
Antisense Oligonucleotide: A Potential Therapeutic Intervention for Chronic Kidney Disease

 ,
,
Abstract
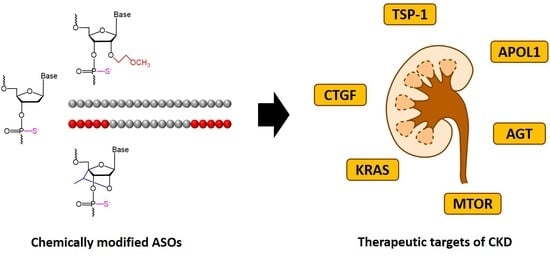
1. Introduction
1.1. Antisense Oligonucleotide
1.2. Chronic Kidney Disease
1.3. Cardiorenal Syndromes
2. Conventional Therapies and Their Limitations
3. Antisense Oligonucleotide as Therapeutics
3.1. Mechanisms of Action
3.2. Chemical Modification and Rational Design of ASO
3.3. ASO, siRNA, and miRNA
4. Antisense Oligonucleotides Targeting Chronic Kidney Disease
4.1. Thrombospondin-1 (TSP1)
4.2. Connective Tissue Growth Factor (CTGF)
4.3. Kirsten Rat Sarcoma Viral Oncogene Homolog (KRAS)
4.4. Mammalian Target of Rapamycin (mTOR)
4.5. Angiotensinogen (AGT)
4.6. Apolipoprotein L1 (APOL1)
5. Potential Problems of ASO-Based CKD Therapy and Possible Solutions
- (1)
- (2)
- Exploration of target genes that are newly evolved or less conserved, such as APOL1 [118], so that inhibition of such genes is probably less risky compared with the genes that are functionally conserved.
- (3)
- (4)
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alama, A.; Barbieri, F.; Cagnoli, M.; Schettini, G. Antisense oligonucleotides as therapeutic agents. Pharmacol. Res. 1997, 36, 171–178. [Google Scholar] [CrossRef]
- Dias, N.; Stein, C.A. Antisense oligonucleotides: Basic concepts and mechanisms. Mol. Cancer Ther. 2002, 1, 347–355. [Google Scholar]
- Crooke, S.T.; Baker, B.F.; Crooke, R.M.; Liang, X.H. Antisense technology: An overview and prospectus. Nat. Rev. Drug Discov. 2021, 20, 427–453. [Google Scholar] [CrossRef]
- Chan, J.H.; Lim, S.; Wong, W.S. Antisense oligonucleotides: From design to therapeutic application. Clin. Exp. Pharmacol. Physiol. 2006, 33, 533–540. [Google Scholar] [CrossRef]
- Paterson, B.M.; Roberts, B.E.; Kuff, E.F. Structural gene identification and mapping by DNA-mRNA hybrid-arrested cell-free translation. Proc. Natl. Acad. Sci. USA 1977, 74, 4370–4374. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef]
- Stephenson, M.L.; Zamecnik, P.C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 285–288. [Google Scholar] [CrossRef]
- Chen, S.; Sbuh, N.; Veedu, R.N. Antisense oligonucleotides as potential therapeutics for type 2 diabetes. Nucleic Acid Ther. 2021, 31, 39–57. [Google Scholar] [CrossRef]
- Roehr, B. Fomivirsen approved for CMV retinitis. J. Int. Assoc. Physicians AIDS Care 1998, 4, 14–16. [Google Scholar]
- De Smet, M.D.; Meenken, C.J.; van den Horn, G.J. Fomivirsen—A phosphorothioate oligonucleotide for the treatment of CMV retinitis. Ocul. Immunol. Inflamm. 1999, 7, 189–198. [Google Scholar] [CrossRef]
- Hair, P.; Cameron, F.; McKeage, K. Mipomersen sodium: First global approval. Drugs 2013, 73, 487–493. [Google Scholar] [CrossRef]
- Wong, E.; Goldberg, T. Mipomersen (kynamro): A novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. Pharmacol. Ther. 2014, 39, 119–122. [Google Scholar]
- Syed, Y.Y. Eteplirsen: First global approval. Drugs 2016, 76, 1699–1704. [Google Scholar] [CrossRef]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef]
- Baker, D.E. Eteplirsen. Hosp. Pharm. 2017, 52, 302–305. [Google Scholar] [CrossRef]
- Charleston, J.S.; Schnell, F.J.; Dworzak, J.; Donoghue, C.; Lewis, S.; Chen, L.; Young, D.; Milici, A.; Voss, J.; DeAlwis, U.; et al. Eteplirsen treatment for Duchenne muscular dystrophy: Exon skipping and dystrophin production. Neurology 2018, 90, e2146–e2154. [Google Scholar] [CrossRef]
- Hoy, S.M. Nusinersen: First global approval. Drugs 2017, 77, 473–479. [Google Scholar] [CrossRef]
- Corey, D.R. Nusinersen, an antisense oligonucleotide drug for spinal muscular atrophy. Nat. Neurosci. 2017, 20, 497–499. [Google Scholar] [CrossRef]
