pathways, including mitogen-activated protein kinase and phosphatidylinositol-4,5-bisphosphate 3-kinase/Akt. This, in turn, curtails cancer cell proliferation, differentiation, adhesion, migration, and survival [73]. Necitumumab is intended to inhibit the progression of squamous NSCLC, a subtype of lung cancer that is often associated with a poorer prognosis. It is specifically indicated for patients with metastatic squamous NSCLC who have not previously received medication for their metastatic disease.
Clinical trials, such as the SQUIRE trial (NCT00981058), have played a pivotal role in establishing the efficacy and safety of necitumumab. In the SQUIRE trial, the addition of necitumumab to gemcitabine and cisplatin demonstrated a significant improvement in overall survival in patients with advanced squamous NSCLC. Necitumumab is typically administered intravenously and may be used in combination with other chemotherapy agents [74]. In 2015, necitumumab was approved by the US FDA for the treatment of metastatic squamous NSCLC when combined with gemcitabine and cisplatin. Research efforts continue to explore novel combination therapies to enhance treatment outcomes and address resistance mechanisms. Given the dynamic nature of lung cancer treatment, ongoing clinical trials may reveal new insights into combinational approaches involving necitumumab.
4.2.5. Ramucirumab
Marketed under the brand name Cyramza, ramucirumab is a monoclonal antibody designed for the treatment of various cancers [45,46]. As a fully human IgG1 monoclonal antibody, it specifically targets VEGFR-2, inhibiting angiogenesis, the process by which new blood vessels form for tumor growth.
Extensive clinical assessments spanning various cancer categories have underscored the therapeutic potential of ramucirumab. In the international, randomized, double-blind, placebo-controlled phase 3 REGARD trial (NCT00917384), patients aged 24–87 years with advanced gastric or gastro-esophageal junction (GEJ) adenocarcinoma, experiencing disease progression after initial platinum-containing or fluoropyrimidine-containing chemotherapy, were randomized to receive either ramucirumab or placebo. The results indicated a median overall survival of 5.2 months in the ramucirumab group versus 3.8 months in the placebo group, whereas the rates of other adverse events were mostly similar between groups (94% vs. 88%). These findings affirmed the efficacy of remucirumab in metastatic gastric or GEJ adenocarcinoma [75]. Similarly, the REVEL phase III trial (NCT01168973) demonstrated improved outcomes with the combination of ramucirumab and paclitaxel in patients with metastatic NSCLC following or during a first-line platinum-based chemotherapy regimen [76]. Moreover, investigations in the REACH and REACH-2 trials revealed the benefits of ramucirumab in advanced HCC [77]. Presently, ramucirumab holds approval for use in metastatic gastric or GEJ adenocarcinoma, metastatic NSCLC, metastatic colorectal cancer, HCC, and advanced or metastatic urothelial carcinoma [78].
Beyond its approved indications, ramucirumab has undergone testing across various cancer types and stages. Often combined with chemotherapy or other targeted therapies, it has exhibited promising anti-cancer effects. In the phase II trial NCT02581215, patients with recurrent/metastatic pancreatic ductal adenocarcinoma received either mFOLFIRINOX/ramucirumab or mFOLFIRINOX/placebo. At 9 months, the progression-free survival rates were 25.1% and 35.0% for the mFOLFIRINOX/ramucirumab and mFOLFIRINOX/placebo groups, respectively. The median overall survival was 10.3 months for the mFOLFIRINOX/ramucirumab group and 9.7 months for the mFOLFIRINOX/placebo group [79]. Furthermore, ramucirumab has been explored in the treatment of patients with HER2-negative, unresectable, locally recurrent, or metastatic breast cancer (NCT00703326) and patients with locally advanced or unresectable or metastatic urothelial carcinoma (NCT02426125), exhibiting promising outcomes when combined with chemotherapies [80,81].
5. Discussion of Challenges and Future Prospectives
Cancer, a complex and multifaceted group of diseases characterized by uncontrolled cell growth, remains a significant global health challenge. The development of effective cancer treatment drugs is crucial in addressing this pervasive issue [82,83,84]. Among the diverse arsenal of therapeutic approaches, antibodies play a pivotal role due to their precision in targeting specific molecules associated with cancer cells. In the contemporary landscape, phage display stands out as one of the most prevalent and robust systems for the discovery and development of antibodies. Its widespread adoption is attributed to several advantages, such as the manipulable size of the phage’s genome, the efficient nature of phage infection, the cost effectiveness and safety associated with phage preparation and propagation, and the ability to perform high-throughput screening for peptides and antibodies. Additionally, the technology allows for rapid screening, contributing to its appeal [85]. Despite these numerous advantages, it is crucial to acknowledge that there are still areas within this technology that demand further enhancement and refinement to meet evolving needs.
