
Renal Mechanisms of Diuretic Resistance in Congestive Heart Failure

Abstract
1. Introduction
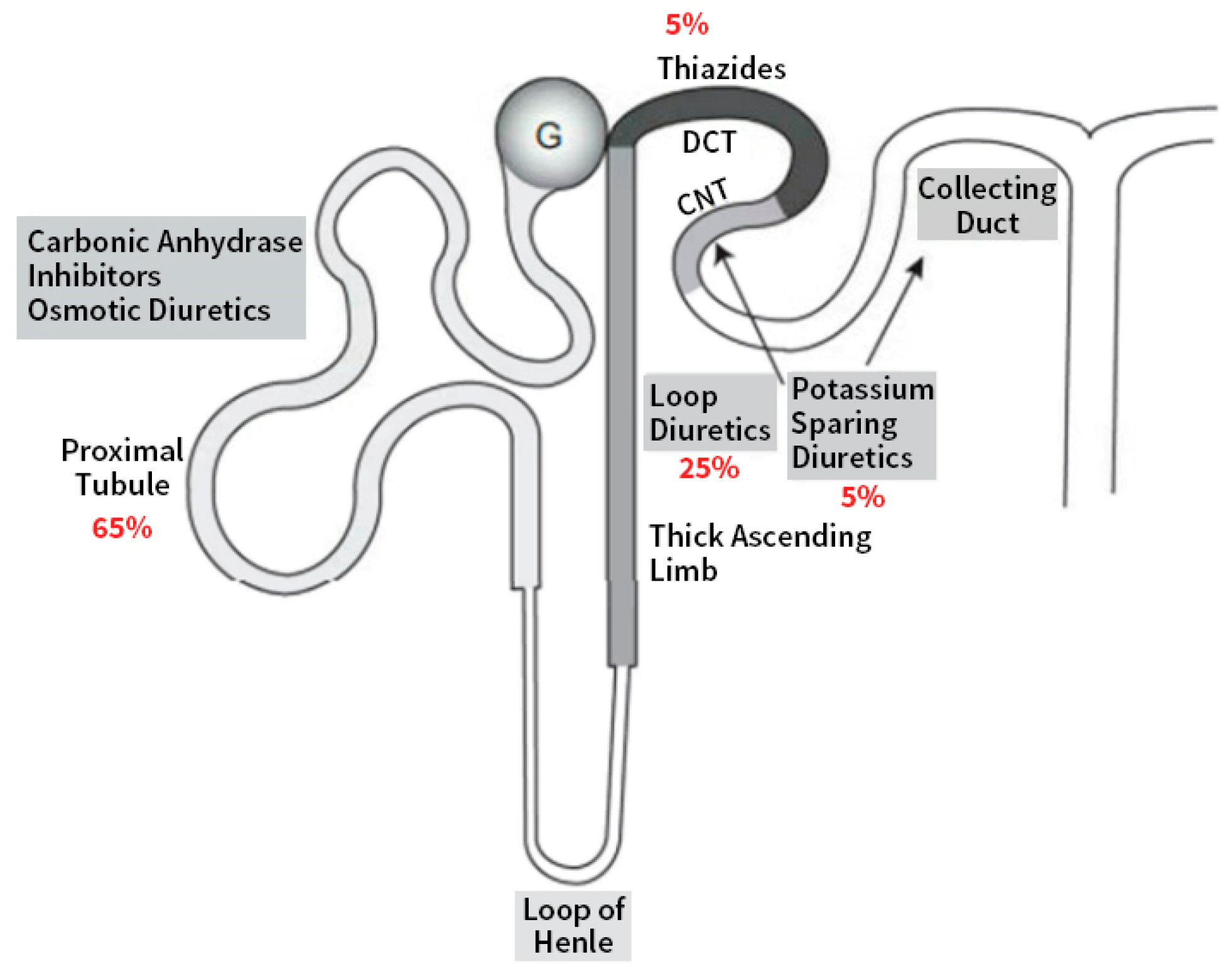
2. Diuretics Acting on the Proximal Tubule (PT)
2.1. Acetazolamide
2.2. SGLT2 Inhibitors (SGLT2i)
3. Diuretics Acting in the Loop of Henle
4. The Distal Nephron Acting Diuretics
5. Diuretics Mainly Acting on the Collecting Duct
5.1. The Potassium-Sparing Diuretics
5.2. Aldosterone Antagonists
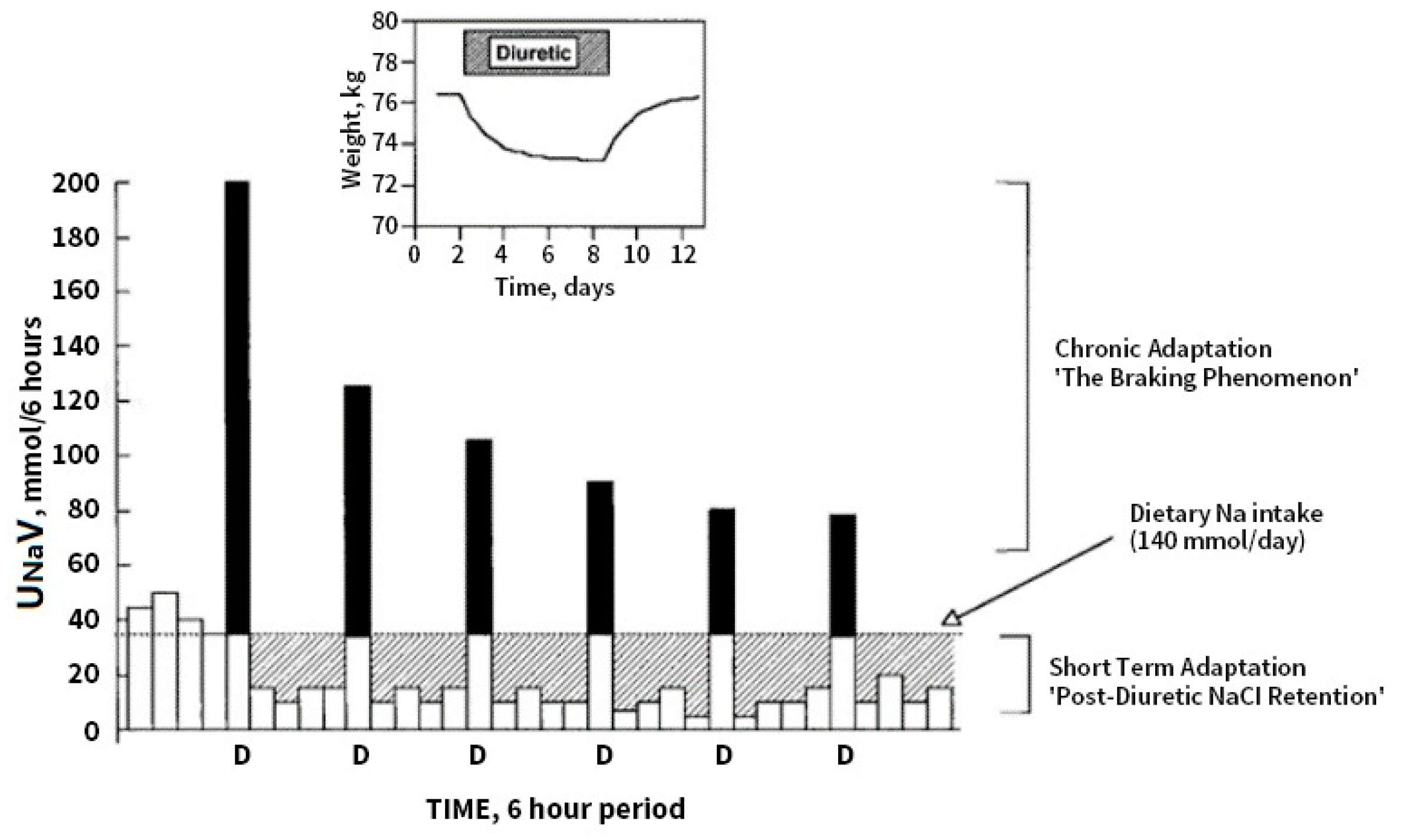
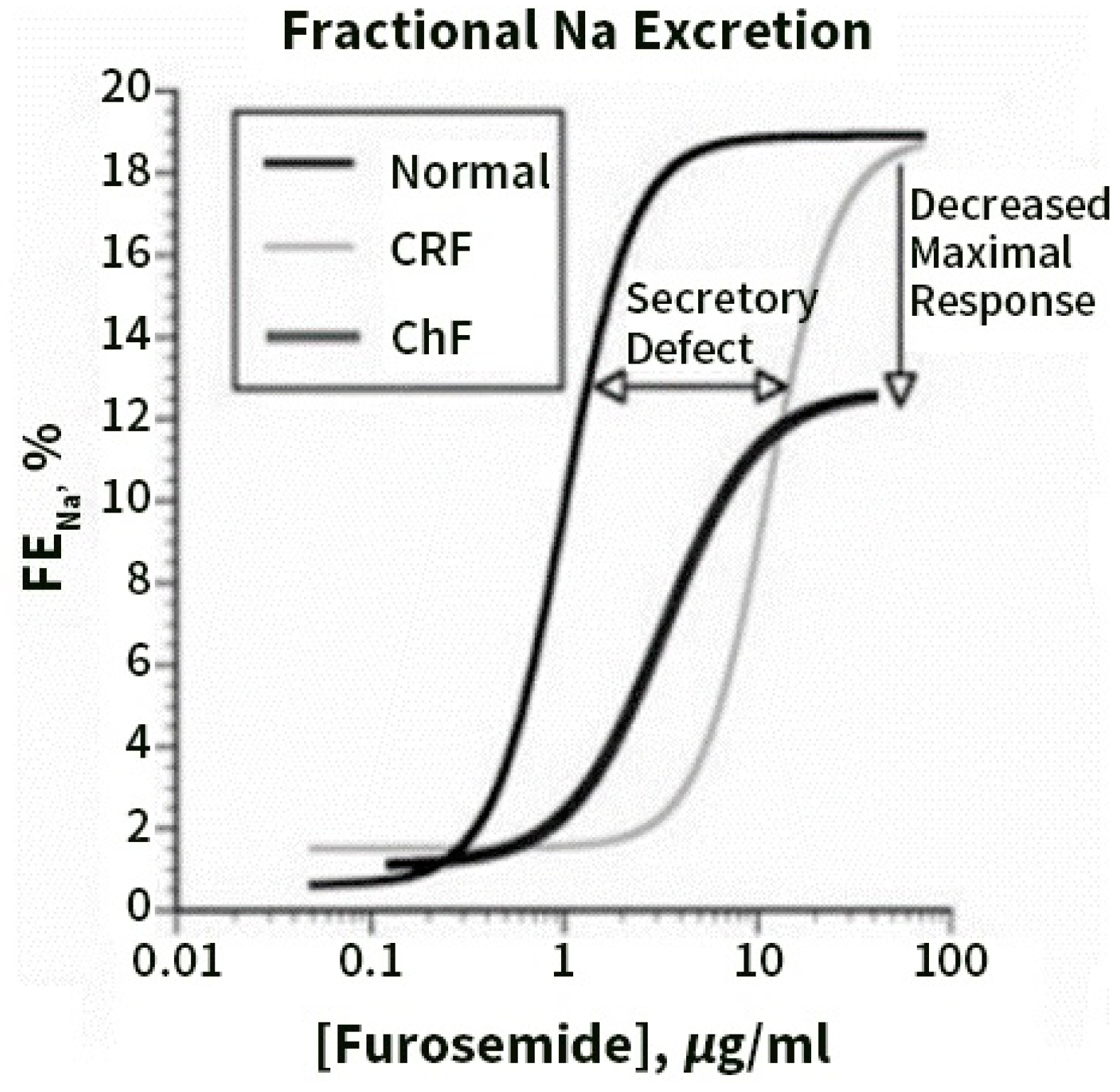
6. Diuretic Resistance (DR) in Congestive Heart Failure
7. Pre- Distal/Tubular Mechanisms of Diuretic Resistance
8. Contribution of Proximal versus Distal Tubular Mechanisms of Diuretic Resistance (DR)
9. Distal Tubular Mechanisms in DR
10. Diagnosing Diuretic Resistance
11. Diagnosis of Congestion/Decongestion
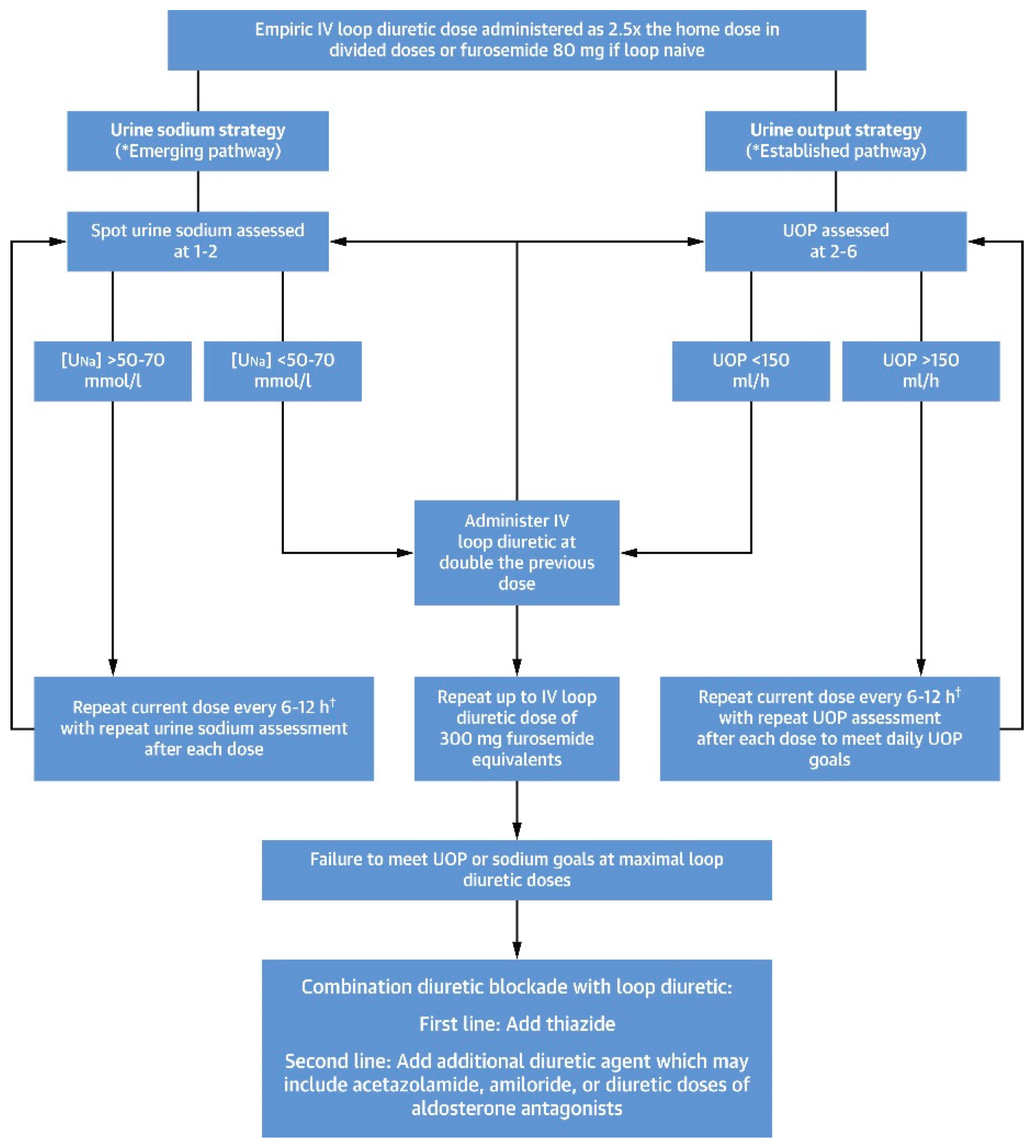
12. Role of Diuretics in the Treatments of Diuretic Resistance
13. Co-Administration of Diuretics with Albumin
14. Continuous Infusion of Loop Diuretics
15. Hypertonic Saline Infusion (HSS)
16. Combination Diuretic Therapy
16.1. Thiazides
16.2. Aldosterone Antagonists
16.3. Acetazolamide
16.4. Multi-Nephron Segment Diuretic Therapy (MSDT)
16.4.1. SGLT2 Inhibitors
16.4.2. Aquaretics
16.4.3. Ultrafiltration
17. Discussion
18. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoorn, E.J.; Ellison, D.H. Diuretic Resistance. Am. J. Kidney Dis. 2017, 69, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Martens, P.; Testani, J.M.; Tang, W.H.W.; Skouri, H.; Verbrugge, F.H.; Fudim, M.; Iacoviello, M.; Franke, J.; Flammer, A.J.; et al. Renal effects of guideline-directed medical therapies in heart failure: A consensus document from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2022, 24, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, C.S.; Testani, J.M.; Pitt, B. Pathophysiology of Diuretic Resistance and Its Implications for the Management of Chronic Heart Failure. Hypertension 2020, 76, 1045–1054. [Google Scholar] [CrossRef]
- Ellison, D.H. Clinical Pharmacology in Diuretic Use. Clin. J. Am. Soc. Nephrol. 2019, 14, 1248–1257. [Google Scholar] [CrossRef]
- Ellison, D.H.; Subramanya, A.R. Clinical Use of Diuretics. In Oxford Textbook of Clinical Nephrology, 4th ed.; Turner, N., Lameire, N., Goldsmith, D.J., Winearls, C.G., Himmelfarb, J., Remuzzi, G., Eds.; Oxford University Press: Oxford, UK, 2016; pp. 299–322. [Google Scholar]
- Wu, W.; Bush, K.T.; Nigam, S.K. Key Role for the Organic Anion Transporters, OAT1 and OAT3, in the in vivo Handling of Uremic Toxins and Solutes. Sci. Rep. 2017, 7, 4939. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M. SGLT2 Inhibitors: Physiology and Pharmacology. Kidney360 2021, 2, 2027–2037. [Google Scholar] [CrossRef]
- Brater, D.C.; Ellison, D.H. Mechanism of Action of Diuretics; Post, T.W., Ed.; UpToDate: Waltham, MA, USA, 2020; Volume 2020. [Google Scholar]
- Novak, J.E.; Ellison, D.H. Diuretics in States of Volume Overload: Core Curriculum 2022. Am. J. Kidney Dis. 2022, 80, 264–276. [Google Scholar] [CrossRef]
- Vallon, V.; Thomson, S.C. The tubular hypothesis of nephron filtration and diabetic kidney disease. Nat. Rev. Nephrol. 2020, 16, 317–336. [Google Scholar] [CrossRef]
- Onishi, A.; Fu, Y.; Patel, R.; Darshi, M.; Crespo-Masip, M.; Huang, W.; Song, P.; Freeman, B.; Kim, Y.C.; Soleimani, M.; et al. A role for tubular Na(+)/H(+) exchanger NHE3 in the natriuretic effect of the SGLT2 inhibitor empagliflozin. Am. J. Physiol. Renal. Physiol. 2020, 319, F712–F728. [Google Scholar] [CrossRef]
