
Virus-like Particles (VLPs) as Important Tools for Flavivirus Vaccine Development


Abstract
1. Introduction
2. Viral Vaccine Types
3. Virus-like Particles (VLPs) and Flavivirus-like Particles (FVLPs)
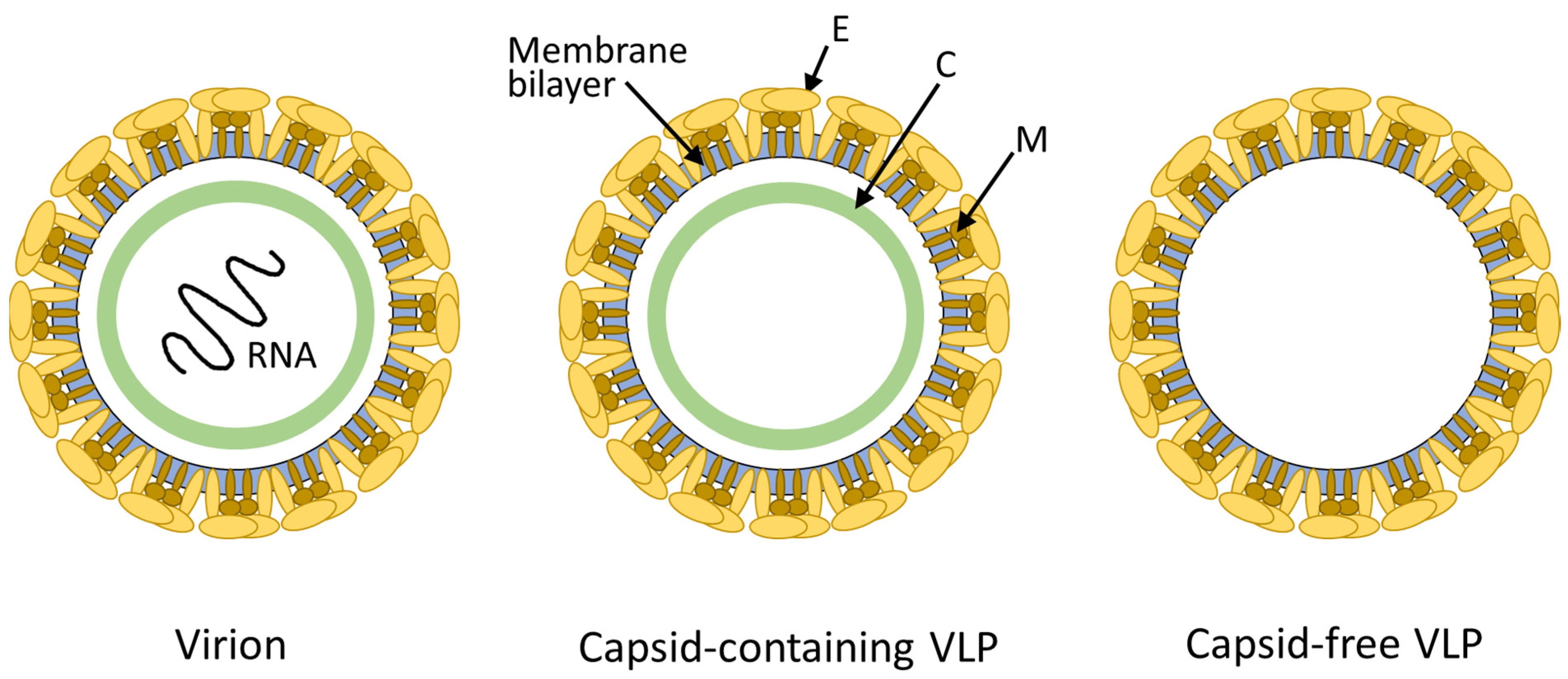
3.1. Molecular Features of FVLPs
3.2. Technologies Proposed for the Production of Flavivirus-like Particles

{kind=link}
| Expression System | Cell Type Used | FVLPs or Particles Containing a Flaviviral Antigen | References |
|---|---|---|---|
| Mammalian cells | Chinese hamster ovary (CHO) cells | DENV | [82] |
| Baby hamster kidney (BHK) cells | JEV | [105] | |
| Human embryonic kidney (HEK293) cells | ZIKV | [70,78,106] | |
| YFV | [68] | ||
| DENV | [107] | ||
| Insect cells | Sf-9 | JEV | [108] |
| High Five | JEV | [108] | |
| Mosquito cells | JEV | [109] | |
| Plant cells | Nicotiana benthamiana | DENV | [110] |
| Microbial cells | E. coli | DENV “virus-sized particles” | [111] |
| P. pastoris | HBsAg *-ZIKV EDIII | [112] | |
| HBsAg *-DENV EDIII | [113] |
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Collins, M.H.; Metz, S.W. Progress and works in progress: Update on flavivirus vaccine development. Clin. Ther. 2017, 39, 1519–1536. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.C.; Diamond, M.S. The continued threat of emerging flaviviruses. Nat. Microbiol. 2020, 5, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Payne, S.L. Family Flaviviridae. In Viruses: From Understanding to Investigation; Payne, S.L., Ed.; Academic Press: Cambridge, MA, USA, 2017; Chapter 15; pp. 129–139. [Google Scholar] [CrossRef]
- Lobigs, M. Flavivirus premembrane protein cleavage and spike heterodimer secretion require the function of the viral proteinase NS3. Proc. Natl. Acad. Sci. USA 1993, 90, 6218–6222. [Google Scholar] [CrossRef] [PubMed]
- Hansen, C.A.; Barrett, A.D.T. The present and future of yellow fever vaccines. Pharmaceuticals 2021, 14, 891. [Google Scholar] [CrossRef]
- Hegde, N.R.; Gore, M.M. Japanese encephalitis vaccines: Immunogenicity, protective efficacy, effectiveness, and impact on the burden of disease. Hum. Vaccin. Immunother. 2017, 13, 1320–1337. [Google Scholar] [CrossRef]
- Prompetchara, E.; Ketloy, C.; Thomas, S.J.; Ruxrungtham, K. Dengue vaccine: Global development update. Asian Pac. J. Allergy Immunol. 2020, 38, 178–185. [Google Scholar] [CrossRef]
- Rendi-Wagner, P. Advances in vaccination against tick-borne encephalitis. Expert Rev. Vaccines 2008, 7, 589–596. [Google Scholar] [CrossRef]