An advantageous aspect of the phage display library, in comparison to other antibody development technologies, lies in its swift and convenient retrieval of peptide sequence information [14]. Typically, the assessment focuses on the most abundant sequences obtained from the panning output. However, a challenge arises when very few specific clones, carrying a small percentage of the sequence information, are acquired. This limitation is a result of the amplification bias of the desirable antibody-displaying phages during the panning cycles, causing certain sequences to become increasingly rare throughout the process. Furthermore, it is essential to acknowledge the potential divergence in conformation between chemically synthesized peptides and their original state on the phage display. The presence of multiple copies of exogenously displayed peptides on the phages can lead to robust binding during the selection process. Therefore, maintaining the authentic conformation of phage-derived peptides during chemical synthesis becomes crucial to ensure that their binding strength remains comparable to the original phages. Additionally, a recurring challenge in this technology is the occurrence of false positives during screening. This issue arises from the non-specific absorption that can preoccupy the phages before positive selection, emphasizing the need for strategies to mitigate false positives and enhance the accuracy of the screening process [86]. To address this issue, recent advancements have integrated next-generation sequencing technologies into the phage display library. This integration aims to identify the rare sequences holding potential binding affinity and predict the binding features of sequences obtained during the panning process [87,88].
The phage display library finds another compelling application in the development of tumor-targeting peptides, particularly for diagnostic purposes [89,90]. These peptides play a pivotal role in creating imaging agents designed to specifically illuminate cancerous lesions. By associating with nanoparticle conjugation, these peptides not only enhance sensitivity in cancer diagnosis, but also contribute to improved contrast imaging capabilities. An inherent advantage of leveraging phage display libraries for tumor-targeting peptide development lies in the vast and diverse pool of peptide sequences that can be explored. This unique feature enables the discovery of peptides with the capacity to selectively recognize and bind to the distinctive molecular signatures present on the surface of cancer cells [48,91]. While fluorescent antibodies are currently employed in molecular imaging for their exceptional affinity and selectivity, peptides emerge as superior carriers for guiding imaging probes to tumor sites due to their effective tissue penetration—an attribute not easily achievable with antibodies. Despite its recognized potential, the clinical use of tumor-targeting peptides has been limited, primarily due to challenges related to immunogenicity, clearance issues, and a lack of a comprehensive understanding of their behavior within the human body. Addressing these hurdles requires ongoing efforts to unlock the full potential of tumor-targeting peptides in the realm of cancer diagnostics, ensuring their effective and safe utilization in clinical settings.
While phage display has matured into a well-established platform for antibody development, the commercial utilization of major phage display platforms, as outlined in Table 1, has predominantly been limited to a handful of biopharmaceutical companies holding intellectual property rights. Consequently, a majority of the antibodies developed today are owned by these companies with proprietary access to the underlying technologies. While many patents have expired in Europe and the United States, there is a notable opportunity for academic groups or startups to create their libraries and contribute to the development of antibodies as translational products. Encouragingly, some companies, such as Bio-Rad, now provide cost-effective antibody discovery services. Conversely, some phage display antibody libraries established prior to 2010 remain accessible to academic laboratories, notably at The University of Cambridge and the Scottish Biologics Facility. Of note among these libraries are Tomlinson’s libraries, which have yielded high-affinity antibodies for numerous targets [92,93].
Antibody engineering through phage-display technology is a powerful but intricate process, facing several challenges that researchers have actively addressed to enhance its efficacy. One significant difficulty lies in achieving optimal binding affinities and specificities. The diverse and complex nature of target antigens often necessitates the meticulous optimization of phage display conditions to isolate antibodies with the desired properties. This involves multiple rounds of selection, sometimes requiring extensive screening and affinity maturation to enhance the binding capabilities of the displayed antibodies. Meanwhile, the issue of immunogenicity poses another obstacle in antibody engineering [13]. Antibodies derived from non-human sources, like murine antibodies, may trigger an immune response in humans. Humanization techniques, involving the grafting of CDRs onto human frameworks, have been developed to mitigate this challenge and create antibodies with reduced immunogenic potential [94]. Additionally, achieving functional antibodies with diverse isotypes and formats adds complexity to the engineering process. Scientists have explored different display formats, such as scFvs or Fab fragments, to optimize the selection of antibodies based on their intended applications [23]. Advancements in screening and selection methodologies have significantly improved the efficiency and throughput of antibody discovery using phage display. Techniques such as high-throughput screening, fluorescence-activated cell sorting, and next-generation sequencing allow for the rapid identification and characterization of antibodies with desired binding specificities and functional properties. Moreover, advances in library construction, selection methodologies, and molecular design have significantly improved the efficiency of isolating antibodies with high affinity, specificity, and reduced immunogenicity. Continuous innovations in phage display technology will help to overcome these obstructions, facilitating the development of antibodies for diagnostics, therapeutics, and various biotechnological applications.
Author Contributions
T.Z. contributed to the literature review and writing. T.Z. and Z.W. contributed to the table design. T.Z. contributed to the conception and design. T.Z. and Z.W. contributed to the proofreading and edition. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Parts of the figures were drawn by using pictures from Servier Medical Art accessed on 6 February 2024. Servier Medical Art by Servier is licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Conflicts of Interest
All other authors declared no conflicts of interest.
