
mRNA and Synthesis-Based Therapeutic Proteins: A Non-Recombinant Affordable Option


Abstract
:1. Introduction
- Barney S Graham;
- Hamilton Bennett;
- Brett Leav;
- Paul Bates;
- Giuseppe Ciaramella;
- Scott E Hensley;
- Kizzmekia S Corbett;
- Nicole A Doria-Rose;
- Ingmar Hoerr;
- Florian Krammer.
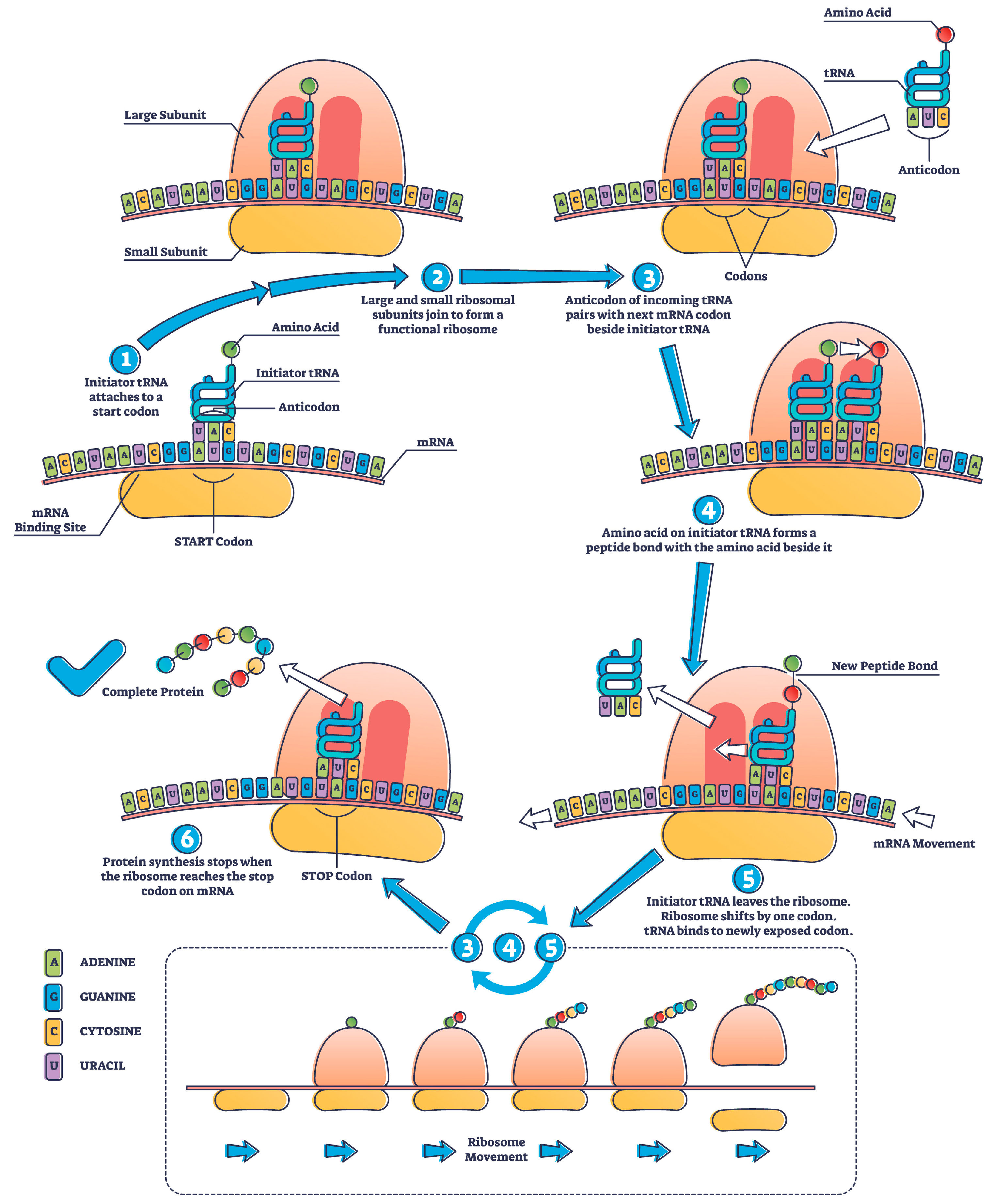
2. mRNA Protein Translation
3. Post-Translational Modification (PTM)
4. mRNA Production
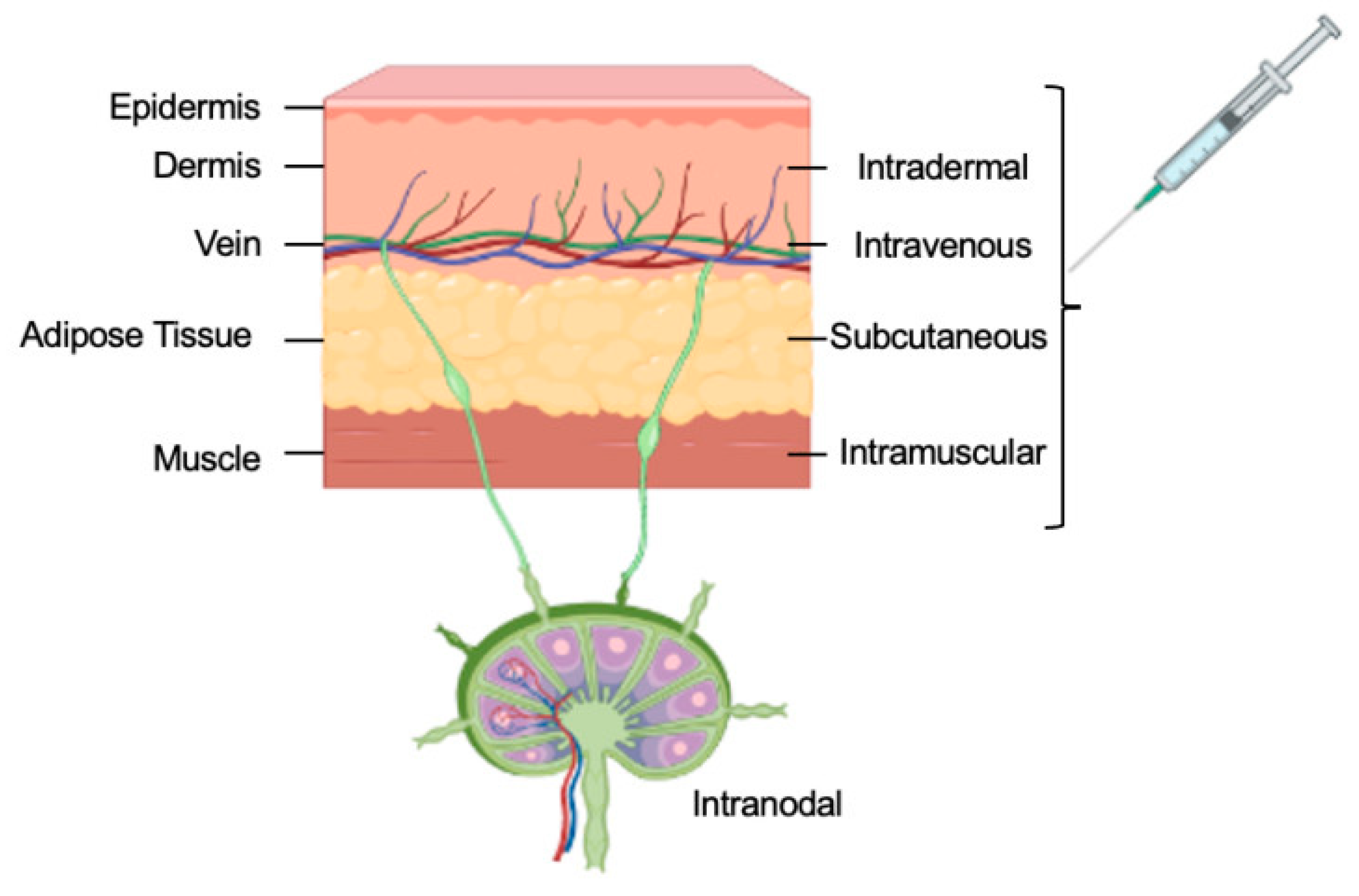
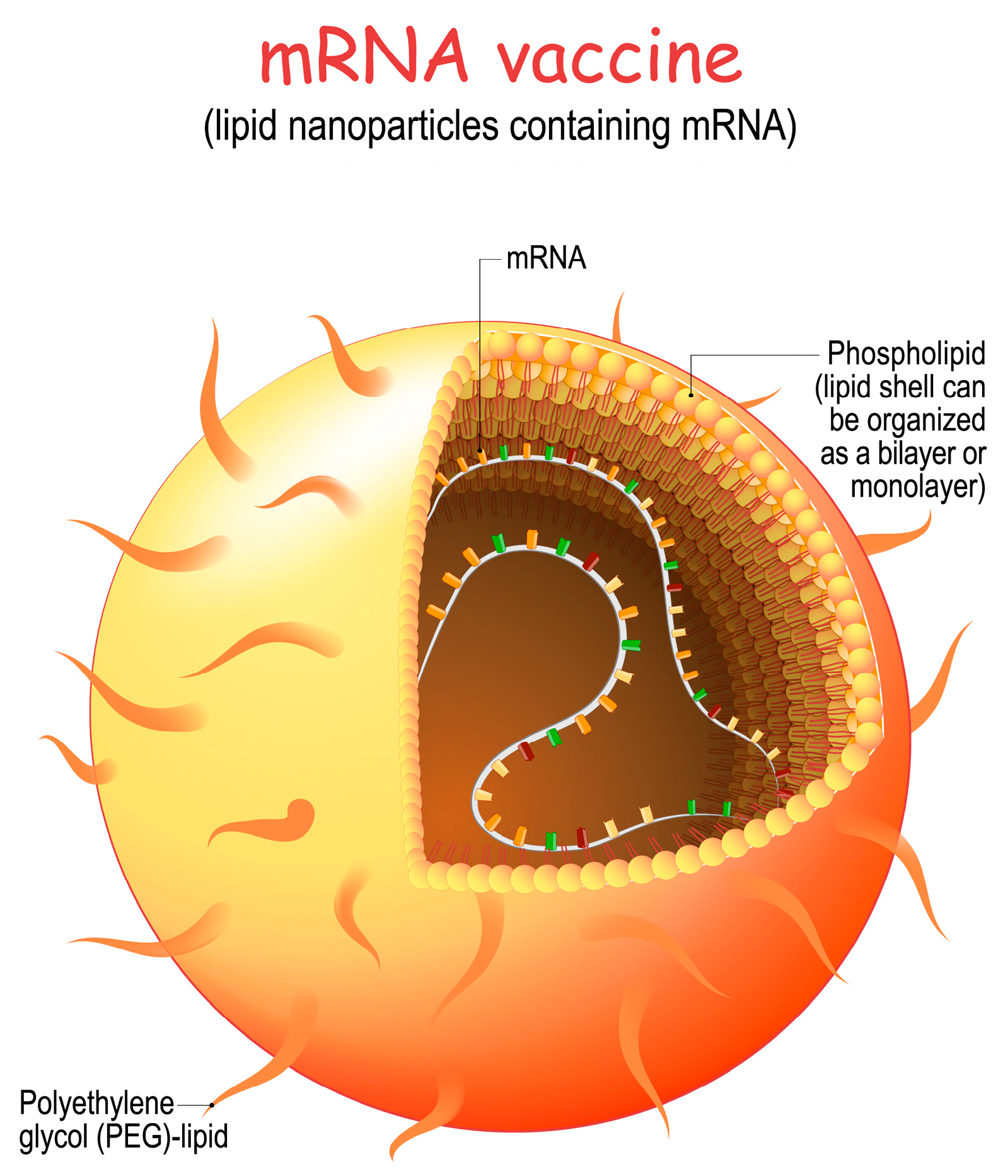
5. mRNA Delivery
- Lipid Selection: Lipids are carefully selected to form the core structure of the nanoparticle. These lipids are chosen for their ability to self-assemble into nanoparticles and to protect the mRNA from degradation.
- Encapsulation of mRNA: The synthetic mRNA encoding the target viral protein is mixed with the selected lipids. This mixture is then subjected to microfluidization or homogenization, which helps encapsulate the mRNA within the lipid nanoparticles.
- Surface Modifications: The surface of the lipid nanoparticles can be modified with polyethylene glycol (PEG) or other molecules to improve stability, reduce clearance by the immune system, and enhance cellular uptake.
- Stability and Sterilization: The LNP formulation is rigorously tested to ensure the mRNA remains intact during storage and transportation. The formulation is also sterilized to ensure that it is free from contaminants.
- Cellular Uptake: After vaccination, the LNPs are injected into the body, where they encounter host cells at the injection site. These LNPs are taken up by antigen-presenting cells such as dendritic cells and macrophages.