- Shah, S.Z.; Jabbar, B.; Ahmed, N.; Rehman, A.; Nasir, H.; Nadeem, S.; Jabbar, I.; Rahman, Z.U.; Azam, S. Epidemiology, Pathogenesis, and Control of a Tick-Borne Disease-Kyasanur Forest Disease: Current Status and Future Directions. Front. Cell Infect. Microbiol. 2018, 8, 149. [Google Scholar] [CrossRef]
- Montalvo Zurbia-Flores, G.; Rollier, C.S.; Reyes-Sandoval, A. Re-thinking yellow fever vaccines: Fighting old foes with new generation vaccines. Hum. Vaccin. Immunother. 2022, 18, 1895644. [Google Scholar] [CrossRef]
- Roldão, A.; Mellado, M.C.; Castilho, L.R.; Carrondo, M.J.; Alves, P.M. Virus-like particles in vaccine development. Expert Rev. Vaccines 2010, 9, 1149–1176. [Google Scholar] [CrossRef]
- Boigard, H.; Alimova, A.; Martin, G.R.; Katz, A.; Gottlieb, P.; Galarza, J.M. Zika virus-like particle (VLP) based vaccine. PLoS Negl. Trop. Dis. 2017, 11, e0005608. [Google Scholar] [CrossRef] [PubMed]
- Vang, L.; Morello, C.S.; Mendy, J.; Thompson, D.; Manayani, D.; Guenther, B.; Julander, J.; Sanford, D.; Jain, A.; Patel, A.; et al. Zika virus-like particle vaccine protects AG129 mice and rhesus macaques against Zika virus. PLoS Negl. Trop. Dis. 2021, 15, e0009195. [Google Scholar] [CrossRef] [PubMed]
- Hilleman, M.R. Yeast recombinant hepatitis B vaccine. Infection 1987, 15, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Huzair, F.; Sturdy, S. Biotechnology and the transformation of vaccine innovation: The case of the hepatitis B vaccines 1968-2000. Stud. Hist. Philos. Biol. Biomed. Sci. 2017, 64, 11–21. [Google Scholar] [CrossRef]
- Gary, E.N.; Weiner, D.B. DNA vaccines: Prime time is now. Curr Opin Immunol. 2020, 65, 21–27. [Google Scholar] [CrossRef]
- Halstead, S.B.; Thomas, S.J. New Japanese encephalitis vaccines: Alternatives to production in mouse brain. Expert Rev. Vaccines 2011, 10, 355–364. [Google Scholar] [CrossRef]
- Mendonça, S.A.; Lorincz, R.; Boucher, P.; Curiel, D.T. Adenoviral vector vaccine platforms in the SARS-CoV-2 pandemic. NPJ Vaccines 2021, 6, 97. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics--developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Wolf, J.; Jannat, R.; Dubey, S.; Troth, S.; Onorato, M.T.; Coller, B.A.; Hanson, M.E.; Simon, J.K. Development of pandemic vaccines: ERVEBO case study. Vaccines 2021, 9, 190. [Google Scholar] [CrossRef]
- FDA. Vaccines Licensed for Use in the United States. 2022. Available online: https://www.fda.gov/vaccines-blood-biologics/vaccines/vaccines-licensed-use-united-states (accessed on 25 July 2022).
- WHO. Prequalified Vaccines. 2022. Available online: https://extranet.who.int/pqweb/vaccines/prequalified-vaccines (accessed on 25 July 2022).