- Goodkey, K.; Aslesh, T.; Maruyama, R.; Yokota, T. Nusinersen in the treatment of spinal muscular atrophy. Methods Mol. Biol. 2018, 1828, 69–76. [Google Scholar]
- Neil, E.E.; Bisaccia, E.K. Nusinersen: A novel antisense oligonucleotide for the treatment of spinal muscular atrophy. J. Pediatr. Pharmacol. Ther. 2019, 24, 194–203. [Google Scholar] [CrossRef]
- Keam, S.J. Inotersen: First global approval. Drugs 2018, 78, 1371–1376. [Google Scholar] [CrossRef]
- Gales, L. Tegsedi (inotersen): An antisense oligonucleotide approved for the treatment of adult patients with hereditary transthyretin amyloidosis. Pharmaceuticals 2019, 12, 78. [Google Scholar] [CrossRef]
- Heo, Y.A. Golodirsen: First approval. Drugs 2020, 80, 329–333. [Google Scholar] [CrossRef]
- Dhillon, S. Viltolarsen: First approval. Drugs 2020, 80, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Casimersen: First Approval. Drugs 2021, 81, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Lv, J.C.; Zhang, L.X. Prevalence and disease burden of chronic kidney disease. Adv. Exp. Med. Biol. 2019, 1165, 3–15. [Google Scholar] [PubMed]
- Li, P.K.T.; Garcia-Garcia, G.; Lui, S.F.; Andreoli, S.; Fung, W.W.S.; Hradsky, A.; Kumaraswami, L.; Liakopoulos, V.; Rakhimova, Z.; Saadi, G.; et al. Kidney health for everyone everywhere: From prevention to detection and equitable access to care. Am. J. Hypertens. 2020, 33, 282–289. [Google Scholar] [CrossRef]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic kidney disease. Lancet 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Foreman, K.J.; Marquez, N.; Dolgert, A.; Fukutaki, K.; Fullman, N.; McGaughey, M.; Pletcher, M.A.; Smith, A.; Tang, K.; Yuan, C.; et al. Forecasting life expectancy, years of life lost, and all-cause and cause-specific mortality for 250 causes of death: Reference and alternative scenarios for 2016-40 for 195 countries and territories. Lancet 2018, 392, 2052–2090. [Google Scholar] [CrossRef]
- Coresh, J.; Selvin, E.; Stevens, L.A.; Manzi, J.; Kusek, J.W.; Eggers, P.; Van Lente, F.; Levey, A.S. Prevalence of chronic kidney disease in the United States. JAMA 2007, 298, 2038–2047. [Google Scholar] [CrossRef]
- Saran, R.; Li, Y.; Robinson, B.; Abbott, K.C.; Agodoa, L.Y.; Ayanian, J.; Bragg-Gresham, J.; Balkrishnan, R.; Chen, J.L.; Cope, E.; et al. US renal data system 2015 annual data report: Epidemiology of kidney disease in the United States. Am. J. Kidney Dis. 2016, 67, A7–A8. [Google Scholar] [CrossRef]
- Levin, A.; Stevens, P.E.; Bilous, R.W.; Coresh, J.; De Francisco, A.L.M.; De Jong, P.E.; Griffith, K.E.; Hemmelgarn, B.R.; Iseki, K.; Edmund, L.; et al. Kidney disease: Improving global outcomes (KDIGO) CKD work group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar]
- Forbes, A.; Gallagher, H. Chronic kidney disease in adults: Assessment and management. Clin. Med. 2020, 20, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Liyanage, T.; Ninomiya, T.; Jha, V.; Neal, B.; Patrice, H.M.; Okpechi, I.; Zhao, M.H.; Lv, J.; Garg, A.X.; Knight, J.; et al. Worldwide access to treatment for end-stage kidney disease: A systematic review. Lancet 2015, 385, 1975–1982. [Google Scholar] [CrossRef]
- Fishman, J.A. Infection in renal transplant recipients. Semin. Nephrol. 2007, 27, 445–461. [Google Scholar] [CrossRef]
- Dantal, J.; Soulillou, J.P. Immunosuppressive drugs and the risk of cancer after organ transplantation. N. Engl. J. Med. 2005, 352, 1371–1373. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.C.; Cooper, M.E.; Zimmet, P. Changing epidemiology of type 2 diabetes mellitus and associated chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 73–81. [Google Scholar] [CrossRef]
- Foley, R.N.; Collins, A.J. The growing economic burden of diabetic kidney disease. Curr. Diabetes Rep. 2009, 9, 460–465. [Google Scholar] [CrossRef]