- Translation of mRNA: Once inside the host cells, the mRNA is released from the LNPs and enters the cytoplasm. The cell’s ribosomes then use the mRNA as a template to synthesize the target viral protein, such as the spike protein of the SARS-CoV-2 virus.
- Immune Response: The newly synthesized viral protein is displayed on the cell’s surface, triggering an immune response. This includes the production of antibodies and activating T cells, which recognize and remember the viral protein.
- Immune Memory: The immune system “learns” to recognize the viral protein, allowing it to mount a rapid and effective immune response if the actual virus is encountered in the future.
5.1. Cell-Based Delivery
5.2. Extracellular Vesicles
5.3. Biomimetic Delivery
5.4. Tissue Targeting
6. Challenges of mRNA Therapeutics
7. Cell-Free Protein Synthesis (CFPS)
8. Regulatory
9. Intellectual Property Issues
10. Advantages of mRNA Technology
- Innate Translation: Proteins translated by ribosomes inside the cell are innate and almost impossible to replicate in any ex vivo system; this includes the protein structure and the post-translational modification that are the most significant hurdles in manufacturing therapeutic proteins.
- Fast-to-Market: mRNA-based methods for manufacturing therapeutic proteins present a more expeditious and effective alternative than recombinant technology or cell-free protein synthesis (CFPS). The conventional process of recombination technology necessitates the cloning and subsequent expression of genetic material within host cells, which is both intricate and time-intensive. On the other hand, mRNA technology entails artificially generating mRNA molecules outside of the cellular environment and subsequently introducing them into cells to facilitate the translation process. Therefore, the process of mRNA transfection is characterized by enhanced speed and efficacy. mRNA is transported directly to the cytoplasm and undergoes expression therein. It possesses a smaller molecular size compared to plasmid DNA and does not traverse the nuclear membrane. This methodology facilitates mRNA’s effective and scalable production, making it an appealing option for synthesizing therapeutic proteins defined by accelerated development timelines. The production of therapeutic proteins necessitates utilizing cell culture techniques and implementing purification procedures that are particular to the protein of interest and are both labor-intensive and time-consuming.
- Multiple Applications: When considering recombinant technology and in vitro translation, it can be shown that mRNA technology exhibits greater flexibility and adaptability. Introducing or eliminating specific sequences within mRNA molecules makes it readily feasible to generate a diverse range of therapeutic proteins possessing distinct attributes. Due to its inherent versatility, mRNA technology presents a favorable option for synthesizing intricate proteins, especially those that pose difficulties in expression via recombinant methodologies. Moreover, the utilization of mRNA technology presents a versatile framework to produce therapeutic proteins, as it offers a straightforward means of customization to facilitate the synthesis of novel proteins following evolving medical requirements.
- Less Contamination Hazard: mRNA technology does not require living cells, unlike recombinant technology, which uses genetically modified organisms (GMOs) to produce proteins. As a result, there is less chance that manufacturing systems using cells may become contaminated with undesirable or hazardous chemicals, such as endotoxins or adventitious agents. This reduces safety and regulatory compliance concerns, making mRNA technology safer for manufacturing therapeutic proteins.
- Lowest Cost of Good (COGs): mRNA technology can potentially decrease the financial costs related to the manufacturing of therapeutic proteins compared to recombinant technology or in vitro translation methods. The standard approach to recombination technology sometimes requires additional processing steps, such as protein purification and refolding, which can result in substantial costs and time requirements. In contrast, utilizing mRNA technology eliminates the need for labor-intensive methods, as proteins are generated directly from mRNA molecules within biological entities. Irrespective of the coding sequence, mRNA synthesis is accomplished via a consistent methodology within a conventional single-container process [108]. Generating synthetic mRNA through mRNA technology allows for producing molecules that mimic the composition of naturally occurring cytoplasmic molecules. This enables the transitory delivery of specific proteins into cells [109]. This leads to a more economical manufacturing process with lower manufacturing costs.
- Better Safety: The utilization of mRNA technology presents a heightened level of safety in comparison to traditional recombinant or in vitro translation methods. Due to mRNA molecules’ non-infectious nature and inability to integrate into the host genome, the likelihood of insertional mutagenesis or unintended genetic modifications is reduced. Additionally, the precise control of therapeutic protein expression made possible by mRNA technology lowers the possibility of overexpression or off-target effects. Inadequately changed or misfolded proteins can cause problems and trigger the immune system. Plasmid DNA transfection is less effective in inactive cells because a specific promoter is required, and penetrating the nuclear membrane is difficult [110]. Because of this, producing therapeutic proteins using mRNA has fewer security risks.
- Scaling Options: When compared to in vitro translation techniques, mRNA technology has advantages. The availability of suitable cell-free systems can place restrictions on in vitro translation procedures and result in reduced yields. On the other hand, mRNA technology may be scaled up to meet higher production demands through producing and introducing more mRNA into the cells. The intricacy of protein synthesis usually requires long development cycles, making GMP compliance challenging. Because of its scalability, mRNA technology can commercially produce therapeutic proteins on a big scale.
11. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cohen, S.N.; Chang, A.C.; Boyer, H.W.; Helling, R.B. Construction of biologically functional bacterial plasmids in vitro. Proc. Natl. Acad. Sci. USA 1973, 70, 3240–3244. [Google Scholar] [CrossRef] [PubMed]
- FDA-Licensed Biological Products. Inxight Drugs. National Center for Advancing Translational Sciences. Available online: https://drugs.ncats.io/ (accessed on 9 August 2023).
- Crick, F.H. On protein synthesis. Symp. Soc. Exp. Biol. 1958, 12, 138–163. [Google Scholar] [PubMed]
- Brenner, S.; Jacob, F.; Meselson, M. An unstable intermediate carrying information from genes to ribosomes for protein synthesis. Nature 1961, 190, 576–581. [Google Scholar] [CrossRef]
- Wayment-Steele, H.K.; Kim, D.S.; Choe, C.A. Theoretical Basis for Stabilizing Messenger RNA through Secondary Structure Design. Nucleic Acids Res. 2021, 49, 10604–10617. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Patel, A.K.; Kauffman, K.J.; Fenton, O.S.; Webber, M.J.; Anderson, D.G. Polymer–lipid nanoparticles for systemic delivery of mRNA to the lungs. Angew. Chem. Int. Ed. 2016, 55, 13808–13812. [Google Scholar] [CrossRef]
- Tang, D.C.; DeVit, M.; Johnston, S.A. Genetic immunization is a simple method for eliciting an immune response. Nature 1992, 356, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.A.; Ulmer, J.B. Human clinical trials of plasmid DNA vaccines. Adv. Genet. 2005, 55, 25–40. [Google Scholar]
- Gurdon, J.B.; Lane, C.D.; Woodland, H.R.; Marbaix, G. Use of frog eggs and oocytes for the study of messenger RNA and its translation in living cells. Nature 1971, 233, 177–182. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.G.; Lévy, J.P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef]
- FDA COVID-19 Vaccine Approval. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-covid-19-vaccine (accessed on 12 September 2023).