- EMA. Vaxzevria (Previously COVID-19 Vaccine AstraZeneca). 2022. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/vaxzevria-previously-covid-19-vaccine-astrazeneca (accessed on 25 July 2022).
- Ministry of Science and Technology of India. DBT-BIRAC supported ZyCoV-D developed by Zydus Cadila Receives Emergency Use Authorization. 2021. Available online: https://pib.gov.in/PressReleasePage.aspx?PRID=1747669 (accessed on 25 July 2022).
- Oxford University, Vaccine Knowledge Project. Types of Vaccine. 2021. Available online: https://vk.ovg.ox.ac.uk/vk/types-of-vaccine (accessed on 24 July 2022).
- Cheng, L.; Wang, Y.; Du, J. Human papillomavirus vaccines: An updated review. Vaccines 2020, 8, 391. [Google Scholar] [CrossRef]
- Li, S.W.; Zhao, Q.; Wu, T.; Chen, S.; Zhang, J.; Xia, N.S. The development of a recombinant hepatitis E vaccine HEV 239. Hum. Vaccin. Immunother. 2015, 11, 908–914. [Google Scholar] [CrossRef] [PubMed]
- Cox, M.M.J. Innovations in the insect cell expression system for industrial recombinant vaccine antigen production. Vaccines 2021, 9, 1504. [Google Scholar] [CrossRef]
- EMA. First Malaria Vaccine Receives Positive Scientific Opinion from EMA. 2015. Available online: https://www.ema.europa.eu/en/news/first-malaria-vaccine-receives-positive-scientific-opinion-ema (accessed on 25 July 2022).
- Laurens, M.B. RTS,S/AS01 vaccine (Mosquirix™): An overview. Hum. Vaccin. Immunother. 2020, 16, 480–489. [Google Scholar] [CrossRef] [PubMed]
- WHO. WHO Recommends Groundbreaking Malaria Vaccine for Children at Risk. 2021. Available online: https://www.who.int/news/item/06-10-2021-who-recommends-groundbreaking-malaria-vaccine-for-children-at-risk (accessed on 25 July 2022).
- Bharucha, T.; Ming, D.; Breuer, J. A critical appraisal of ‘Shingrix’, a novel herpes zoster subunit vaccine (HZ/Su or GSK1437173A) for varicella zoster virus. Hum. Vaccin. Immunother. 2017, 13, 1789–1797. [Google Scholar] [CrossRef]
- Estrada, J.A.; Cheng, C.Y.; Ku, S.Y.; Hu, H.C.; Yeh, H.W.; Lin, Y.C.; Chen, C.P.; Cheng, S.H.; Janssen, R.; Lin, I.F. An Immunobridging Study to Evaluate the Neutralizing Antibody Titer in Adults Immunized with Two Doses of Either ChAdOx1-nCov-19 (AstraZeneca) or MVC-COV1901. Vaccines 2022, 10, 655. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: First Approval of the Protein-Based Adjuvanted Nuvaxovid (NVX-CoV2373) Novavax Vaccine for SARS-CoV-2 Could Increase Vaccine Uptake and Provide Immune Protection from Viral Variants. Med. Sci. Monit. 2022, 28, e936523. [Google Scholar] [CrossRef]
- Mohsen, M.O.; Augusto, G.; Bachmann, M.F. The 3Ds in virus-like particle based-vaccines: “Design, Delivery and Dynamics”. Immunol. Rev. 2020, 296, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Draper, S.J.; Heeney, J.L. Viruses as vaccine vectors for infectious diseases and cancer. Nat. Rev. Microbiol. 2010, 8, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Rauch, S.; Jasny, E.; Schmidt, K.E.; Petsch, B. New Vaccine Technologies to Combat Outbreak Situations. Front. Immunol. 2018, 9, 1963. [Google Scholar] [CrossRef] [PubMed]
- Sanofi. Dengvaxia®, World’s First Dengue Vaccine, Approved in Mexico. 2015. Available online: https://www.sanofi.com/en/media-room/press-releases/2015/2015-12-09-15-30-00-1684331 (accessed on 19 July 2022).