- Aguilar-Gallardo, J.S.; Correa, A.; Contreras, J.P. Cardio-renal benefits of sodium-glucose co-transporter 2 inhibitors in heart failure with reduced ejection fraction: Mechanisms and clinical evidence. Eur. Heart J. Cardiovasc. Pharmacother. 2022, 8, 311–321. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Szarek, M.; Steg, P.G.; Cannon, C.P.; Leiter, L.A.; McGuire, D.K.; Lewis, J.B.; Riddle, M.C.; Voors, A.A.; Metra, M.; et al. Sotagliflozin in Patients with Diabetes and Recent Worsening Heart Failure. N. Engl. J. Med. 2021, 384, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Maddox, T.M.; Januzzi, J.L., Jr.; Allen, L.A.; Breathett, K.; Butler, J.; Davis, L.L.; Fonarow, G.C.; Ibrahim, N.E.; Lindenfeld, J.; Masoudi, F.A.; et al. 2021 Update to the 2017 ACC Expert Consensus Decision Pathway for Optimization of Heart Failure Treatment: Answers to 10 Pivotal Issues About Heart Failure with Reduced Ejection Fraction: A Report of the American College of Cardiology Solution Set Oversight Committee. J. Am. Coll. Cardiol. 2021, 77, 772–810. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- McMurray, J.J.V.; Solomon, S.D.; Inzucchi, S.E.; Køber, L.; Kosiborod, M.N.; Martinez, F.A.; Ponikowski, P.; Sabatine, M.S.; Anand, I.S.; Bělohlávek, J.; et al. Dapagliflozin in Patients with Heart Failure and Reduced Ejection Fraction. N. Engl. J. Med. 2019, 381, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Mordi, N.A.; Mordi, I.R.; Singh, J.S.; McCrimmon, R.J.; Struthers, A.D.; Lang, C.C. Renal and Cardiovascular Effects of SGLT2 Inhibition in Combination with Loop Diuretics in Patients with Type 2 Diabetes and Chronic Heart Failure: The RECEDE-CHF Trial. Circulation 2020, 142, 1713–1724. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Verma, S. Effects of SGLT2 Inhibitors on Kidney and Cardiovascular Function. Annu. Rev. Physiol. 2021, 83, 503–528. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.H.; Felker, G.M. Diuretic Treatment in Heart Failure. N. Engl. J. Med. 2017, 377, 1964–1975. [Google Scholar] [CrossRef]
- Schnermann, J.; Briggs, J.P. Synthesis and secretion of renin in mice with induced genetic mutations. Kidney Int. 2012, 81, 529–538. [Google Scholar] [CrossRef]
- Ellison, D.H. Mechanistic Insights into Loop Diuretic Responsiveness in Heart Failure. Clin. J. Am. Soc. Nephrol. 2019, 14, 650–652. [Google Scholar] [CrossRef]
- Felker, G.M.; Ellison, D.H.; Mullens, W.; Cox, Z.L.; Testani, J.M. Diuretic Therapy for Patients with Heart Failure. JACC State Art Rev. 2019, 75, 1178–1195. [Google Scholar] [CrossRef]
- Brater, D.C.; Day, B.; Burdette, A.; Anderson, S. Bumetanide and furosemide in heart failure. Kidney Int. 1984, 26, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Testani, J.M.; Brisco, M.A.; Turner, J.M.; Spatz, E.S.; Bellumkonda, L.; Parikh, C.R.; Tang, W.H. Loop diuretic efficiency: A metric of diuretic responsiveness with prognostic importance in acute decompensated heart failure. Circ. Heart Fail. 2014, 7, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Vargo, D.L.; Kramer, W.G.; Black, P.K.; Smith, W.B.; Serpas, T.; Brater, D.C. Bioavailability, pharmacokinetics, and pharmacodynamics of torsemide and furosemide in patients with congestive heart failure. Clin. Pharmacol. Ther. 1995, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Pearce, D.; Manis, A.D.; Nesterov, V.; Korbmacher, C. Regulation of distal tubule sodium transport: Mechanisms and roles in homeostasis and pathophysiology. Pflugers Arch. 2022, 474, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Pearce, D.; Soundararajan, R.; Trimpert, C.; Kashlan, O.B.; Deen, P.M.; Kohan, D.E. Collecting duct principal cell transport processes and their regulation. Clin. J. Am. Soc. Nephrol. 2015, 10, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Wall, S.M.; Verlander, J.W.; Romero, C.A. The Renal Physiology of Pendrin-Positive Intercalated Cells. Physiol. Rev. 2020, 100, 1119–1147. [Google Scholar] [CrossRef]