Abbreviations
BC: Breast Cancer; CCL: Chemokine (C-C motif) Ligand; CC: Colorectal Cancer; CD: Cluster of Differentiation; CLL: chronic lymphocytic leukemia; EGFR: Epidermal Growth Factor Receptor; ErbB3: Erb-B2 Receptor Tyrosine Kinase 3; EpCAM: Epithelial Cell Adhesion Molecule; GC: Gastric Carcinoma; HCL: Hairy Cell Leukemia; IGF-1R: Insulin-Like Growth Factor 1 Receptor; IgG: Immunoglobulin G; MIF: Macrophage Migration Inhibitory Factor; MCC: Merkel Cell Carcinoma; MCP: Monocyte Chemoattractant Protein; MM: Multiple Myeloma; NHL: Non-Hodgkin Lymphoma; NSCLC: Non-Small Cell Lung Cancer; PD-L1: Programmed Death-Ligand 1; RCC: Renal Cell Carcinoma; TRAIL: Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand; UBC: Urothelial Bladder Cancer; UC: Urothelial Carcinoma; VEGF: Vascular Endothelial Growth Factor.
References
- Stanfield, R.L.; Wilson, I.A. Antibody Structure. Microbiol. Spectr. 2014, 2, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Chiu, M.L.; Gilliland, G.L. Engineering antibody therapeutics. Curr. Opin. Struct. Biol. 2016, 38, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Shim, H. Antibody Phage Display. Adv. Exp. Med. Biol. 2017, 1053, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Filamentous fusion phage: Novel expression vectors that display cloned antigens on the virion surface. Science 1985, 228, 1315–1317. [Google Scholar] [CrossRef] [PubMed]
- McCafferty, J.; Griffiths, A.D.; Winter, G.; Chiswell, D.J. Phage antibodies: Filamentous phage displaying antibody variable domains. Nature 1990, 348, 552–554. [Google Scholar] [CrossRef] [PubMed]
- Franca, R.K.A.; Studart, I.C.; Bezerra, M.R.L.; Pontes, L.Q.; Barbosa, A.M.A.; Brigido, M.M.; Furtado, G.P.; Maranhao, A.Q. Progress on Phage Display Technology: Tailoring Antibodies for Cancer Immunotherapy. Viruses 2023, 15, 1903. [Google Scholar] [CrossRef] [PubMed]
- Domm, W.; Brewer, M.; Baker, S.F.; Feng, C.; Martinez-Sobrido, L.; Treanor, J.; Dewhurst, S. Use of bacteriophage particles displaying influenza virus hemagglutinin for the detection of hemagglutination-inhibition antibodies. J. Virol. Methods 2014, 197, 47–50. [Google Scholar] [CrossRef]
- Chikaev, A.N.; Chikaev, A.N.; Rudometov, A.P.; Merkulyeva, Y.A.; Karpenko, L.I. Phage display as a tool for identifying HIV-1 broadly neutralizing antibodies. Vavilovskii Zhurnal Genet. Sel. 2021, 25, 562–572. [Google Scholar] [CrossRef]
- Peltomaa, R.; Benito-Pena, E.; Barderas, R.; Moreno-Bondi, M.C. Phage Display in the Quest for New Selective Recognition Elements for Biosensors. ACS Omega 2019, 4, 11569–11580. [Google Scholar] [CrossRef]
- Jones, M.L.; Alfaleh, M.A.; Kumble, S.; Zhang, S.; Osborne, G.W.; Yeh, M.; Arora, N.; Hou, J.J.; Howard, C.B.; Chin, D.Y.; et al. Targeting membrane proteins for antibody discovery using phage display. Sci. Rep. 2016, 6, 26240. [Google Scholar] [CrossRef]
- Parmley, S.F.; Smith, G.P. Antibody-selectable filamentous fd phage vectors: Affinity purification of target genes. Gene 1988, 73, 305–318. [Google Scholar] [CrossRef] [PubMed]
- Bashir, S.; Paeshuyse, J. Construction of Antibody Phage Libraries and Their Application in Veterinary Immunovirology. Antibodies 2020, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Bazan, J.; Calkosinski, I.; Gamian, A. Phage display--a powerful technique for immunotherapy: 1. Introduction and potential of therapeutic applications. Hum. Vaccin. Immunother. 2012, 8, 1817–1828. [Google Scholar] [CrossRef] [PubMed]
- Anand, T.; Virmani, N.; Bera, B.C.; Vaid, R.K.; Vashisth, M.; Bardajatya, P.; Kumar, A.; Tripathi, B.N. Phage Display Technique as a Tool for Diagnosis and Antibody Selection for Coronaviruses. Curr. Microbiol. 2021, 78, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Saw, P.E.; Song, E.W. Phage display screening of therapeutic peptide for cancer targeting and therapy. Protein Cell 2019, 10, 787–807. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.K.; Smith, G.P. Searching for peptide ligands with an epitope library. Science 1990, 249, 386–390. [Google Scholar] [CrossRef]