- Yadav, T.; Kumar, S.; Mishra, G.; Saxena, S.K. Tracking the COVID-19 vaccines: The global landscape. Hum. Vaccines Immunother. 2023, 19, 2191577. [Google Scholar] [CrossRef] [PubMed]
- FDA COVID-19 Vaccine Preparedness. Available online: https://www.fda.gov/emergency-preparedness-and-response/counterterrorism-and-emerging-threats/coronavirus-disease-2019-covid-19 (accessed on 1 September 2023).
- Vaccine Tracker. Available online: https://covid19.trackvaccines.org (accessed on 1 September 2023).
- WHO COVID-19 Vaccine Landscape. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed on 1 September 2023).
- WHO Covid Dashboard. Available online: https://covid19.who.int (accessed on 1 September 2023).
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N(1)-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control Release 2015, 217, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Svitkin, Y.V.; Cheng, Y.M.; Chakraborty, T.; Presnyak, V.; John, M.; Sonenberg, N. N1-methyl-pseudouridine in mRNA enhances translation through eIF2α-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res. 2017, 45, 6023–6036. [Google Scholar] [CrossRef]
- Nobel Prize 2023 Medicine or Physiology. Available online: https://www.nobelprize.org/prizes/medicine/2023/press-release/ (accessed on 1 September 2023).
- Qu, L.; Yi, Z.; Shen, Y.; Lin, L.; Chen, F.; Xu, Y.; Wu, Z.; Tang, H.; Zhang, X.; Tian, F.; et al. Circular RNA vaccines against SARS-CoV-2 and emerging variants. Cell 2022, 185, 1728–1744.e16. [Google Scholar] [CrossRef] [PubMed]
- Erasmus, J.H.; Khandhar, A.P.; O’Connor, M.A.; Walls, A.C.; Hemann, E.A.; Murapa, P.; Archer, J.; Leventhal, S.; Fuller, J.T.; Lewis, T.B.; et al. An Alphavirus-derived replicon RNA vaccine induces SARS-CoV-2 neutralizing antibody and T cell responses in mice and nonhuman primates. Sci. Transl. Med. 2020, 12, eabc9396. [Google Scholar] [CrossRef]
- Lee, J.; Woodruff, M.C.; Kim, E.H.; Nam, J.H. Knife’s edge: Balancing immunogenicity and reactogenicity in mRNA vaccines. Exp. Mol. Med. 2023, 55, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Jirikowski, G.F.; Sanna, P.P.; Maciejewski-Lenoir, D.; Bloom, F.E. Reversal of diabetes insipidus in Brattleboro rats: Intrahypothalamic injection of vasopressin mRNA. Science 1992, 255, 996–998. [Google Scholar] [CrossRef]
- Rojas, L.A.; Sethna, Z.; Soares, K.C.; Olcese, C.; Pang, N.; Patterson, E.; Lihm, J.; Ceglia, N.; Guasp, P.; Chu, A.; et al. Personalized RNA neoantigen vaccines stimulate T cells in pancreatic cancer. Nature 2023, 618, 144–150. [Google Scholar] [CrossRef]
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017. [Google Scholar] [CrossRef]
- Barbier, A.J.; Jiang, A.Y.; Zhang, P.; Wooster, R.; Anderson, D.G. The clinical progress of mRNA vaccines and immunotherapies. Nat. Biotechnol. 2022, 40, 840–854. [Google Scholar] [CrossRef]
- Liu, C.; Shi, Q.; Huang, X.; Koo, S.; Kong, N.; Tao, W. mRNA-based cancer therapeutics. Nat. Rev. Cancer 2023, 23, 526–543. [Google Scholar] [CrossRef] [PubMed]
- Pubmed Search. Available online: https://pubmed.ncbi.nlm.nih.gov/?term=mrna+vaccine&filter=datesearch.y_5&sort=pubdate (accessed on 1 September 2023).
- Musa, H.H.; Musa, T.H. A systematic and thematic analysis of the top 100 cited articles on mRNA vaccine indexed in Scopus database. Hum. Vaccines Immunother. 2022, 18, 2135927. [Google Scholar] [CrossRef] [PubMed]
- Davis, F.F.; Allen, F.W. Ribonucleic acids from yeast which contain a fifth nucleotide. J. Biol. Chem. 1957, 227, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.C.; Keck, J.G. The clustering of ribosomes on homogeneous mRNA in the erythrocyte cell-free system. J. Biol. Chem. 1982, 257, 4259–4264. [Google Scholar]
- Bloom, K.; van den Berg, F.; Arbuthnot, P. Self-amplifying RNA vaccines for infectious diseases. Gene Ther. 2021, 28, 117–129. [Google Scholar] [CrossRef]
- Li, M.; Wang, Z.; Xie, C.; Xia, X. Advances in mRNA vaccines. Int. Rev. Cell Mol. Biol. 2022, 372, 295–316. [Google Scholar]
- US Licensed Biological Products. Inxight Drug Database. Available online: https://drugs.ncats.io/substances?facet=Development%20Status%2FUS%20Approved%20Rx&facet=Substance%20Class%2Fprotein&facet=Substance%20Form%2FPrincipal%20Form&page=1 (accessed on 25 April 2023).