- Tully, D.; Griffiths, C.L. Dengvaxia: The world’s first vaccine for prevention of secondary dengue. Ther. Adv. Vaccines Immunother. 2021, 9, 25151355211015839. [Google Scholar] [CrossRef]
- Bassi, M.R.; Larsen, M.A.; Kongsgaard, M.; Rasmussen, M.; Buus, S.; Stryhn, A.; Thomsen, A.R.; Christensen, J.P. Vaccination with Replication Deficient Adenovectors Encoding YF-17D Antigens Induces Long-Lasting Protection from Severe Yellow Fever Virus Infection in Mice. PLoS Negl. Trop. Dis. 2016, 10, e0004464. [Google Scholar] [CrossRef] [PubMed]
- Brandler, S.; Lucas-Hourani, M.; Moris, A.; Frenkiel, M.P.; Combredet, C.; Février, M.; Bedouelle, H.; Schwartz, O.; Desprès, P.; Tangy, F. Pediatric measles vaccine expressing a dengue antigen induces durable serotype-specific neutralizing antibodies to dengue virus. PLoS Negl. Trop. Dis. 2007, 1, e96. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, A.; Toriniwa, H.; Komiya, T.; Nakayama, T. Recombinant measles AIK-C vaccine strain expressing the prM-E antigen of Japanese Encephalitis Virus. PLoS ONE 2016, 11, e0150213. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.H.; Chen, H.W.; Hsiao, Y.J.; Yan, J.Y.; Chiang, C.Y.; Chen, M.Y.; Hu, H.M.; Wu, S.H.; Pan, C.H. Immunodomination of Serotype-Specific CD4+ T-Cell Epitopes Contributed to the Biased Immune Responses Induced by a Tetravalent Measles-Vectored Dengue Vaccine. Front. Immunol. 2020, 11, 546. [Google Scholar] [CrossRef] [PubMed]
- Nuernberger, C.; Bodmer, B.S.; Fiedler, A.H.; Gabriel, G.; Muehlebach, M.D. A measles virus-based vaccine candidate mediates protection against Zika virus in an allogeneic mouse pregnancy model. J. Virol. 2019, 93, e01485-18. [Google Scholar] [CrossRef]
- National Library of Medicine (US). ClinicalTrials.gov. NCT02996890. Zika-Vaccine Dose Finding Study Regarding Safety, Immunogenicity and Tolerability (V186-001). 2016. Available online: https://clinicaltrials.gov/ct2/show/NCT02996890 (accessed on 19 July 2022).
- Henao-Restrepo, A.M.; Camacho, A.; Longini, I.M.; Watson, C.H.; Edmunds, W.J.; Egger, M.; Carroll, M.W.; Dean, N.E.; Diatta, I.; Doumbia, M.; et al. Efficacy and effectiveness of an rVSV-vectored vaccine in preventing Ebola virus disease: Final results from the Guinea ring vaccination, open-label, cluster-randomised trial (Ebola Ça Suffit!). Lancet 2017, 389, 505–518. [Google Scholar] [CrossRef]
- Prevec, L.; Campbell, J.B.; Christie, B.S.; Belbeck, L.; Graham, F.L. A recombinant human adenovirus vaccine against rabies. J. Infect. Dis. 1990, 161, 27–30. [Google Scholar] [CrossRef]
- Tatsis, N. Ertl HC. Adenoviruses as vaccine vectors. Mol. Ther. 2004, 10, 616–629. [Google Scholar] [CrossRef]
- Folegatti, P.M.; Bittaye, M.; Flaxman, A.; Lopez, F.R.; Bellamy, D.; Kupke, A.; Mair, C.; Makinson, R.; Sheridan, J.; Rohde, C.; et al. Safety and immunogenicity of a candidate Middle East respiratory syndrome coronavirus viral-vectored vaccine: A dose-escalation, open-label, non-randomised, uncontrolled, phase 1 trial. Lancet Infect. Dis. 2020, 20, 816–826. [Google Scholar] [CrossRef]
- Ledgerwood, J.E.; DeZure, A.D.; Stanley, D.A.; Coates, E.E.; Novik, L.; Enama, M.E.; Berkowitz, N.M.; Hu, Z.; Joshi, G.; Ploquin, A.; et al. Chimpanzee adenovirus vector Ebola vaccine. N. Engl. J. Med. 2017, 376, 928–938. [Google Scholar] [CrossRef]
- Sheridan, C. First COVID-19 DNA Vaccine Approved, Others in Hot Pursuit. 2021. Available online: https://www.nature.com/articles/d41587-021-00023-5 (accessed on 24 July 2022).
- Gaudinski, M.R.; Houser, K.V.; Morabito, K.M.; Hu, Z.; Yamshchikov, G.; Rothwell, R.S.; Berkowitz, N.; Mendoza, F.; Saunders, J.G.; Novik, L.; et al. Safety, tolerability, and immunogenicity of two Zika virus DNA vaccine candidates in healthy adults: Randomised, open-label, phase 1 clinical trials. Lancet 2018, 391, 552–562. [Google Scholar] [CrossRef]
- Ledgerwood, J.E.; Pierson, T.C.; Hubka, S.A.; Desai, N.; Rucker, S.; Gordon, I.J.; Enama, M.E.; Nelson, S.; Nason, M.; Gu, W.; et al. A West Nile virus DNA vaccine utilizing a modified promoter induces neutralizing antibody in younger and older healthy adults in a phase I clinical trial. J. Infect. Dis. 2011, 203, 1396–1404. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef] [PubMed]
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520. [Google Scholar] [CrossRef]
- Dolgin, E. The Tangled History of mRNA Vaccines. 2021. Available online: https://www.nature.com/articles/d41586-021-02483-w (accessed on 24 July 2022).