- Nascimento, A.; Mullerpatan, A.; Azevedo, A.M.; Karande, P.; Cramer, S. Development of phage biopanning strategies to identify affinity peptide ligands for kappa light chain Fab fragments. Biotechnol. Prog. 2019, 35, e2884. [Google Scholar] [CrossRef]
- Tulika, T.; Pedersen, R.W.; Rimbault, C.; Ahmadi, S.; Rivera-de-Torre, E.; Fernandez-Quintero, M.L.; Loeffler, J.R.; Bohn, M.F.; Ljungars, A.; Ledsgaard, L.; et al. Phage display assisted discovery of a pH-dependent anti-alpha-cobratoxin antibody from a natural variable domain library. Protein Sci. 2023, 32, e4821. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, H.; Feng, N.; Li, Y.; Gu, J.; Wang, Z. Developability assessment at early-stage discovery to enable development of antibody-derived therapeutics. Antib. Ther. 2023, 6, 13–29. [Google Scholar] [CrossRef]
- Li, W.; Caberoy, N.B. New perspective for phage display as an efficient and versatile technology of functional proteomics. Appl. Microbiol. Biotechnol. 2010, 85, 909–919. [Google Scholar] [CrossRef]
- Ledsgaard, L.; Kilstrup, M.; Karatt-Vellatt, A.; McCafferty, J.; Laustsen, A.H. Basics of Antibody Phage Display Technology. Toxins 2018, 10, 236. [Google Scholar] [CrossRef] [PubMed]
- Alfaleh, M.A.; Alsaab, H.O.; Mahmoud, A.B.; Alkayyal, A.A.; Jones, M.L.; Mahler, S.M.; Hashem, A.M. Phage Display Derived Monoclonal Antibodies: From Bench to Bedside. Front. Immunol. 2020, 11, 1986. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Iorno, N.; Sierro, F.; Christ, D. Selection of human antibody fragments by phage display. Nat. Protoc. 2007, 2, 3001–3008. [Google Scholar] [CrossRef] [PubMed]
- Prassler, J.; Thiel, S.; Pracht, C.; Polzer, A.; Peters, S.; Bauer, M.; Norenberg, S.; Stark, Y.; Kolln, J.; Popp, A.; et al. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J. Mol. Biol. 2011, 413, 261–278. [Google Scholar] [CrossRef] [PubMed]
- Gram, H.; Marconi, L.A.; Barbas, C.F., 3rd; Collet, T.A.; Lerner, R.A.; Kang, A.S. In vitro selection and affinity maturation of antibodies from a naive combinatorial immunoglobulin library. Proc. Natl. Acad. Sci. USA 1992, 89, 3576–3580. [Google Scholar] [CrossRef] [PubMed]
- Bao, G.; Tang, M.; Zhao, J.; Zhu, X. Nanobody: A promising toolkit for molecular imaging and disease therapy. EJNMMI Res. 2021, 11, 6. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Ding, Z.; Yang, X.; Zhao, X.; Zhao, M.; Gao, L.; Chen, Q.; Xie, S.; Liu, A.; Yin, S.; et al. Nanobody: A Small Antibody with Big Implications for Tumor Therapeutic Strategy. Int. J. Nanomed. 2021, 16, 2337–2356. [Google Scholar] [CrossRef]
- Weber, M.; Bujak, E.; Putelli, A.; Villa, A.; Matasci, M.; Gualandi, L.; Hemmerle, T.; Wulhfard, S.; Neri, D. A highly functional synthetic phage display library containing over 40 billion human antibody clones. PLoS ONE 2014, 9, e100000. [Google Scholar] [CrossRef]
- Kumar, R.; Parray, H.A.; Shrivastava, T.; Sinha, S.; Luthra, K. Phage display antibody libraries: A robust approach for generation of recombinant human monoclonal antibodies. Int. J. Biol. Macromol. 2019, 135, 907–918. [Google Scholar] [CrossRef]
- Andre, A.S.; Moutinho, I.; Dias, J.N.R.; Aires-da-Silva, F. In vivo Phage Display: A promising selection strategy for the improvement of antibody targeting and drug delivery properties. Front. Microbiol. 2022, 13, 962124. [Google Scholar] [CrossRef]
- Frenzel, A.; Schirrmann, T.; Hust, M. Phage display-derived human antibodies in clinical development and therapy. MAbs 2016, 8, 1177–1194. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.K.; Rahumatullah, A.; Lai, J.Y.; Lim, T.S. Naive Human Antibody Libraries for Infectious Diseases. Adv. Exp. Med. Biol. 2017, 1053, 35–59. [Google Scholar] [CrossRef] [PubMed]
- Braunagel, M.; Little, M. Construction of a semisynthetic antibody library using trinucleotide oligos. Nucleic Acids Res. 1997, 25, 4690–4691. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pedrioli, A.; Oxenius, A. Single B cell technologies for monoclonal antibody discovery. Trends Immunol. 2021, 42, 1143–1158. [Google Scholar] [CrossRef] [PubMed]
- Ormundo, L.F.; Barreto, C.T.; Tsuruta, L.R. Development of Therapeutic Monoclonal Antibodies for Emerging Arbovirus Infections. Viruses 2023, 15, 2177. [Google Scholar] [CrossRef] [PubMed]