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef]
- Varki, A.; Cummings, R.D.; Esko, J.D.; Stanley, P.; Hart, G.W.; Aebi, M.; Darvill, A.G.; Kinoshita, T.; Packer, N.H.; Prestegard, J.H.; et al. Essentials of Glycobiology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009. [Google Scholar]
- Kouzarides, T. Acetylation: A Regul. Modif. Rival Phosphorylation? EMBO J. 2000, 19, 1176–1179. [Google Scholar] [CrossRef]
- Martin, C.; Zhang, Y. The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol. 2005, 6, 838–849. [Google Scholar] [CrossRef]
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2004, 1695, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef]
- Bergo, M.O.; Leung, G.K.; Ambroziak, P.; Otto, J.C.; Casey, P.J.; Young, S.G. Targeted inactivation of the isoprenylcysteine carboxyl methyltransferase gene causes mislocalization of K-Ras in mammalian cells. J. Biol. Chem. 2001, 276, 9673–9677. [Google Scholar] [CrossRef]
- Steiner, D.F.; Cunningham, D.; Spigelman, L.; Aten, B. The Metabolic and Molecular Bases of Inherited Disease; McGraw-Hill Professional: New York, NY, USA, 2002; pp. 962–1009. [Google Scholar]
- Turnbull, J.E.; Field, R.A.; Brown, J.R. Aliphatic hydroxylation of tyrosine residues in fibrinogen and fibrin: Evidence for the formation of 3, 4-dihydroxyphenylalanine. Biochem. J. 2001, 356, 13–16. [Google Scholar]
- Hottiger, M.O.; Hassa, P.O.; Lüscher, B.; Schüler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Hunter, T. The age of crosstalk: Phosphorylation, ubiquitination, and beyond. Mol. Cell 2007, 28, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Walsh, G.; Jefferis, R. Post-translational modifications in the context of therapeutic proteins. Nat. Biotechnol. 2006, 24, 1241–1252. [Google Scholar] [CrossRef]
- Imperiali, B.; O’Connor, S.E. Effect of N-linked glycosylation on glycopeptide and glycoprotein structure. Curr. Opin. Chem. Biol. 1999, 3, 643–649. [Google Scholar] [CrossRef]
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Damase, T.R.; Sukhovershin, R.; Boada, C.; Taraballi, F.; Pettigrew, R.I.; Cooke, J.P. The Limitless Future of RNA Therapeutics. Front. Bioeng. Biotechnol. 2021, 9, 62813. [Google Scholar] [CrossRef]
- Stepinski, J.; Waddell, C.; Stolarski, R.; Darzynkiewicz, E.; Rhoads, R.E. Synthesis and properties of mRNAs containing the novel “anti-reverse” cap analogs 7-methyl(3′-O-methyl)GpppG and 7-methyl (3′-deoxy)GpppG. RNA 2001, 7, 1486–1495. [Google Scholar]
- mRNA Vaccines. USP. Available online: https://www.usp.org/mrna (accessed on 15 August 2023).
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7, 319–334. [Google Scholar] [CrossRef]
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA Vaccines for Infectious Diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid nanoparticles for mRNA delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Zhang, Y.; Huang, L. mRNA vaccine for cancer immunotherapy. Mol. Cancer 2021, 20, 41. [Google Scholar] [CrossRef] [PubMed]
- Heida, R.; Hinrichs, W.L.; Frijlink, H.W. Inhaled vaccine delivery in the combat against respiratory viruses: A 2021 overview of recent developments and implications for COVID-19. Expert Rev. Vaccines 2021, 21, 957–974. [Google Scholar] [CrossRef]
- Silva-Sanchez, A.; Randall, T.D. Role of iBALT in Respiratory Immunity. In Inducible Lymphoid Organs; Kabashima, K., Egawa, G., Eds.; Springer International Publishing: Cham, Switzerland, 2020; pp. 21–43. [Google Scholar]
- Nitika, J.W.; Hui, A.M. The Delivery of mRNA Vaccines for Therapeutics. Life 2022, 12, 1254. [Google Scholar] [CrossRef]
- Dimitriadis, G.J. Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature 1978, 274, 923–924. [Google Scholar] [CrossRef]
- Ostro, M.J.; Giacomoni, D.; Lavelle, D.; Paxton, W.; Dray, S. Evidence for translation of rabbit globin mRNA after liposome-mediated insertion into a human cell line. Nature 1978, 274, 921–923. [Google Scholar] [CrossRef]
- Schroeder, A.; Levins, C.G.; Cortez, C.; Langer, R.; Anderson, D.G. Lipid-based nanotherapeutics for siRNA delivery. J. Intern. Med. 2010, 267, 9–21. [Google Scholar] [CrossRef]
- Malone, R.W.; Felgner, P.L.; Verma, I.M. Cationic liposome-mediated RNA transfection. Proc. Natl. Acad. Sci. USA 1989, 86, 6077–6081. [Google Scholar] [CrossRef]
- Jeffs, L.B.; Palmer, L.R.; Ambegia, E.G.; Giesbrecht, C.; Ewanick, S.; MacLachlan, I. A scalable, extrusion-free method for efficient liposomal encapsulation of plasmid DNA. Pharm. Res. 2005, 22, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Ganta, S.; Talekar, M.; Singh, A.; Coleman, T.P.; Amiji, M.M. Nanoemulsions in translational research-opportunities and challenges in targeted cancer therapy. AAPS PharmSciTech 2014, 15, 694–708. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M.P.; Langer, R.; Jensen, K.F. Intracellular Delivery by Membrane Disruption: Mechanisms, Strategies, and Concepts. Chem. Rev. 2018, 118, 7409–7531. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.L.; Drane, D.; Gowans, E.J. Long-term storage of DNA-free RNA for use in vaccine studies. Biotechniques 2007, 43, 675–681. [Google Scholar] [CrossRef]
- Singer, D.F.; Linderman, J.J. The relationship between antigen concentration, antigen internalization, and antigenic complexes: Modeling insights into antigen processing and presentation. J. Cell Biol. 1990, 111, 55–68. [Google Scholar] [CrossRef]
- Mbongue, J.; Nicholas, D.; Firek, A.; Langridge, W. The role of dendritic cells in tissue-specific autoimmunity. J. Immunol. Res. 2014, 2014, 857143. [Google Scholar] [CrossRef]
- Garg, A.D.; Coulie, P.G.; Van den Eynde, B.J.; Agostinis, P. Integrating Next-Generation Dendritic Cell Vaccines into the Current Cancer Immunotherapy Landscape. Trends Immunol. 2017, 38, 577–593. [Google Scholar] [CrossRef]
- Batich, K.A.; Reap, E.A.; Archer, G.E.; Sanchez-Perez, L.; Nair, S.K.; Schmittling, R.J.; Norberg, P.; Xie, W.; Herndon, J.E., II; Healy, P.; et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin. Cancer Res. 2017, 23, 1898–1909. [Google Scholar] [CrossRef]
- Grau, M.; Walker, P.R.; Derouazi, M. Mechanistic insights into the efficacy of cell penetrating peptide-based cancer vaccines. Cell. Mol. Life Sci. 2018, 75, 2887–2896. [Google Scholar] [CrossRef] [PubMed]