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838. [Google Scholar] [CrossRef] [PubMed]
- National Library of Medicine (US). ClinicalTrials.gov. NCT04917861. A Study of Zika Vaccine mrna-1893 in Adult Participants Living in Endemic and Non-Endemic Flavivirus Areas. 2021. Available online: https://clinicaltrials.gov/ct2/show/NCT04917861 (accessed on 24 July 2022).
- Zhang, C.; Maruggi, G.; Shan, H.; Li, J. Advances in mRNA vaccines for infectious diseases. Front. Immunol. 2019, 10, 594. [Google Scholar] [CrossRef]
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571. [Google Scholar] [CrossRef]
- Graham, B.S.; Corbett, K.S. Prototype pathogen approach for pandemic preparedness: World on fire. J. Clin. Investig. 2020, 130, 3348–3349. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef] [PubMed]
- Zepeda-Cervantes, J.; Ramírez-Jarquín, J.O.; Vaca, L. Interaction between virus-like particles (VLPs) and pattern recognition receptors (PRRs) from dendritic cells (DCs): Toward better engineering of VLPs. Front. Immunol. 2020, 11, 1100. [Google Scholar] [CrossRef] [PubMed]
- Metz, S.W.; Thomas, A.; White, L.; Stoops, M.; Corten, M.; Hannemann, H.; de Silva, A.M. Dengue virus-like particles mimic the antigenic properties of the infectious dengue virus envelope. Virol. J. 2018, 15, 60. [Google Scholar] [CrossRef]
- Alvim, R.G.F.; Lima, T.M.; Silva, J.L.; de Oliveira, G.A.P.; Castilho, L.R. Process intensification for the production of yellow fever virus-like particles as potential recombinant vaccine antigen. Biotechnol Bioeng. 2021, 118, 3581–3592. [Google Scholar] [CrossRef]
- Dowd, K.A.; Ko, S.Y.; Morabito, K.M.; Yang, E.S.; Pelc, R.S.; DeMaso, C.R.; Castilho, L.R.; Abbink, P.; Boyd, M.; Nityanandam, R.; et al. Rapid development of a DNA vaccine for Zika virus. Science 2016, 354, 237–240. [Google Scholar] [CrossRef]
- Garg, H.; Mehmetoglu-Gurbuz, T.; Ruddy, G.M.; Joshi, A. Capsid containing virus like particle vaccine against Zika virus made from a stable cell line. Vaccine 2019, 37, 7123–7131. [Google Scholar] [CrossRef]
- Fahad, A.S.; Timm, M.R.; Madan, B.; Burgomaster, K.E.; Dowd, K.A.; Normandin, E.; Gutiérrez-González, M.F.; Pennington, J.M.; De Souza, M.O.; Henry, A.R.; et al. Functional Profiling of Antibody Immune Repertoires in Convalescent Zika Virus Disease Patients. Front. Immunol. 2021, 12, 615102. [Google Scholar] [CrossRef]
- Rana, J.; Slon Campos, J.L.; Leccese, G.; Francolini, M.; Bestagno, M.; Poggianella, M.; Burrone, O.R. Role of Capsid Anchor in the Morphogenesis of Zika Virus. J. Virol. 2018, 92, e01174-18. [Google Scholar] [CrossRef]
- Wong, S.H.; Jassey, A.; Wang, J.Y.; Wang, W.C.; Liu, C.H.; Lin, L.T. Virus-like particle systems for vaccine development against viruses in the flaviviridae family. Vaccines 2019, 7, 123. [Google Scholar] [CrossRef]
- Lee, E.; Stocks, C.E.; Amberg, S.M.; Rice, C.M.; Lobigs, M. Mutagenesis of the signal sequence of yellow fever virus prM protein: Enhancement of signalase cleavage In vitro is lethal for virus production. J. Virol. 2000, 74, 24–32. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, R.; Miner, J.J.; Gorman, M.J.; Rausch, K.; Ramage, H.; White, J.P.; Zuiani, A.; Zhang, P.; Fernandez, E.; Zhang, Q.; et al. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature 2016, 535, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.J.J.; Davis, B.S.; Hunt, A.R.; Holmes, D.A.; Kuno, G. Flavivirus DNA Vaccines. Ann. N. Y. Acad. Sci. 2001, 951, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.S.; Chang, G.J.; Cropp, B.; Roehrig, J.T.; Martin, D.A.; Mitchell, C.J.; Bowen, R.; Bunning, M.L. West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme-linked immunosorbent assays. J. Virol. 2001, 75, 4040–4047. [Google Scholar] [CrossRef]
- Alvim, R.G.F.; Itabaiana, I., Jr.; Castilho, L.R. Zika virus-like particles (VLPs): Stable cell lines and continuous perfusion processes as a new potential vaccine manufacturing platform. Vaccine 2019, 37, 6970–6977. [Google Scholar] [CrossRef]