- Cil, O.; Haggie, P.M.; Phuan, P.W.; Tan, J.A.; Verkman, A.S. Small-Molecule Inhibitors of Pendrin Potentiate the Diuretic Action of Furosemide. J. Am. Soc. Nephrol. JASN 2016, 27, 3706–3714. [Google Scholar] [CrossRef]
- Soleimani, M. The multiple roles of pendrin in the kidney. Nephrol. Dial. Transplant. 2015, 30, 1257–1266. [Google Scholar] [CrossRef]
- Brown, R.; Quirk, J.; Kirkpatrick, P. Eplerenone. Nat. Rev. Drug Discov. 2003, 2, 177–178. [Google Scholar] [CrossRef]
- Weinberger, M.H.; Roniker, B.; Krause, S.L.; Weiss, R.J. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am. J. Hypertens. 2002, 15, 709–716. [Google Scholar] [CrossRef]
- Agarwal, R.; Kolkhof, P.; Bakris, G.; Bauersachs, J.; Haller, H.; Wada, T.; Zannad, F. Steroidal and non-steroidal mineralocorticoid receptor antagonists in cardiorenal medicine. Eur. Heart J. 2021, 42, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Rico-Mesa, J.S.; White, A.; Ahmadian-Tehrani, A.; Anderson, A.S. Mineralocorticoid Receptor Antagonists: A Comprehensive Review of Finerenone. Curr. Cardiol. Rep. 2020, 22, 140. [Google Scholar] [CrossRef] [PubMed]
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Filippatos, G.; Agarwal, R.; Anker, S.D.; Bakris, G.L.; Rossing, P.; Joseph, A.; Kolkhof, P.; Nowack, C.; Schloemer, P.; et al. Cardiovascular Events with Finerenone in Kidney Disease and Type 2 Diabetes. N. Engl. J. Med. 2021, 385, 2252–2263. [Google Scholar] [CrossRef] [PubMed]
- Cox, Z.L.; Testani, J.M. Loop diuretic resistance complicating acute heart failure. Heart Fail. Rev. 2020, 25, 133–145. [Google Scholar] [CrossRef]
- Ellison, D.H. Diuretic resistance: Physiology and therapeutics. Semin. Nephrol. 1999, 19, 581–597. [Google Scholar]
- Ahmad, T.; Jackson, K.; Rao, V.S.; Tang, W.H.W.; Brisco-Bacik, M.A.; Chen, H.H.; Felker, G.M.; Hernandez, A.F.; O’Connor, C.M.; Sabbisetti, V.S.; et al. Worsening Renal Function in Patients with Acute Heart Failure Undergoing Aggressive Diuresis Is Not Associated with Tubular Injury. Circulation 2018, 137, 2016–2028. [Google Scholar] [CrossRef]
- Fudim, M.; Loungani, R.; Doerfler, S.M.; Coles, A.; Greene, S.J.; Cooper, L.B.; Fiuzat, M.; O’Connor, C.M.; Rogers, J.G.; Mentz, R.J. Worsening renal function during decongestion among patients hospitalized for heart failure: Findings from the Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness (ESCAPE) trial. Am. Heart J. 2018, 204, 163–173. [Google Scholar] [CrossRef]
- Metra, M.; Davison, B.; Bettari, L.; Sun, H.; Edwards, C.; Lazzarini, V.; Piovanelli, B.; Carubelli, V.; Bugatti, S.; Lombardi, C.; et al. Is worsening renal function an ominous prognostic sign in patients with acute heart failure? The role of congestion and its interaction with renal function. Circ. Heart Fail. 2012, 5, 54–62. [Google Scholar] [CrossRef]
- Rao, V.S.; Ahmad, T.; Brisco-Bacik, M.A.; Bonventre, J.V.; Wilson, F.P.; Siew, E.D.; Felker, G.M.; Anstrom, K.K.; Mahoney, D.D.; Bart, B.A.; et al. Renal Effects of Intensive Volume Removal in Heart Failure Patients with Preexisting Worsening Renal Function. Circ. Heart Fail. 2019, 12, e005552. [Google Scholar] [CrossRef]