- Stitz, L.; Vogel, A.; Schnee, M.; Voss, D.; Rauch, S.; Mutzke, T.; Ketterer, T.; Kramps, T.; Petsch, B. A thermostable messenger RNA based vaccine against rabies. PLoS Negl. Trop. Dis. 2017, 11, e0006108. [Google Scholar] [CrossRef] [PubMed]
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkic-Zrna, S.; Probst, J.; Kallen, K.J. Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J. Immunother. 2011, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Calvo, Á.; Saiz, J.-C.; McCullough, K.C.; Sobrino, F.; Martín-Acebes, M.A. Acid-dependent viral entry. Virus Res. 2012, 167, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Replicon RNA Viral Vectors as Vaccines. Vaccines 2016, 4, 39. [Google Scholar] [CrossRef]
- Grankvist, R.; Jensen-Urstad, M.; Clarke, J.; Lehtinen, M.; Little, P.; Lundberg, J.; Arnberg, F.; Jonsson, S.; Chien, K.R.; Holmin, S. Superselective endovascular tissue access using trans-vessel wall technique: Feasibility study for treatment applications in heart, pancreas and kidney in swine. J. Intern. Med. 2019, 285, 398–406. [Google Scholar] [CrossRef]
- Mali, S. Delivery systems for gene therapy. Indian J. Hum. Genet. 2013, 19, 3–8. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines—A new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Jackson, N.A.C.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. NPJ Vaccines 2020, 5, 11. [Google Scholar] [CrossRef]
- Le, T.T.; Andreadakis, Z.; Kumar, A.; Román, R.G.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306. [Google Scholar] [CrossRef]
- Larson, H.J.; Jarrett, C.; Eckersberger, E.; Smith DM, D.; Paterson, P. Understanding vaccine hesitancy around vaccines and vaccination from a global perspective: A systematic review of published literature, 2007–2012. Vaccine 2014, 32, 2150–2159. [Google Scholar] [CrossRef]
- Lurie, N.; Saville, M.; Hatchett, R.; Halton, J. Developing Covid-19 Vaccines at Pandemic Speed. N. Engl. J. Med. 2020, 382, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford KH, D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.; Álvarez-Vallina, L. Engineered mRNA and the Rise of Next-Generation Antibodies. Antibodies 2021, 10, 37. [Google Scholar] [CrossRef] [PubMed]
- Vlatkovic, I. Non-immunotherapy application of LNP-mRNA: Maximizing efficacy and safety. Biomedicines 2021, 9, 530. [Google Scholar] [CrossRef]
- Cafuir, L.A.; Kempton, C.L. Current and emerging factor VIII replacement products for hemophilia A. Ther. Adv. Hematol. 2017, 8, 303–313. [Google Scholar] [CrossRef]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-engineered mRNA without chemical nucleoside modifications enables an effective protein therapy in large animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef]
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 2017, 8, 14630. [Google Scholar] [CrossRef]
- Pardi, N.; Muramatsu, H.; Weissman, D.; Karikó, K. In vitro transcription of long RNA containing modified nucleosides. Methods Mol. Biol. 2013, 969, 29–42. [Google Scholar]
- Kallen, K.-J.; Theß, A. A Development That May Evolve into a Revolution in Medicine: MRNA as the Basis for Novel, Nucleotide Based Vaccines and Drugs. Ther. Adv. Vaccines 2014, 2, 10–31. [Google Scholar] [CrossRef]
- mRNA Production Reinvented. Quantoom Biosciences. Available online: https://quantoom.com/ (accessed on 10 April 2023).
- Zahnd, C.; Amstutz, P.; Plückthu, A. Ribosome display: Selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 2007, 4, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Ginsburg, J. Deciphering the Genetic Code: A National Historic Chemical Landmark. American Chemical Society. 2009. Available online: https://www.acs.org/content/acs/en/education/whatischemistry/landmarks/geneticcode.html#poly-u-experiment (accessed on 1 September 2023).
- Chong, S. Overview of Cell-Free Protein Synthesis: Historic Landmarks, Commercial Systems, and Expanding Applications. Curr. Protoc. Mol. Biol. 2014, 108, 16–30. [Google Scholar] [CrossRef] [PubMed]
- He, M. Cell-free protein synthesis: Applications in proteomics and biotechnology. New Biotech. 2008, 25, 126–132. [Google Scholar] [CrossRef]
- Carlson, E.D.; Gan, R.; Hodgman, C.E.; Jewett, M.C. Cell-Free Protein Synthesis: Applications Come of Age. Biotechnol. Adv. 2012, 30, 1185–1194. [Google Scholar] [CrossRef]
- Frey, S.; Haslbeck, M.; Hainzl, O.; Buchner, J. Synthesis and characterization of a functional intact IgG in a prokaryotic cell-free expression system. Biol. Chem. 2007, 389, 37–45. [Google Scholar] [CrossRef]
- Perez, J.G.; Stark, J.C.; Jewett, M.C. Cell-Free Synthetic Biology: Engineering Beyond the Cell. Cold Spring Harb. Perspect. Biol. 2016, 8, a023853. [Google Scholar] [CrossRef] [PubMed]
- Lambrinakos-Raymond, K.; Robillard, N. Regulatory considerations for the development of mRNA vaccines. Curr. Opin. Virol. 2019, 36, 100–107. [Google Scholar]
- Ich Harmonised Tripartite Guideline. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. 1999. Available online: https://www.gmp-compliance.org/files/guidemgr/3-1-16.pdf (accessed on 1 September 2023).
- Guerriaud, M.; Kohli, E. RNA-based drugs and regulation: Toward a necessary evolution of the definitions issued from the European union legislation. Front. Med. 2022, 9, 1012497. [Google Scholar] [CrossRef]
- FDA Report: Guidance for Human Somatic Cell Therapy and Gene Therapy. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/guidance-human-somatic-cell-therapy-and-gene-therapy (accessed on 12 September 2023).
- Congressional Research Service. Follow-on Biologics. Available online: https://www2.law.umaryland.edu/marshall/crsreports/crsdocuments/R41483_01242012.pdf (accessed on 12 September 2023).
- Rai, A.K.; Eisenberg, R.S. Bayh-Dole Reform and the Progress of Biomedicine. Law Contemp. Probl. 2003, 66, 289–314. [Google Scholar]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.J. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—Developing a new class of drugs. Nat Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Zhang, S.; Fu, R.; Zhang, L.; Huang, K.; Peng, H.; Dai, L.; Chen, Q. Therapeutic Prospects of mRNA-Based Gene Therapy for Glioblastoma. Front. Oncol. 2019, 9, 1208. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.genecards.org (accessed on 1 September 2023).