- Moraes, J.Z.; Hamaguchi, B.; Braggion, C.; Speciale, E.R.; Cesar, F.B.V.; Soares, G.d.F.d.S.; Osaki, J.H.; Pereira, T.M.; Aguiar, R.B. Hybridoma technology: Is it still useful? Curr. Res. Immunol. 2021, 2, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Callanan, D.; Kunimoto, D.; Maturi, R.K.; Patel, S.S.; Staurenghi, G.; Wolf, S.; Cheetham, J.K.; Hohman, T.C.; Kim, K.; Lopez, F.J.; et al. Double-Masked, Randomized, Phase 2 Evaluation of Abicipar Pegol (an Anti-VEGF DARPin Therapeutic) in Neovascular Age-Related Macular Degeneration. J. Ocul. Pharmacol. Ther. 2018, 34, 700–709. [Google Scholar] [CrossRef]
- Markham, A. Atezolizumab: First Global Approval. Drugs 2016, 76, 1227–1232. [Google Scholar] [CrossRef]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Pastan, I. Antibody fusion proteins: Anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin. Cancer Res. 2011, 17, 6398–6405. [Google Scholar] [CrossRef]
- Ho, M.; Nagata, S.; Pastan, I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc. Natl. Acad. Sci. USA 2006, 103, 9637–9642. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Moxetumomab Pasudotox: First Global Approval. Drugs 2018, 78, 1763–1767. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Kussie, P.; Ferguson, K.M. Structural basis for EGF receptor inhibition by the therapeutic antibody IMC-11F8. Structure 2008, 16, 216–227. [Google Scholar] [CrossRef]
- Garnock-Jones, K.P. Necitumumab: First Global Approval. Drugs 2016, 76, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Shen, J.; Vil, M.D.; Zhang, H.; Jimenez, X.; Bohlen, P.; Witte, L.; Zhu, Z. Tailoring in vitro selection for a picomolar affinity human antibody directed against vascular endothelial growth factor receptor 2 for enhanced neutralizing activity. J. Biol. Chem. 2003, 278, 43496–43507. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Jimenez, X.; Zhang, H.; Bohlen, P.; Witte, L.; Zhu, Z. Selection of high affinity human neutralizing antibodies to VEGFR2 from a large antibody phage display library for antiangiogenesis therapy. Int. J. Cancer 2002, 97, 393–399. [Google Scholar] [CrossRef]
- Kim, E.S. Avelumab: First Global Approval. Drugs 2017, 77, 929–937. [Google Scholar] [CrossRef]
- Nixon, A.E.; Sexton, D.J.; Ladner, R.C. Drugs derived from phage display: From candidate identification to clinical practice. MAbs 2014, 6, 73–85. [Google Scholar] [CrossRef]
- Kindler, H.L.; Richards, D.A.; Garbo, L.E.; Garon, E.B.; Stephenson, J.J., Jr.; Rocha-Lima, C.M.; Safran, H.; Chan, D.; Kocs, D.M.; Galimi, F.; et al. A randomized, placebo-controlled phase 2 study of ganitumab (AMG 479) or conatumumab (AMG 655) in combination with gemcitabine in patients with metastatic pancreatic cancer. Ann. Oncol. 2012, 23, 2834–2842. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Capanu, M.; O’Reilly, E.M.; Ma, J.; Chou, J.F.; Gansukh, B.; Shia, J.; Kalin, M.; Katz, S.; Abad, L.; et al. A phase II study of cixutumumab (IMC-A12, NSC742460) in advanced hepatocellular carcinoma. J. Hepatol. 2014, 60, 319–324. [Google Scholar] [CrossRef]
- Liu, J.F.; Ray-Coquard, I.; Selle, F.; Poveda, A.M.; Cibula, D.; Hirte, H.; Hilpert, F.; Raspagliesi, F.; Gladieff, L.; Harter, P.; et al. Randomized Phase II Trial of Seribantumab in Combination with Paclitaxel in Patients with Advanced Platinum-Resistant or -Refractory Ovarian Cancer. J. Clin. Oncol. 2016, 34, 4345–4353. [Google Scholar] [CrossRef]
- Younes, A.; Vose, J.M.; Zelenetz, A.D.; Smith, M.R.; Burris, H.A.; Ansell, S.M.; Klein, J.; Halpern, W.; Miceli, R.; Kumm, E.; et al. A Phase 1b/2 trial of mapatumumab in patients with relapsed/refractory non-Hodgkin’s lymphoma. Br. J. Cancer 2010, 103, 1783–1787. [Google Scholar] [CrossRef] [PubMed]
- Pienta, K.J.; Machiels, J.P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Scheulen, M.E.; Dittrich, C.; Obrist, P.; Marschner, N.; Dirix, L.; Schmidt, M.; Ruttinger, D.; Schuler, M.; Reinhardt, C.; et al. An open-label, randomized phase II study of adecatumumab, a fully human anti-EpCAM antibody, as monotherapy in patients with metastatic breast cancer. Ann. Oncol. 2010, 21, 275–282. [Google Scholar] [CrossRef]