- Lo, K.B.; Rangaswami, J. Mechanistic Insights in Cardiorenal Syndrome. NEJM Evid. 2022, 1, EVIDra2200053. [Google Scholar] [CrossRef]
- Verbrugge, F.H. Editor’s Choice-Diuretic resistance in acute heart failure. Eur. Heart J. Acute Cardiovasc. Care 2018, 7, 379–389. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Abrahams, Z.; Francis, G.S.; Sokos, G.; Taylor, D.O.; Starling, R.C.; Young, J.B.; Tang, W.H.W. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J. Am. Coll. Cardiol. 2009, 53, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, F.H.; Dupont, M.; Steels, P.; Grieten, L.; Malbrain, M.; Tang, W.H.; Mullens, W. Abdominal contributions to cardiorenal dysfunction in congestive heart failure. J. Am. Coll. Cardiol. 2013, 62, 485–495. [Google Scholar] [CrossRef]
- Cox, Z.L.; Rao, V.S.; Testani, J.M. Classic and Novel Mechanisms of Diuretic Resistance in Cardiorenal Syndrome. Kidney360 2022, 3, 954–967. [Google Scholar]
- Grodin, J.L.; Simon, J.; Hachamovitch, R.; Wu, Y.; Jackson, G.; Halkar, M.; Starling, R.C.; Testani, J.M.; Tang, W.H. Prognostic Role of Serum Chloride Levels in Acute Decompensated Heart Failure. J. Am. Coll. Cardiol. 2015, 66, 659–666. [Google Scholar] [CrossRef]
- Grodin, J.L.; Testani, J.M.; Pandey, A.; Sambandam, K.; Drazner, M.H.; Fang, J.C.; Tang, W.H.W. Perturbations in serum chloride homeostasis in heart failure with preserved ejection fraction: Insights from TOPCAT. Eur. J. Heart Fail. 2018, 20, 1436–1443. [Google Scholar] [CrossRef]
- Grodin, J.L.; Verbrugge, F.H.; Ellis, S.G.; Mullens, W.; Testani, J.M.; Tang, W.H. Importance of Abnormal Chloride Homeostasis in Stable Chronic Heart Failure. Circ. Heart Fail. 2016, 9, e002453. [Google Scholar] [CrossRef]
- Testani, J.M.; Hanberg, J.S.; Arroyo, J.P.; Brisco, M.A.; Ter Maaten, J.M.; Wilson, F.P.; Bellumkonda, L.; Jacoby, D.; Tang, W.H.; Parikh, C.R. Hypochloraemia is strongly and independently associated with mortality in patients with chronic heart failure. Eur. J. Heart Fail. 2016, 18, 660–668. [Google Scholar] [CrossRef]
- Briggs, J. The macula densa sensing mechanism for tubuloglomerular feedback. Fed. Proc. 1981, 40, 99–103. [Google Scholar]
- Kotchen, T.A.; Luke, R.G.; Ott, C.E.; Galla, J.H.; Whitescarver, S. Effect of chloride on renin and blood pressure responses to sodium chloride. Ann. Intern. Med. 1983, 98 Pt 2, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Wesson, D.E. Glomerular filtration effects of acute volume expansion: Importance of chloride. Kidney Int. 1987, 32, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Piala, A.T.; Moon, T.M.; Akella, R.; He, H.; Cobb, M.H.; Goldsmith, E.J. Chloride sensing by WNK1 involves inhibition of autophosphorylation. Sci. Signal. 2014, 7, ra41. [Google Scholar] [CrossRef] [PubMed]
- Ponce-Coria, J.; San-Cristobal, P.; Kahle, K.T.; Vazquez, N.; Pacheco-Alvarez, D.; de Los Heros, P.; Juárez, P.; Muñoz, E.; Michel, G.; Bobadilla, N.A.; et al. Regulation of NKCC2 by a chloride-sensing mechanism involving the WNK3 and SPAK kinases. Proc. Natl. Acad. Sci. USA 2008, 105, 8458–8463. [Google Scholar] [CrossRef]
- Hanberg, J.S.; Rao, V.; Ter Maaten, J.M.; Laur, O.; Brisco, M.A.; Perry Wilson, F.; Grodin, J.L.; Assefa, M.; Samuel Broughton, J.; Planavsky, N.J.; et al. Hypochloremia and Diuretic Resistance in Heart Failure: Mechanistic Insights. Circ. Heart Fail. 2016, 9, e003180. [Google Scholar] [CrossRef]
- Kataoka, H. Chloride in Heart Failure Syndrome: Its Pathophysiologic Role and Therapeutic Implication. Cardiol. Ther. 2021, 10, 407–428. [Google Scholar] [CrossRef]