- Wollner, C.J.; Richner, M.; Hassert, M.A.; Pinto, A.K.; Brien, J.D.; Richner, J.M. A dengue virus serotype 1 mRNA-LNP vaccine elicits protective immune responses. J. Virol. 2021, 95, e02482-20. [Google Scholar] [CrossRef]
- Zhang, S.; Liang, M.; Gu, W.; Li, C.; Miao, F.; Wang, X.; Jin, C.; Zhang, L.; Zhang, F.; Zhang, Q.; et al. Vaccination with dengue virus-like particles induces humoral and cellular immune responses in mice. Virol. J. 2011, 8, 333. [Google Scholar] [CrossRef]
- Chang, G.J.; Hunt, A.R.; Holmes, D.A.; Springfield, T.; Chiueh, T.S.; Roehrig, J.T.; Gubler, D.J. Enhancing biosynthesis and secretion of premembrane and envelope proteins by the chimeric plasmid of dengue virus type 2 and Japanese encephalitis virus. Virology 2003, 306, 170–180. [Google Scholar] [CrossRef]
- Purdy, D.E.; Chang, G.J. Secretion of noninfectious dengue virus-like particles and identification of amino acids in the stem region involved in intracellular retention of envelope protein. Virology 2005, 333, 239–250. [Google Scholar] [CrossRef]
- Luisi, K.; Morabito, K.M.; Burgomaster, K.E.; Sharma, M.; Kong, W.P.; Foreman, B.M.; Patel, S.; Fisher, B.; Aleshnick, M.A.; Laliberte, J.; et al. Development of a potent Zika virus vaccine using self-amplifying messenger RNA. Sci. Adv. 2020, 6, eaba5068. [Google Scholar] [CrossRef]
- Rodenhuis-Zybert, I.A.; Moesker, B.; da Silva Voorham, J.M.; van der Ende-Metselaar, H.; Diamond, M.S.; Wilschut, J.; Smit, J.M. A fusion-loop antibody enhances the infectious properties of immature flavivirus particles. J. Virol. 2011, 85, 11800–11808. [Google Scholar] [CrossRef] [PubMed]
- Dowd, K.A.; Mukherjee, S.; Kuhn, R.J.; Pierson, T.C. Combined effects of the structural heterogeneity and dynamics of flaviviruses on antibody recognition. J. Virol. 2014, 88, 11726–11737. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Sirohi, D.; Dowd, K.A.; Chen, Z.; Diamond, M.S.; Kuhn, R.J.; Pierson, T.C. Enhancing dengue virus maturation using a stable furin over-expressing cell line. Virology 2016, 497, 33–40. [Google Scholar] [CrossRef]
- Maciejewski, S.; Ruckwardt, T.J.; Morabito, K.M.; Foreman, B.M.; Burgomaster, K.E.; Gordon, D.N.; Pelc, R.S.; DeMaso, C.R.; Ko, S.Y.; Fisher, B.E.; et al. Distinct neutralizing antibody correlates of protection among related Zika virus vaccines identify a role for antibody quality. Sci. Transl. Med. 2020, 12, eaaw9066. [Google Scholar] [CrossRef] [PubMed]
- Wollner, C.J.; Richner, J.M. mRNA vaccines against flaviviruses. Vaccines 2021, 9, 148. [Google Scholar] [CrossRef] [PubMed]
- Katzelnick, L.C.; Gresh, L.; Halloran, M.E.; Mercado, J.C.; Kuan, G.; Gordon, A.; Balmaseda, A.; Harris, E. Antibody-dependent enhancement of severe dengue disease in humans. Science 2017, 358, 929–932. [Google Scholar] [CrossRef]
- Plotkin, S.A. Dengue vaccines: The road to failure or to success? Hum. Vaccin. Immunother. 2020, 16, 2677–2679. [Google Scholar] [CrossRef]
- Halstead, S.B. Dengvaxia sensitizes seronegatives to vaccine enhanced disease regardless of age. Vaccine 2017, 35, 6355–6358. [Google Scholar] [CrossRef]
- Shukla, R.; Ramasamy, V.; Shanmugam, R.K.; Ahuja, R.; Khanna, N. Antibody-Dependent Enhancement: A Challenge for Developing a Safe Dengue Vaccine. Front. Cell Infect. Microbiol. 2020, 10, 572681. [Google Scholar] [CrossRef]
- Dai, L.; Xu, K.; Li, J.; Huang, Q.; Song, J.; Han, Y.; Zheng, T.; Gao, P.; Lu, X.; Yang, H.; et al. Protective Zika vaccines engineered to eliminate enhancement of dengue infection via immunodominance switch. Nat. Immunol. 2021, 22, 958–968. [Google Scholar] [CrossRef]