- Dowlati, A.; Vlahovic, G.; Natale, R.B.; Rasmussen, E.; Singh, I.; Hwang, Y.C.; Rossi, J.; Bass, M.B.; Friberg, G.; Pickett, C.A. A Phase I, First-in-Human Study of AMG 780, an Angiopoietin-1 and -2 Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 4574–4584. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Knost, J.A.; Chiorean, E.G.; Kambhampati, S.R.; Yu, D.; Pytowski, B.; Qin, A.; Kauh, J.S.; O’Neil, B.H. Phase 1 study of the anti-vascular endothelial growth factor receptor 3 monoclonal antibody LY3022856/IMC-3C5 in patients with advanced and refractory solid tumors and advanced colorectal cancer. Cancer Chemother. Pharmacol. 2016, 78, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Mahalingam, D.; Patel, M.R.; Sachdev, J.C.; Hart, L.L.; Halama, N.; Ramanathan, R.K.; Sarantopoulos, J.; Volkel, D.; Youssef, A.; de Jong, F.A.; et al. Phase I study of imalumab (BAX69), a fully human recombinant antioxidized macrophage migration inhibitory factor antibody in advanced solid tumours. Br. J. Clin. Pharmacol. 2020, 86, 1836–1848. [Google Scholar] [CrossRef] [PubMed]
- Raab, M.S.; Engelhardt, M.; Blank, A.; Goldschmidt, H.; Agis, H.; Blau, I.W.; Einsele, H.; Ferstl, B.; Schub, N.; Rollig, C.; et al. MOR202, a novel anti-CD38 monoclonal antibody, in patients with relapsed or refractory multiple myeloma: A first-in-human, multicentre, phase 1-2a trial. Lancet Haematol. 2020, 7, e381–e394. [Google Scholar] [CrossRef]
- Golfier, S.; Kopitz, C.; Kahnert, A.; Heisler, I.; Schatz, C.A.; Stelte-Ludwig, B.; Mayer-Bartschmid, A.; Unterschemmann, K.; Bruder, S.; Linden, L.; et al. Anetumab ravtansine: A novel mesothelin-targeting antibody-drug conjugate cures tumors with heterogeneous target expression favored by bystander effect. Mol. Cancer Ther. 2014, 13, 1537–1548. [Google Scholar] [CrossRef]
- Smith, D.C.; Chugh, R.; Patnaik, A.; Papadopoulos, K.P.; Wang, M.; Kapoun, A.M.; Xu, L.; Dupont, J.; Stagg, R.J.; Tolcher, A. A phase 1 dose escalation and expansion study of Tarextumab (OMP-59R5) in patients with solid tumors. Investig. New Drugs 2019, 37, 722–730. [Google Scholar] [CrossRef]
- Diamond, J.R.; Becerra, C.; Richards, D.; Mita, A.; Osborne, C.; O’Shaughnessy, J.; Zhang, C.; Henner, R.; Kapoun, A.M.; Xu, L.; et al. Phase Ib clinical trial of the anti-frizzled antibody vantictumab (OMP-18R5) plus paclitaxel in patients with locally advanced or metastatic HER2-negative breast cancer. Breast Cancer Res. Treat. 2020, 184, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Cuesta-Mateos, C.; Alcaraz-Serna, A.; Somovilla-Crespo, B.; Munoz-Calleja, C. Monoclonal Antibody Therapies for Hematological Malignancies: Not Just Lineage-Specific Targets. Front. Immunol. 2017, 8, 1936. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A.; Jotte, R.M.; Cappuzzo, F.; Orlandi, F.; Stroyakovskiy, D.; Nogami, N.; Rodriguez-Abreu, D.; Moro-Sibilot, D.; Thomas, C.A.; Barlesi, F.; et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 2018, 378, 2288–2301. [Google Scholar] [CrossRef] [PubMed]
- Fehrenbacher, L.; Spira, A.; Ballinger, M.; Kowanetz, M.; Vansteenkiste, J.; Mazieres, J.; Park, K.; Smith, D.; Artal-Cortes, A.; Lewanski, C.; et al. Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): A multicentre, open-label, phase 2 randomised controlled trial. Lancet 2016, 387, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jin, J.; Guo, D.; Tao, Z.; Hu, X. Immune Checkpoint Inhibitors Combined with Targeted Therapy: The Recent Advances and Future Potentials. Cancers 2023, 15, 2858. [Google Scholar] [CrossRef]
- Pelizzaro, F.; Farinati, F.; Trevisani, F. Immune Checkpoint Inhibitors in Hepatocellular Carcinoma: Current Strategies and Biomarkers Predicting Response and/or Resistance. Biomedicines 2023, 11, 1020. [Google Scholar] [CrossRef] [PubMed]
- Uchimiak, K.; Badowska-Kozakiewicz, A.M.; Sobiborowicz-Sadowska, A.; Deptala, A. Current State of Knowledge on the Immune Checkpoint Inhibitors in Triple-Negative Breast Cancer Treatment: Approaches, Efficacy, and Challenges. Clin. Med. Insights Oncol. 2022, 16, 11795549221099869. [Google Scholar] [CrossRef]