- Ter Maaten, J.M.; Rao, V.S.; Hanberg, J.S.; Perry Wilson, F.; Bellumkonda, L.; Assefa, M.; Sam Broughton, J.; D’Ambrosi, J.; Wilson Tang, W.H.; Damman, K.; et al. Renal tubular resistance is the primary driver for loop diuretic resistance in acute heart failure. Eur. J. Heart Fail. 2017, 19, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.S.; Planavsky, N.; Hanberg, J.S.; Ahmad, T.; Brisco-Bacik, M.A.; Wilson, F.P.; Jacoby, D.; Chen, M.; Tang, W.H.W.; Cherney, D.Z.I.; et al. Compensatory Distal Reabsorption Drives Diuretic Resistance in Human Heart Failure. J. Am. Soc. Nephrol. JASN 2017, 28, 3414–3424. [Google Scholar] [CrossRef]
- Brater, D.C.; Seiwell, R.; Anderson, S.; Burdette, A.; Dehmer, G.J.; Chennavasin, P. Absorption and disposition of furosemide in congestive heart failure. Kidney Int. 1982, 22, 171–176. [Google Scholar] [CrossRef]
- Bockenhauer, D. Over- or underfill: Not all nephrotic states are created equal. Pediatr. Nephrol. 2013, 28, 1153–1156. [Google Scholar] [CrossRef]
- Svenningsen, P.; Bistrup, C.; Friis, U.G.; Bertog, M.; Haerteis, S.; Krueger, B.; Stubbe, J.; Jensen, O.N.; Thiesson, H.C.; Uhrenholt, T.R.; et al. Plasmin in nephrotic urine activates the epithelial sodium channel. J. Am. Soc. Nephrol. JASN 2009, 20, 299–310. [Google Scholar] [CrossRef]
- Hinrichs, G.R.; Mortensen, L.A.; Jensen, B.L.; Bistrup, C. Amiloride resolves resistant edema and hypertension in a patient with nephrotic syndrome; a case report. Physiol. Rep. 2018, 6, e13743. [Google Scholar] [CrossRef]
- Yamaguchi, E.; Yoshikawa, K.; Nakaya, I.; Kato, K.; Miyasato, Y.; Nakagawa, T.; Kakizoe, Y.; Mukoyama, M.; Soma, J. Liddle’s-like syndrome associated with nephrotic syndrome secondary to membranous nephropathy: The first case report. BMC Nephrol. 2018, 19, 122. [Google Scholar] [CrossRef] [PubMed]
- Cox, Z.L.; Fleming, J.; Ivey-Miranda, J.; Griffin, M.; Mahoney, D.; Jackson, K.; Hodson, D.Z.; Thomas, D., Jr.; Gomez, N.; Rao, V.S.; et al. Mechanisms of Diuretic Resistance Study: Design and rationale. ESC Heart Fail. 2020, 7, 4458–4464. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.H. Diuretic therapy and resistance in congestive heart failure. Cardiology 2001, 96, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Ellison, D.H.; Velázquez, H.; Wright, F.S. Adaptation of the distal convoluted tubule of the rat. Structural and functional effects of dietary salt intake and chronic diuretic infusion. J. Clin. Investig. 1989, 83, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.A.; Kaissling, B. Regulation of renal ion transport and cell growth by sodium. Am. J. Physiol. 1989, 257 Pt 2, F1–F10. [Google Scholar] [CrossRef]
- Wilcox, C.S.; Mitch, W.E.; Kelly, R.A.; Skorecki, K.; Meyer, T.W.; Friedman, P.A.; Souney, P.F. Response of the kidney to furosemide. I. Effects of salt intake and renal compensation. J. Lab. Clin. Med. 1983, 102, 450–458. [Google Scholar]
- Cox, Z.L.; Rao, V.S.; Ivey-Miranda, J.B.; Moreno-Villagomez, J.; Mahoney, D.; Ponikowski, P.; Biegus, J.; Turner, J.M.; Maulion, C.; Bellumkonda, L.; et al. Compensatory post-diuretic renal sodium reabsorption is not a dominant mechanism of diuretic resistance in acute heart failure. Eur. Heart J. 2021, 42, 4468–4477. [Google Scholar] [CrossRef]
- Mullens, W.; Damman, K.; Harjola, V.P.; Mebazaa, A.; Brunner-La Rocca, H.P.; Martens, P.; Testani, J.M.; Tang, W.H.W.; Orso, F.; Rossignol, P.; et al. The use of diuretics in heart failure with congestion—A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2019, 21, 137–155. [Google Scholar] [CrossRef]