- Dai, C.; Füllgrabe, A.; Pfeuffer, J.; Solovyeva, E.M.; Deng, J.; Moreno, P.; Kamatchinathan, S.; Kundu, D.J.; George, N.; Fexova, S.; et al. A proteomics sample metadata representation for multiomics integration and big data analysis. Nat. Commun. 2021, 12, 5854. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5′UTR cap |
| GAGAATAAACTAGTATTCTTCTGGTCCCCACAGACTCAGAGAGAACCCGCCACATGTTCGTGTTCCTGGTGCTGCTGCCTCTGGTGTCCA |
| Start codon (Kozak) |
| GCAGCCAGTGCGTGAACCTGACCACCCGGACCCAGCTGCCACCAGCCTACACCAACAGCTTCACCCGGGGCGTCTACTACCCCGACAAGGT |
| Coding Sequence (CDS) |
| UGGCGGGCCCGGCGACCCAGAGCCCGAUGAAACUGAUGGCGCUGCAGCUGCUGCUGUGGCAUAGCGCGCUGUGGACCGUGCAGGAAGCGACCCCGCUGGGCCCGGCGAGCAGCCUGCCGCAGAGCUUUCUGCUGAAAUGCCUGGAACAGGUGCGCAAAAUUCAGGGCGAUGGCGCGGCGCUGCAGGAAAAACUGGUGAGCGAAUGCGCGACCUAUAAACUGUGCCAUCCGGAAGAACUGGUGCUGCUGGGCCAUAGCCUGGGCAUUCCGUGGGCGCCGCUGAGCAGCUGCCCGAGCCAGGCGCUGCAGCUGGCGGGCUGCCUGAGCCAGCUGCAUAGCGGCCUGUUUCUGUAUCAGGGCCUGCUGCAGGCGCUGGAAGGCAUUAGCCCGGAACUGGGCCCGACCCUGGAUACCCUGCAGCUGGAUGUGGCGGAUUUUGCGACCACCAUUUGGCAGCAGAUGGAAGAACUGGGCAUGGCGCCGGCGCUGCAGCCGACCCAGGGCGCGAUGCCGGCGUUUGCGAGCGCGUUUCAGCGCCGCGCGGGCGGCGUGCUGGUGGCGAGCCAUCUGCAGAGCUUUCUGGAAGUGAGCUAUCGCGUGCUGCGCCAUCUGGCGCAGCCG |
| 3′UTR |
| GCCCCTTTCCCGTCCTGGGTACCCCGAGTCTCCCCCGACCTCGGGTCCCAGGTATGCTCCCACCTCCACCTGCCCCACTCACCACCTCTGCTAGTTCCAGACACCTCCCAAGCACGCAGCAATGCAGCTCAAAACGCTTAGCCTAGCCACACCCCCACGGGAAACAGCAGTGATAACCTTTAGCAATAAACGAAAGTTTAACTAAGCTATACTAACCCCAGGGTTGGTCAATTTCGTGCCAGCCACACCCTGGAGCTAGCA |
| poly-A chain |
| Therapeutic Protein | Numbers |
|---|---|
| Monoclonal antibody | 94 |
| Hormone | 10 |
| Enzyme | 8 |
| Cytokine | 4 |
| Bispecific antibody | 3 |
| Coagulation factor | 3 |
| Growth factor | 3 |
| Peptide | 3 |
| Carrier protein | 1 |
| Enzyme inhibitor | 1 |
| Fab | 1 |
| Fusion proteins | 1 |
| Single-domain antibody | 1 |
| Toxin | 1 |
| Protein | Brand Name (Product) |
|---|---|
| Erythropoietin (EPO) | Epogen, Procrit (Epoetin alfa) |
| Insulin | Humalog, NovoLog (Insulin lispro); Lantus, Levemir (Insulin glargine) |
| Factor VIII | Advate, Kogenate FS, Eloctate (Recombinant factor VIII) |
| Granulocyte-Colony Stimulating Factor (G-CSF) | Neupogen, Neulasta (Filgrastim, Pegfilgrastim) |
| Tissue Plasminogen Activator (tPA) | Activase, Cathflo Activase (Alteplase) |
| Growth Hormones | Genotropin, Humatrope, Norditropin (Somatropin) |
| Interleukins | Proleukin (Aldesleukin) |
| Monoclonal Antibodies | Herceptin (Trastuzumab); Rituxan (Rituximab); Humira (Adalimumab) |
| Enzyme Replacement Therapies | Cerezyme (Imiglucerase); Fabrazyme (Agalsidase beta) |
| Hormone Replacement Therapies | Premarin (Conjugated estrogens); AndroGel (Testosterone) |
| Therapeutic Protein | Use |
|---|---|
| Abatacept (Orencia) | Rheumatoid arthritis and juvenile idiopathic arthritis |
| Abciximab (ReoPro) | Prevention of platelet aggregation in angioplasty |
| Acalabrutinib (Calquence) | Mantle cell lymphoma and chronic lymphocytic leukemia |
| Adalimumab (Humira) | Various autoimmune disorders |
| Aducanumab (Aduhelm) | Alzheimer’s disease (monoclonal antibody) |
| Aflibercept (Eylea) | Wet age-related macular degeneration |
| Alemtuzumab (Campath) | Chronic lymphocytic leukemia |
| Alpelisib (Piqray) | Breast cancer with PIK3CA mutations |
| Alteplase (Activase) | Thrombolytic drug for acute myocardial infarction, acute ischemic stroke, and PE |
| Anakinra (Kineret) | Rheumatoid arthritis |
| Atezolizumab (Tecentriq) | Various types of cancer |
| Avelumab (Bavencio) | Various types of cancer |
| Basiliximab (Simulect) | Prevention of organ rejection in transplantation |
| Belatacept (Nulojix) | Immunosuppressive therapy for kidney transplantation |
| Belimumab (Benlysta) | Systemic lupus erythematosus |
| Bevacizumab (Avastin) | Various cancers by inhibiting angiogenesis |
| Bivalirudin (Angiomax) | Anticoagulant for patients undergoing percutaneous coronary intervention |
| Blinatumomab (Blincyto) | Acute lymphoblastic leukemia |
| Cabotegravir (Vocabria) | Long-acting HIV-1 integrase inhibitor for prevention |
| Cabozantinib (Cometriq) | Advanced renal cell carcinoma and hepatocellular carcinoma |
| Certolizumab pegol (Cimzia) | Crohn’s disease, rheumatoid arthritis |
| Cetuximab (Erbitux) | Metastatic colorectal cancer, head and neck cancer |
| Daclizumab (Zinbryta) | Multiple sclerosis |
| Daptomycin (Cubicin) | Antibiotic for complicated skin and skin structure infections |
| Daratumumab (Darzalex) | Multiple myeloma |
| Darolutamide (Nubeqa) | Prostate cancer |
| Denosumab (Prolia, Xgeva) | Osteoporosis, prevention of skeletal-related events in cancer patients |
| Dulaglutide (Trulicity) | Type 2 diabetes |