- Zakharia, Y.; Huynh, L.; Du, S.; Chang, R.; Pi, S.; Sundaresan, S.; Duh, M.S.; Zanotti, G.; Thomaidou, D. Impact of Therapy Management on Axitinib-Related Adverse Events in Patients with Advanced Renal Cell Carcinoma Receiving First-Line Axitinib + Checkpoint Inhibitor. Clin. Genitourin. Cancer 2023, 21, e343–e351. [Google Scholar] [CrossRef]
- Conforti, F.; Zucali, P.A.; Pala, L.; Catania, C.; Bagnardi, V.; Sala, I.; Della Vigna, P.; Perrino, M.; Zagami, P.; Corti, C.; et al. Avelumab plus axitinib in unresectable or metastatic type B3 thymomas and thymic carcinomas (CAVEATT): A single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022, 23, 1287–1296. [Google Scholar] [CrossRef]
- Nobre, C.F.; Newman, M.J.; DeLisa, A.; Newman, P. Moxetumomab pasudotox-tdfk for relapsed/refractory hairy cell leukemia: A review of clinical considerations. Cancer Chemother. Pharmacol. 2019, 84, 255–263. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Dearden, C.; Zinzani, P.L.; Delgado, J.; Robak, T.; le Coutre, P.D.; Gjertsen, B.T.; Troussard, X.; Roboz, G.J.; Karlin, L.; et al. Moxetumomab pasudotox in heavily pre-treated patients with relapsed/refractory hairy cell leukemia (HCL): Long-term follow-up from the pivotal trial. J. Hematol. Oncol. 2021, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.N.; Bhojwani, D.; August, K.; Baruchel, A.; Bertrand, Y.; Boklan, J.; Dalla-Pozza, L.; Dennis, R.; Hijiya, N.; Locatelli, F.; et al. Results from an international phase 2 study of the anti-CD22 immunotoxin moxetumomab pasudotox in relapsed or refractory childhood B-lineage acute lymphoblastic leukemia. Pediatr. Blood Cancer 2020, 67, e28112. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Zhang, H.; Koo, H.; Tonra, J.; Balderes, P.; Prewett, M.; Corcoran, E.; Mangalampalli, V.; Bassi, R.; Anselma, D.; et al. A fully human recombinant IgG-like bispecific antibody to both the epidermal growth factor receptor and the insulin-like growth factor receptor for enhanced antitumor activity. J. Biol. Chem. 2005, 280, 19665–19672. [Google Scholar] [CrossRef] [PubMed]
- Fala, L. Portrazza (Necitumumab), an IgG1 Monoclonal Antibody, FDA Approved for Advanced Squamous Non-Small-Cell Lung Cancer. Am. Health Drug Benefits 2016, 9, 119–122. [Google Scholar]
- Fuchs, C.S.; Tomasek, J.; Yong, C.J.; Dumitru, F.; Passalacqua, R.; Goswami, C.; Safran, H.; Dos Santos, L.V.; Aprile, G.; Ferry, D.R.; et al. Ramucirumab monotherapy for previously treated advanced gastric or gastro-oesophageal junction adenocarcinoma (REGARD): An international, randomised, multicentre, placebo-controlled, phase 3 trial. Lancet 2014, 383, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Garon, E.B.; Ciuleanu, T.E.; Arrieta, O.; Prabhash, K.; Syrigos, K.N.; Goksel, T.; Park, K.; Gorbunova, V.; Kowalyszyn, R.D.; Pikiel, J.; et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): A multicentre, double-blind, randomised phase 3 trial. Lancet 2014, 384, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Singal, A.G.; Villanueva, A.; Finn, R.S.; Kudo, M.; Galle, P.R.; Ikeda, M.; Callies, S.; McGrath, L.M.; Wang, C.; et al. Prognostic and Predictive Factors in Patients with Advanced HCC and Elevated Alpha-Fetoprotein Treated with Ramucirumab in Two Randomized Phase III Trials. Clin. Cancer Res. 2022, 28, 2297–2305. [Google Scholar] [CrossRef]
- Debeuckelaere, C.; Murgioni, S.; Lonardi, S.; Girardi, N.; Alberti, G.; Fano, C.; Gallimberti, S.; Magro, C.; Ahcene-Djaballah, S.; Daniel, F.; et al. Ramucirumab: The long and winding road toward being an option for mCRC treatment. Expert Opin. Biol. Ther. 2019, 19, 399–409. [Google Scholar] [CrossRef]
- Shaib, W.L.; Manali, R.; Liu, Y.; El-Rayes, B.; Loehrer, P.; O’Neil, B.; Cohen, S.; Khair, T.; Robin, E.; Huyck, T.; et al. Phase II randomised, double-blind study of mFOLFIRINOX plus ramucirumab versus mFOLFIRINOX plus placebo in advanced pancreatic cancer patients (HCRN GI14-198). Eur. J. Cancer 2023, 189, 112847. [Google Scholar] [CrossRef]