- Testani, J.M.; Hanberg, J.S.; Cheng, S.; Rao, V.; Onyebeke, C.; Laur, O.; Kula, A.; Chen, M.; Wilson, F.P.; Darlington, A.; et al. Rapid and Highly Accurate Prediction of Poor Loop Diuretic Natriuretic Response in Patients with Heart Failure. Circ. Heart Fail. 2016, 9, e002370. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.S.; Ivey-Miranda, J.B.; Cox, Z.L.; Riello, R.; Griffin, M.; Fleming, J.; Soucier, R.; Sangkachand, P.; O’Brien, M.; LoRusso, F.; et al. Natriuretic Equation to Predict Loop Diuretic Response in Patients with Heart Failure. J. Am. Coll. Cardiol. 2021, 77, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Girerd, N.; Seronde, M.F.; Coiro, S.; Chouihed, T.; Bilbault, P.; Braun, F.; Kenizou, D.; Maillier, B.; Nazeyrollas, P.; Roul, G.; et al. Integrative Assessment of Congestion in Heart Failure Throughout the Patient Journey. JACC Heart Fail. 2018, 6, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Kitsios, G.D.; Mascari, P.; Ettunsi, R.; Gray, A.W. Co-administration of furosemide with albumin for overcoming diuretic resistance in patients with hypoalbuminemia: A meta-analysis. J. Crit. Care 2014, 29, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Pasini, A.; Benetti, E.; Conti, G.; Ghio, L.; Lepore, M.; Massella, L.; Molino, D.; Peruzzi, L.; Emma, F.; Fede, C.; et al. The Italian Society for Pediatric Nephrology (SINePe) consensus document on the management of nephrotic syndrome in children: Part I—Diagnosis and treatment of the first episode and the first relapse. Ital. J. Pediatr. 2017, 43, 41. [Google Scholar] [CrossRef]
- Felker, G.M.; Lee, K.L.; Bull, D.A.; Redfield, M.M.; Stevenson, L.W.; Goldsmith, S.R.; LeWinter, M.M.; Deswal, A.; Rouleau, J.L.; Ofili, E.O.; et al. Diuretic strategies in patients with acute decompensated heart failure. N. Engl. J. Med. 2011, 364, 797–805. [Google Scholar] [CrossRef]
- Thomson, M.R.; Nappi, J.M.; Dunn, S.P.; Hollis, I.B.; Rodgers, J.E.; Van Bakel, A.B. Continuous versus intermittent infusion of furosemide in acute decompensated heart failure. J. Card. Fail. 2010, 16, 188–193. [Google Scholar] [CrossRef]
- Monteiro Pacheco, A., Jr.; Martins Coimbra, R.S.; Kreimeier, U.; Frey, L.; Messmer, K. Hypertonic volume therapy: Feasibility in the prevention and treatment of multiple organ failure and sepsis. Sao Paulo Med. J. 1995, 113, 1053–1060. [Google Scholar] [CrossRef][Green Version]
- De Vecchis, R.; Esposito, C.; Ariano, C.; Cantatrione, S. Hypertonic saline plus i.v. furosemide improve renal safety profile and clinical outcomes in acute decompensated heart failure: A meta-analysis of the literature. Herz 2015, 40, 423–435. [Google Scholar] [CrossRef]
- Griffin, M.; Soufer, A.; Goljo, E.; Colna, M.; Rao, V.S.; Jeon, S.; Raghavendra, P.; D’Ambrosi, J.; Riello, R.; Coca, S.G.; et al. Real World Use of Hypertonic Saline in Refractory Acute Decompensated Heart Failure: A U.S. Center’s Experience. JACC Heart Fail. 2020, 8, 199–208. [Google Scholar] [CrossRef]
- Butler, J.; Anstrom, K.J.; Felker, G.M.; Givertz, M.M.; Kalogeropoulos, A.P.; Konstam, M.A.; Mann, D.L.; Margulies, K.B.; McNulty, S.E.; Mentz, R.J.; et al. Efficacy and Safety of Spironolactone in Acute Heart Failure: The ATHENA-HF Randomized Clinical Trial. JAMA Cardiol. 2017, 2, 950–958. [Google Scholar] [CrossRef] [PubMed]
- Dormans, T.P.; Gerlag, P.G. Combination of high-dose furosemide and hydrochlorothiazide in the treatment of refractory congestive heart failure. Eur. Heart J. 1996, 17, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.M.; Konopka, E.; Hyderi, A.F.; Hshieh, S.; Tsuji, Y.; Kim, B.J.; Han, S.Y.; Phan, D.H.; Jeng, A.I.; Lou, M.; et al. Comparison of bumetanide- and metolazone-based diuretic regimens to furosemide in acute heart failure. J. Cardiovasc. Pharmacol. Ther. 2013, 18, 345–353. [Google Scholar] [CrossRef]