| Durvalumab (Imfinzi) | Various types of cancer |
| Eculizumab (Soliris) | Paroxysmal nocturnal hemoglobinuria, atypical hemolytic uremic syndrome |
| Efalizumab (Raptiva) | Psoriasis |
| Elotuzumab (Empliciti) | Multiple myeloma |
| Emapalumab (Gamifant) | Hemophagocytic lymphohistiocytosis (HLH) |
| Eptifibatide (Integrilin) | Antiplatelet drug for acute coronary syndrome |
| Erdafitinib (Balversa) | Urothelial cancer with FGFR mutations |
| Eteplirsen (Exondys 51) | Duchenne muscular dystrophy |
| Fingolimod (Gilenya) | Multiple sclerosis |
| Golimumab (Simponi) | Rheumatoid arthritis, psoriatic arthritis, ankylosing spondylitis |
| Golodirsen (Vyondys 53) | Duchenne muscular dystrophy |
| Guselkumab (Tremfya) | Moderate-to-severe plaque psoriasis |
| Gusperimus (Zavesca) | Gaucher disease and Niemann-Pick disease type C |
| Ibalizumab (Trogarzo) | Multidrug-resistant HIV-1 |
| Icatibant (Firazyr) | Hereditary angioedema attacks |
| Inclisiran (Leqvio) | Hypercholesterolemia |
| Infliximab (Remicade) | Autoimmune diseases like Crohn’s and rheumatoid arthritis |
| Ipilimumab (Yervoy) | Melanoma |
| Isatuximab (Sarclisa) | Multiple myeloma |
| Lanadelumab (Takhzyro) | Prevention of hereditary angioedema attacks |
| Laronidase (Aldurazyme) | Enzyme replacement therapy for Hurler syndrome |
| Lenalidomide (Revlimid) | Multiple myeloma and myelodysplastic syndromes |
| Liraglutide (Victoza) | Type 2 diabetes |
| Margetuximab (Margenza) | HER2-positive breast cancer |
| Natalizumab (Tysabri) | Multiple sclerosis and Crohn’s disease |
| Naxitamab (Danyelza) | Neuroblastoma in children |
| Nivolumab (Opdivo) | Various types of cancer |
| Nusinersen (Spinraza) | Spinal muscular atrophy |
| Obiltoxaximab (Anthim) | Inhalational anthrax |
| Obinutuzumab (Gazyva) | Chronic lymphocytic leukemia, follicular lymphoma |
| Ofatumumab (Arzerra) | Chronic lymphocytic leukemia and multiple sclerosis |
| Olaratumab (Lartruvo) | Soft tissue sarcoma |
| Omalizumab (Xolair) | Asthma and chronic idiopathic urticaria |
| Palifermin (Kepivance) | Prevention of severe oral mucositis in cancer patients |
| Palivizumab (Synagis) | Prevention of respiratory syncytial virus in premature infants |
| Panitumumab (Vectibix) | Metastatic colorectal cancer |
| Panobinostat (Farydak) | Multiple myeloma |
| Pegaspargase (Oncaspar) | Acute lymphoblastic leukemia |
| Pegloticase (Krystexxa) | Refractory gout |
| Pembrolizumab (Keytruda) | Various types of cancer |
| Pemetrexed (Alimta) | Chemotherapy for non-small cell lung cancer and mesothelioma |
| Pertuzumab (Perjeta) | HER2-positive breast cancer |
| Pexidartinib (Turalio) | Tenosynovial giant cell tumor |
| Plasminogen (Ryplazim) | Congenital plasminogen deficiency |
| Ramucirumab (Cyramza) | Stomach cancer, colorectal cancer, and lung cancer |
| Ranibizumab (Lucentis) | Wet age-related macular degeneration |
| Rituximab (Rituxan) | Non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, rheumatoid arthritis, and vasculitis |
| Rucaparib (Rubraca) | Ovarian cancer with BRCA mutations |
| Secukinumab (Cosentyx) | Psoriasis and ankylosing spondylitis |
| Selinexor (Xpovio) | Multiple myeloma and diffuse large B-cell lymphoma |
| Siltuximab (Sylvant) | Multicentric castleman’s disease |
| Tafasitamab (Monjuvi) | Diffuse large B-cell lymphoma |
| Tildrakizumab (Ilumya) | Moderate-to-severe plaque psoriasis |
| Tislelizumab (Bavencio) | Various types of cancer |
| Tocilizumab (Actemra) | Cytokine release syndrome and rheumatoid arthritis |
| Trastuzumab (Herceptin) | HER2-positive breast cancer |
| Ustekinumab (Stelara) | Psoriasis, psoriatic arthritis, and Crohn’s disease |
| Vedolizumab (Entyvio) | Inflammatory bowel diseases (Crohn’s and UC) |
| Vemurafenib (Zelboraf) | BRAF-mutated melanoma |
| Venetoclax (Venclexta) | Chronic lymphocytic leukemia |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niazi, S.K.; Magoola, M. mRNA and Synthesis-Based Therapeutic Proteins: A Non-Recombinant Affordable Option. Biologics 2023, 3, 355-379. https://doi.org/10.3390/biologics3040020
Niazi SK, Magoola M. mRNA and Synthesis-Based Therapeutic Proteins: A Non-Recombinant Affordable Option. Biologics. 2023; 3(4):355-379. https://doi.org/10.3390/biologics3040020
Chicago/Turabian StyleNiazi, Sarfaraz K., and Matthias Magoola. 2023. "mRNA and Synthesis-Based Therapeutic Proteins: A Non-Recombinant Affordable Option" Biologics 3, no. 4: 355-379. https://doi.org/10.3390/biologics3040020
APA StyleNiazi, S. K., & Magoola, M. (2023). mRNA and Synthesis-Based Therapeutic Proteins: A Non-Recombinant Affordable Option. Biologics, 3(4), 355-379. https://doi.org/10.3390/biologics3040020