- Mackey, J.R.; Ramos-Vazquez, M.; Lipatov, O.; McCarthy, N.; Krasnozhon, D.; Semiglazov, V.; Manikhas, A.; Gelmon, K.A.; Konecny, G.E.; Webster, M.; et al. Primary results of ROSE/TRIO-12, a randomized placebo-controlled phase III trial evaluating the addition of ramucirumab to first-line docetaxel chemotherapy in metastatic breast cancer. J. Clin. Oncol. 2015, 33, 141–148. [Google Scholar] [CrossRef]
- de Wit, R.; Powles, T.; Castellano, D.; Necchi, A.; Lee, J.L.; van der Heijden, M.S.; Matsubara, N.; Bamias, A.; Flechon, A.; Sternberg, C.N.; et al. Exposure-response relationship of ramucirumab in RANGE, a randomized phase III trial in advanced urothelial carcinoma refractory to platinum therapy. Br. J. Clin. Pharmacol. 2022, 88, 3182–3192. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Kordish, D.H.; Suryawanshi, Y.R.; Eversole, R.R.; Kohler, S.; Mackenzie, C.D.; Essani, K. Oncolytic Tanapoxvirus Expressing Interleukin-2 is Capable of Inducing the Regression of Human Melanoma Tumors in the Absence of T Cells. Curr. Cancer Drug Targets 2018, 18, 577–591. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, T.; Anderson, A.; Lee, V.; Szymura, S.; Dong, Z.; Kuang, B.; Oh, E.; Liu, J.; Neelapu, S.S.; et al. Immortalized B Cells Transfected with mRNA of Antigen Fused to MITD (IBMAM): An Effective Tool for Antigen-Specific T-Cell Expansion and TCR Validation. Biomedicines 2023, 11, 796. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Jou, T.H.; Hsin, J.; Wang, Z.; Huang, K.; Ye, J.; Yin, H.; Xing, Y. Talimogene Laherparepvec (T-VEC): A Review of the Recent Advances in Cancer Therapy. J. Clin. Med. 2023, 12, 1098. [Google Scholar] [CrossRef]
- Greenwood, J.; Willis, A.E.; Perham, R.N. Multiple display of foreign peptides on a filamentous bacteriophage. Peptides from Plasmodium falciparum circumsporozoite protein as antigens. J. Mol. Biol. 1991, 220, 821–827. [Google Scholar] [CrossRef] [PubMed]
- Lamboy, J.A.; Arter, J.A.; Knopp, K.A.; Der, D.; Overstreet, C.M.; Palermo, E.F.; Urakami, H.; Yu, T.B.; Tezgel, O.; Tew, G.N.; et al. Phage wrapping with cationic polymers eliminates nonspecific binding between M13 phage and high pI target proteins. J Am. Chem. Soc. 2009, 131, 16454–16460. [Google Scholar] [CrossRef]
- Akbar, R.; Bashour, H.; Rawat, P.; Robert, P.A.; Smorodina, E.; Cotet, T.S.; Flem-Karlsen, K.; Frank, R.; Mehta, B.B.; Vu, M.H.; et al. Progress and challenges for the machine learning-based design of fit-for-purpose monoclonal antibodies. MAbs 2022, 14, 2008790. [Google Scholar] [CrossRef]
- Zambrano, N.; Froechlich, G.; Lazarevic, D.; Passariello, M.; Nicosia, A.; De Lorenzo, C.; Morelli, M.J.; Sasso, E. High-Throughput Monoclonal Antibody Discovery from Phage Libraries: Challenging the Current Preclinical Pipeline to Keep the Pace with the Increasing mAb Demand. Cancers 2022, 14, 1325. [Google Scholar] [CrossRef]
- Islam, M.S.; Fan, J.; Pan, F. The power of phages: Revolutionizing cancer treatment. Front. Oncol. 2023, 13, 1290296. [Google Scholar] [CrossRef]
- Brown, K.C. Peptidic tumor targeting agents: The road from phage display peptide selections to clinical applications. Curr. Pharm. Des. 2010, 16, 1040–1054. [Google Scholar] [CrossRef]
- Liu, J.K.; Lubelski, D.; Schonberg, D.L.; Wu, Q.; Hale, J.S.; Flavahan, W.A.; Mulkearns-Hubert, E.E.; Man, J.; Hjelmeland, A.B.; Yu, J.; et al. Phage display discovery of novel molecular targets in glioblastoma-initiating cells. Cell Death Differ. 2014, 21, 1325–1339. [Google Scholar] [CrossRef][Green Version]
- de Wildt, R.M.; Mundy, C.R.; Gorick, B.D.; Tomlinson, I.M. Antibody arrays for high-throughput screening of antibody-antigen interactions. Nat. Biotechnol. 2000, 18, 989–994. [Google Scholar] [CrossRef]
- Munke, A.; Persson, J.; Weiffert, T.; De Genst, E.; Meisl, G.; Arosio, P.; Carnerup, A.; Dobson, C.M.; Vendruscolo, M.; Knowles, T.P.J.; et al. Phage display and kinetic selection of antibodies that specifically inhibit amyloid self-replication. Proc. Natl. Acad. Sci. USA 2017, 114, 6444–6449. [Google Scholar] [CrossRef]
- Rader, C.; Cheresh, D.A.; Barbas, C.F., 3rd. A phage display approach for rapid antibody humanization: Designed combinatorial V gene libraries. Proc. Natl. Acad. Sci. USA 1998, 95, 8910–8915. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).