- Sica, D.A. Metolazone and its role in edema management. Congest. Heart Fail. 2003, 9, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Moranville, M.P.; Choi, S.; Hogg, J.; Anderson, A.S.; Rich, J.D. Comparison of metolazone versus chlorothiazide in acute decompensated heart failure with diuretic resistance. Cardiovasc. Ther. 2015, 33, 42–49. [Google Scholar] [CrossRef]
- Brisco-Bacik, M.A.; Ter Maaten, J.M.; Houser, S.R.; Vedage, N.A.; Rao, V.; Ahmad, T.; Wilson, F.P.; Testani, J.M. Outcomes Associated with a Strategy of Adjuvant Metolazone or High-Dose Loop Diuretics in Acute Decompensated Heart Failure: A Propensity Analysis. J. Am. Heart Assoc. 2018, 7, e009149. [Google Scholar] [CrossRef] [PubMed]
- Pitt, B.; Zannad, F.; Remme, W.J.; Cody, R.; Castaigne, A.; Perez, A.; Palensky, J.; Wittes, J. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N. Engl. J. Med. 1999, 341, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Schepkens, H.; Vanholder, R.; Billiouw, J.M.; Lameire, N. Life-threatening hyperkalemia during combined therapy with angiotensin-converting enzyme inhibitors and spironolactone: An analysis of 25 cases. Am. J. Med. 2001, 110, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Juurlink, D.N.; Mamdani, M.M.; Lee, D.S.; Kopp, A.; Austin, P.C.; Laupacis, A.; Redelmeier, D.A. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N. Engl. J. Med. 2004, 351, 543–551. [Google Scholar] [CrossRef]
- Wongboonsin, J.; Thongprayoon, C.; Bathini, T.; Ungprasert, P.; Aeddula, N.R.; Mao, M.A.; Cheungpasitporn, W. Acetazolamide Therapy in Patients with Heart Failure: A Meta-Analysis. J. Clin. Med. 2019, 8, 349. [Google Scholar] [CrossRef]
- Verbrugge, F.H.; Dupont, M.; Bertrand, P.B.; Nijst, P.; Penders, J.; Dens, J.; Verhaert, D.; Vandervoort, P.; Tang, W.H.; Mullens, W. Determinants and impact of the natriuretic response to diuretic therapy in heart failure with reduced ejection fraction and volume overload. Acta Cardiol. 2015, 70, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, F.H.; Martens, P.; Ameloot, K.; Haemels, V.; Penders, J.; Dupont, M.; Tang, W.H.W.; Droogné, W.; Mullens, W. Acetazolamide to increase natriuresis in congestive heart failure at high risk for diuretic resistance. Eur. J. Heart Fail. 2019, 21, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Dauw, J.; Martens, P.; Verbrugge, F.H.; Nijst, P.; Meekers, E.; Tartaglia, K.; Chenot, F.; Moubayed, S.; Dierckx, R.; et al. Acetazolamide in Acute Decompensated Heart Failure with Volume Overload. N. Engl. J. Med. 2022, 387, 1185–1195. [Google Scholar] [CrossRef] [PubMed]
- Felker, G.M. New Decongestion Strategies in an Evolving Heart Failure Landscape. N. Engl. J. Med. 2022, 387, 1231–1233. [Google Scholar] [CrossRef] [PubMed]
- Cox, Z.L.; Sarrell, B.A.; Cella, M.K.; Tucker, B.; Arroyo, J.P.; Umanath, K.; Tidwell, W.; Guide, A.; Testani, J.M.; Lewis, J.B.; et al. Multinephron Segment Diuretic Therapy to Overcome Diuretic Resistance in Acute Heart Failure: A Single-Center Experience. J. Card. Fail. 2022, 28, 21–31. [Google Scholar] [CrossRef]
- Damman, K.; Beusekamp, J.C.; Boorsma, E.M.; Swart, H.P.; Smilde, T.D.J.; Elvan, A.; van Eck, J.W.M.; Heerspink, H.J.L.; Voors, A.A. Randomized, double-blind, placebo-controlled, multicentre pilot study on the effects of empagliflozin on clinical outcomes in patients with acute decompensated heart failure (EMPA-RESPONSE-AHF). Eur. J. Heart Fail. 2020, 22, 713–722. [Google Scholar] [CrossRef]