- Rey, F.A.; Stiasny, K.; Vaney, M.C.; Dellarole, M.; Heinz, F.X. The bright and the dark side of human antibody responses to flaviviruses: Lessons for vaccine design. EMBO Rep. 2018, 19, 206–224. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Jumnainsong, A.; Onsirisakul, N.; Fitton, P.; Vasanawathana, S.; Limpitikul, W.; Puttikhunt, C.; Edwards, C.; Duangchinda, T.; Supasa, S.; et al. Cross-reacting antibodies enhance dengue virus infection in humans. Science 2010, 328, 745–748. [Google Scholar] [CrossRef] [PubMed]
- Krol, E.; Brzuska, G.; Szewczyk, B. Production and biomedical application of flavivirus-like particles. Trends Biotechnol. 2019, 37, 1202–1216. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.P.S.; St John, A.L. Cross-Reactive Immunity Among Flaviviruses. Front. Immunol. 2020, 11, 334. [Google Scholar] [CrossRef] [PubMed]
- Goo, L.; VanBlargan, L.A.; Dowd, K.A.; Diamond, M.S.; Pierson, T.C. A single mutation in the envelope protein modulates flavivirus antigenicity, stability, and pathogenesis. PLoS Pathog. 2017, 13, e1006178. [Google Scholar] [CrossRef]
- Berneck, B.S.; Rockstroh, A.; Fertey, J.; Grunwald, T.; Ulbert, S. A recombinant zika virus envelope protein with mutations in the conserved fusion loop leads to reduced antibody cross-reactivity upon vaccination. Vaccines 2020, 8, 603. [Google Scholar] [CrossRef]
- Galula, J.U.; Salem, G.M.; Chang, G.J.; Chao, D.Y. Does structurally-mature dengue virion matter in vaccine preparation in post-Dengvaxia era? Hum. Vaccin. Immunother. 2019, 15, 2328–2336. [Google Scholar] [CrossRef]
- Uno, N.; Ross, T.M. Universal dengue vaccine elicits neutralizing antibodies against strains from all four dengue virus serotypes. J. Virol. 2021, 95, e00658-20. [Google Scholar] [CrossRef]
- Garg, H.; Mehmetoglu-Gurbuz, T.; Joshi, A. Virus Like Particles (VLP) as multivalent vaccine candidate against Chikungunya, Japanese Encephalitis, Yellow Fever and Zika Virus. Sci. Rep. 2020, 10, 4017. [Google Scholar] [CrossRef]
- Lima, T.M.; Souza, M.O.; Castilho, L.R. Purification of flavivirus VLPs by a two-step chomatographic process. Vaccine 2019, 37, 7061–7069. [Google Scholar] [CrossRef]
- Garg, H.; Sedano, M.; Plata, G.; Punke, E.B.; Joshi, A. Development of Virus-Like-Particle Vaccine and Reporter Assay for Zika Virus. J. Virol. 2017, 91, e00834-17. [Google Scholar] [CrossRef] [PubMed]
- Hua, R.H.; Li, Y.N.; Chen, Z.S.; Liu, L.K.; Huo, H.; Wang, X.L.; Guo, L.P.; Shen, N.; Wang, J.F.; Bu, Z.G. Generation and characterization of a new mammalian cell line continuously expressing virus-like particles of Japanese encephalitis virus for a subunit vaccine candidate. BMC Biotechnol. 2014, 14, 62. [Google Scholar] [CrossRef] [PubMed]
- Thompson, D.; Guenther, B.; Manayani, D.; Mendy, J.; Smith, J.; Espinosa, D.A.; Harris, E.; Alexander, J.; Vang, L.; Morello, C.S. Zika virus-like particle vaccine fusion loop mutation increases production yield but fails to protect AG129 mice against Zika virus challenge. PLoS Negl. Trop. Dis. 2022, 16, e0010588. [Google Scholar] [CrossRef] [PubMed]
- Urakami, A.; Ngwe Tun, M.M.; Moi, M.L.; Sakurai, A.; Ishikawa, M.; Kuno, S.; Ueno, R.; Morita, K.; Akahata, W. An envelope-modified tetravalent dengue virus-like-particle vaccine has implications for flavivirus vaccine design. J. Virol. 2017, 91, e01181-17. [Google Scholar] [CrossRef]
- Yamaji, H.; Segawa, M.; Nakamura, M.; Katsuda, T.; Kuwahara, M.; Konishi, E. Production of Japanese encephalitis virus-like particles using the baculovirus-insect cell system. J. Biosci. Bioeng. 2012, 114, 657–662. [Google Scholar] [CrossRef]
- Chang, Y.H.; Chiao, D.J.; Hsu, Y.L.; Lin, C.C.; Wu, H.L.; Shu, P.Y.; Chang, S.F.; Chang, J.H.; Kuo, S.C. Mosquito cell-derived japanese encephalitis virus-like particles induce specific humoral and cellular immune responses in mice. Viruses 2020, 12, 336. [Google Scholar] [CrossRef]
- Ponndorf, D.; Meshcheriakova, Y.; Thuenemann, E.C.; Dobon Alonso, A.; Overman, R.; Holton, N.; Dowall, S.; Kennedy, E.; Stocks, M.; Lomonossoff, G.P.; et al. Plant-made dengue virus-like particles produced by co-expression of structural and non-structural proteins induce a humoral immune response in mice. Plant Biotechnol. J. 2021, 19, 745–756. [Google Scholar] [CrossRef]