- Imperatore, G.; Boyle, J.P.; Thompson, T.J.; Case, D.; Dabelea, D.; Hamman, R.F.; Lawrence, J.M.; Liese, A.D.; Liu, L.L.; Mayer-Davis, E.J.; et al. Projections of type 1 and type 2 diabetes burden in the U.S. population aged <20 years through 2050: Dynamic modeling of incidence, mortality, and population growth. Diabetes Care 2012, 35, 2515–2520. [Google Scholar] [CrossRef]
- Haileamlak, A. Chronic kidney disease is on the rise. Ethiop. J. Health Sci. 2018, 28, 681–682. [Google Scholar] [PubMed]
- Barsoum, R.S. Chronic kidney disease in the developing world. N. Engl. J. Med. 2006, 354, 997–999. [Google Scholar] [CrossRef] [PubMed]
- USRDS. 2012 Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States; National Institutes of Health, National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Thompson, S.; James, M.; Wiebe, N.; Hemmelgarn, B.; Manns, B.; Klarenbach, S.; Tonelli, M. Cause of death in patients with reduced kidney function. J. Am. Soc. Nephrol. 2015, 26, 2504–2511. [Google Scholar] [CrossRef] [PubMed]
- Ronco, C. The Cardiorenal syndrome: Basis and common ground for a multidisciplinary patient-oriented therapy. Cardiorenal Med. 2011, 1, 3–4. [Google Scholar] [CrossRef]
- Gnanaraj, J.; Radhakrishnan, J. Cardio-renal syndrome. F1000Research 2016, 5, 2123. [Google Scholar] [CrossRef]
- Ruggenenti, P.; Cravedi, P.; Remuzzi, G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat. Rev. Nephrol. 2010, 6, 319–330. [Google Scholar] [CrossRef]
- Hou, F.F.; Zhang, X.; Zhang, G.H.; Xie, D.; Chen, P.Y.; Zhang, W.R.; Jiang, J.P.; Liang, M.; Wang, G.B.; Liu, Z.R.; et al. Efficacy and safety of benazepril for advanced chronic renal insufficiency. N. Engl. J. Med. 2006, 354, 131–140. [Google Scholar] [CrossRef]
- Xie, X.; Liu, Y.; Perkovic, V.; Li, X.; Ninomiya, T.; Hou, W.; Zhao, N.; Liu, L.; Lv, J.; Zhang, H.; et al. Renin-angiotensin system inhibitors and kidney and cardiovascular outcomes in patients with CKD: A bayesian network meta-analysis of randomized clinical trials. Am. J. Kidney Dis. 2016, 67, 728–741. [Google Scholar] [CrossRef]
- Brenner, B.M.; Cooper, M.E.; De Zeeuw, D.; Keane, W.F.; Mitch, W.E.; Parving, H.-H.; Remuzzi, G.; Snapinn, S.M.; Zhang, Z.; Shahinfar, S. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N. Engl. J. Med. 2001, 345, 861–869. [Google Scholar] [CrossRef]
- Zatz, R.; Dunn, B.R.; Meyer, T.W.; Anderson, S.; Rennke, H.G.; Brenner, B.M. Prevention of diabetic glomerulopathy by pharmacological amelioration of glomerular capillary hypertension. J. Clin. Investig. 1986, 77, 1925–1930. [Google Scholar] [CrossRef]
- Holtkamp, F.A.; de Zeeuw, D.; Thomas, M.C.; Cooper, M.E.; de Graeff, P.A.; Hillege, H.J.; Parving, H.-H.; Brenner, B.M.; Shahinfar, S.; Heerspink, H.J.L. An acute fall in estimated glomerular filtration rate during treatment with losartan predicts a slower decrease in long-term renal function. Kidney Int. 2011, 80, 282–287. [Google Scholar] [CrossRef]
- Breyer, M.D.; Susztak, K. Developing treatments for chronic kidney disease in the 21st century. Semin. Nephrol. 2016, 36, 436–447. [Google Scholar] [CrossRef]
- Parving, H.-H.; Brenner, B.M.; Mcmurray, J.; De Zeeuw, D.; Haffner, S.M.; Solomon, S.D.; Chaturvedi, N.; Persson, F.; Desai, A.S.; Nicolaides, M.; et al. Cardiorenal end points in a trial of aliskiren for type 2 diabetes. N. Engl. J. Med. 2012, 367, 2204–2213. [Google Scholar] [CrossRef] [PubMed]
- Fried, L.F.; Emanuele, N.; Zhang, J.H.; Brophy, M.; Conner, T.A.; Duckworth, W.; Leehey, D.J.; McCullough, P.A.; O’Connor, T.; Palevsky, P.; et al. Combined angiotensin inhibition for the treatment of diabetic nephropathy. N. Engl. J. Med. 2013, 369, 1892–1903. [Google Scholar] [CrossRef] [PubMed]