- Wilcox, C.S.; Shen, W.; Boulton, D.W.; Leslie, B.R.; Griffen, S.C. Interaction Between the Sodium-Glucose-Linked Transporter 2 Inhibitor Dapagliflozin and the Loop Diuretic Bumetanide in Normal Human Subjects. J. Am. Heart Assoc. 2018, 7, e007046. [Google Scholar] [CrossRef]
- Griffin, M.; Riello, R.; Rao, V.S.; Ivey-Miranda, J.; Fleming, J.; Maulion, C.; McCallum, W.; Sarnak, M.; Collins, S.; Inzucchi, S.E.; et al. Sodium glucose cotransporter 2 inhibitors as diuretic adjuvants in acute decompensated heart failure: A case series. ESC Heart Fail. 2020, 7, 1966–1971. [Google Scholar] [CrossRef]
- Konstam, M.A.; Gheorghiade, M.; Burnett, J.C., Jr.; Grinfeld, L.; Maggioni, A.P.; Swedberg, K.; Udelson, J.E.; Zannad, F.; Cook, T.; Ouyang, J.; et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: The EVEREST Outcome Trial. JAMA 2007, 297, 1319–1331. [Google Scholar] [CrossRef]
- Felker, G.M.; Mentz, R.J.; Cole, R.T.; Adams, K.F.; Egnaczyk, G.F.; Fiuzat, M.; Patel, C.B.; Echols, M.; Khouri, M.G.; Tauras, J.M.; et al. Efficacy and Safety of Tolvaptan in Patients Hospitalized with Acute Heart Failure. J. Am. Coll. Cardiol. 2017, 69, 1399–1406. [Google Scholar] [CrossRef]
- Konstam, M.A.; Kiernan, M.; Chandler, A.; Dhingra, R.; Mody, F.V.; Eisen, H.; Haught, W.H.; Wagoner, L.; Gupta, D.; Patten, R.; et al. Short-Term Effects of Tolvaptan in Patients with Acute Heart Failure and Volume Overload. J. Am. Coll. Cardiol. 2017, 69, 1409–1419. [Google Scholar] [CrossRef] [PubMed]
- Bart, B.A.; Goldsmith, S.R.; Lee, K.L.; Givertz, M.M.; O’Connor, C.M.; Bull, D.A.; Redfield, M.M.; Deswal, A.; Rouleau, J.L.; LeWinter, M.M.; et al. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N. Engl. J. Med. 2012, 367, 2296–2304. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.R.; Negoianu, D.; Jaski, B.E.; Bart, B.A.; Heywood, J.T.; Anand, I.S.; Smelser, J.M.; Kaneshige, A.M.; Chomsky, D.B.; Adler, E.D.; et al. Aquapheresis versus Intravenous Diuretics and Hospitalizations for Heart Failure. JACC Heart Fail. 2016, 4, 95–105. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Natriuretics | Aquaretics | |||
|---|---|---|---|---|
| Proximal diuretics | Loop diuretics | Distal convoluted tubule | Cortical collecting tubule | Collecting duct |
| Carbonic anhydrase inhibitors Sodium-glucose cotransporter 2 inhibitors | Na-K-CI (NKCC2) transport inhibitors | Na-CI (NCC) cotransporter inhibitors | Na channel blockers (ENAC inhibitors) Aldosterone antagonists | Vasopressor receptor blockers |
| Acetazolamide Empagliflozin Dapagliflozin Canagliflozin Others... | Furosemide Bumetanide Torasemide Ethacrynic acid a | Hydrochlorothiazide Metolazone Chlorthalidone Indapamide b Others | Amiloride Triameterene Spironolactone Eplerenone | Conivaptan Tolvaptan |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lameire, N. Renal Mechanisms of Diuretic Resistance in Congestive Heart Failure. Kidney Dial. 2023, 3, 56-72. https://doi.org/10.3390/kidneydial3010005
Lameire N. Renal Mechanisms of Diuretic Resistance in Congestive Heart Failure. Kidney and Dialysis. 2023; 3(1):56-72. https://doi.org/10.3390/kidneydial3010005
Chicago/Turabian StyleLameire, Norbert. 2023. "Renal Mechanisms of Diuretic Resistance in Congestive Heart Failure" Kidney and Dialysis 3, no. 1: 56-72. https://doi.org/10.3390/kidneydial3010005
APA StyleLameire, N. (2023). Renal Mechanisms of Diuretic Resistance in Congestive Heart Failure. Kidney and Dialysis, 3(1), 56-72. https://doi.org/10.3390/kidneydial3010005