- Hirsch, J.; Faber, B.W.; Crowe, J.E., Jr.; Verstrepen, B.; Cornelissen, G. E. coli production process yields stable dengue 1 virus-sized particles (VSPs). Vaccine 2020, 38, 3305–3312. [Google Scholar] [CrossRef]
- Shanmugam, R.K.; Ramasamy, V.; Shukla, R.; Arora, U.; Swaminathan, S.; Khanna, N. Pichia pastoris-expressed Zika virus envelope domain III on a virus-like particle platform: Design, production and immunological evaluation. Pathog Dis. 2019, 77, ftz026. [Google Scholar] [CrossRef]
- Ramasamy, V.; Arora, U.; Shukla, R.; Poddar, A.; Shanmugam, R.K.; White, L.J.; Mattocks, M.M.; Raut, R.; Perween, A.; Tyagi, P.; et al. A tetravalent virus-like particle vaccine designed to display domain III of dengue envelope proteins induces multi-serotype neutralizing antibodies in mice and macaques which confer protection against antibody dependent enhancement in AG129 mice. PLoS Negl. Trop. Dis. 2018, 12, e0006191. [Google Scholar] [CrossRef]
- Mali, D.N.; Bondre, V.P. Japanese encephalitis genotype I virus-like particles stably expressed in BHK-21 cells serves as potential antigen in JE IgM ELISA. Appl. Microbiol. Biotechnol. 2022, 106, 1945–1955. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.A.; Skinner, B.; Davis, B.S.; Biggerstaff, B.J.; Robb, L.; Gordon, E.; de Souza, W.M.; Fumagalli, M.J.; Calvert, A.E.; Chang, G.J. Development of HEK-293 cell lines constitutively expressing flaviviral antigens for use in diagnostics. Microbiol. Spectr. 2022, 10, e0059222. [Google Scholar] [CrossRef] [PubMed]

| Viral Vaccine Type | Active Pharmaceutical Ingredient | Examples of Vaccines Approved for Human Use | Example of Country or Region Where Approved|Reference | ||
|---|---|---|---|---|---|
| Target | Trade Name | Manufacturer | |||
| Whole virus vaccines | Inactivated virus | Inflluenza | Fluad | Seqirus | USA [21] |
| Japanese encephalitis | Ixiaro | Valneva | USA [21] | ||
| Live-attenuated virus | Yellow fever | YF-Vax|Stamaril | Sanofi | USA [21] | |
| Measles-mumps-rubella | MMR II | Merck | USA [21] | ||
| Subunit vaccines | Recombinant viral protein | Herpes Zoster | Shingrix | GSK | USA [21] |
| Virus-like particle | Cervical cancer caused by certain HPV strains | Gardasil | Merck | USA [21] | |
| Cervarix | GSK | USA [21] | |||
| Chimeric and vectored vaccines | Chimeric virus | Japanese encephalitis | Imojev | Sanofi | WHO prequalified [22] |
| Virus carrying immunogenic sequence of unrelated virus | Ebola | Ervebo | Merck | USA [21] | |
| COVID-19 | Vaxzevria|Covishield | AstraZeneca | European Union [23] | ||
| Nucleic acid vaccines | DNA | COVID-19 | ZyCoV-D | Zydus Cadila | India * [24] |
| mRNA/lipid nanoparticle | COVID-19 | Comirnaty | Pfizer-BioNTech | USA [21] | |
| COVID-19 | Spikevax | Moderna | USA [21] | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castilho, L.R.; Mattos, N.R.; Abreu, W.S.; Gutarra, M.L.E. Virus-like Particles (VLPs) as Important Tools for Flavivirus Vaccine Development. Biologics 2022, 2, 226-242. https://doi.org/10.3390/biologics2040018
Castilho LR, Mattos NR, Abreu WS, Gutarra MLE. Virus-like Particles (VLPs) as Important Tools for Flavivirus Vaccine Development. Biologics. 2022; 2(4):226-242. https://doi.org/10.3390/biologics2040018
Chicago/Turabian StyleCastilho, Leda R., Nathalia R. Mattos, Wallace S. Abreu, and Melissa L. E. Gutarra. 2022. "Virus-like Particles (VLPs) as Important Tools for Flavivirus Vaccine Development" Biologics 2, no. 4: 226-242. https://doi.org/10.3390/biologics2040018
APA StyleCastilho, L. R., Mattos, N. R., Abreu, W. S., & Gutarra, M. L. E. (2022). Virus-like Particles (VLPs) as Important Tools for Flavivirus Vaccine Development. Biologics, 2(4), 226-242. https://doi.org/10.3390/biologics2040018