- Le, B.T.; Raguraman, P.R.; Kosbar, T.R.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Antisense oligonucleotides targeting angiogenic factors as potential cancer therapeutics. Mol. Ther. Nucleic Acids 2019, 14, 142–157. [Google Scholar] [CrossRef]
- Scoles, D.R.; Minikel, E.V.; Pulst, S.M. Antisense oligonucleotides: A primer. Neurol. Genet. 2019, 5, e323. [Google Scholar] [CrossRef] [PubMed]
- Crooke, S.T. Molecular mechanisms of antisense oligonucleotides. Nucleic Acid Ther. 2017, 27, 70–77. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Kole, R.; Krainer, A.R.; Altman, S. RNA therapeutics: Beyond RNA interference and antisense oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef]
- Good, L. Translation repression by antisense sequence. Cell Mol. Life Sci. 2003, 60, 854–861. [Google Scholar] [CrossRef]
- Bennett, C.F.; Cowsert, L.M. Antisense oligonucleotides as a tool for gene functionalization and target validation. Biochim. Biophys. Acta 1999, 1489, 19–30. [Google Scholar] [CrossRef]
- Yin, W.; Rogge, M. Targeting RNA: A transformative therapeutic strategy. Clin. Transl. Sci. 2019, 12, 98–112. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; van Ommen, G.J. Antisense-mediated exon skipping: A versatile tool with therapeutic and research applications. RNA 2007, 13, 1609–1624. [Google Scholar] [CrossRef] [PubMed]
- Kurreck, J. Antisense technologies. Improvement through novel chemical modifications. Eur. J. Biochem. 2003, 270, 1628–1644. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Nakatani, M.; Narukawa, K.; Obika, S. Antisense drug discovery and development. Future Med. Chem. 2011, 3, 339–365. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Sharmab, R.K.; Singh, S.K. Antisense oligonucleotides: Modifications and clinical trials. Med. Chem. Commun. 2014, 5, 1454–1471. [Google Scholar] [CrossRef]
- Wan, W.B.; Seth, P.P. The medicinal chemistry of therapeutic oligonucleotides. J. Med. Chem. 2016, 59, 9645–9667. [Google Scholar] [CrossRef]
- Khvorova, A.; Watts, J.K. The chemical evolution of oligonucleotide therapies of clinical utility. Nat. Biotechnol. 2017, 35, 238–248. [Google Scholar] [CrossRef]
- Shen, X.; Corey, D.R. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018, 46, 1584–1600. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef]
- Ghosh, M.K.; Ghosh, K.; Dahl, O.; Cohen, J.S. Evaluation of some properties of a phosphorodithioate oligodeoxyribonucleotide for antisense application. Nucleic Acids Res. 1993, 21, 5761–5766. [Google Scholar] [CrossRef][Green Version]
- Miller, P.S. Oligonucleoside methylphosphonates as antisense reagents. Biotechnology 1991, 9, 358–362. [Google Scholar] [CrossRef]
- Rait, V.; Sergueev, D.; Summers, J.; He, K.; Huang, F.; Krzyzanowska, B.; Shaw, B.R. Boranophosphate nucleic acids—A versatile DNA backbone. Nucleosides Nucleotides 1999, 18, 1379–1380. [Google Scholar] [CrossRef]
- Sheehan, D.; Lunstad, B.; Yamada, C.M.; Stell, B.G.; Caruthers, M.H.; Dellinger, D.J. Biochemical properties of phosphonoacetate and thiophosphonoacetate oligodeoxyribonucleotides. Nucleic Acids Res. 2003, 31, 4109–4118. [Google Scholar] [CrossRef] [PubMed]
- Miroshnichenko, S.K.; Patutina, O.A.; Burakova, E.A.; Chelobanov, B.P.; Fokina, A.A.; Vlassov, V.V.; Altman, S.; Zenkova, M.A.; Stetsenko, D.A. Mesyl phosphoramidate antisense oligonucleotides as an alternative to phosphorothioates with improved biochemical and biological properties. Proc. Natl. Acad. Sci. USA 2019, 116, 1229–1234. [Google Scholar] [CrossRef]
- Patutina, O.A.; Gaponova Miroshnichenko, S.K.; Sen’kova, A.V.; Savin, I.A.; Gladkikh, D.V.; Burakova, E.A.; Fokina, A.A.; Maslov, M.A.; Shmendel, E.V.; Wood, M.; et al. Mesyl phosphoramidate backbone modified antisense oligonucleotides targeting miR-21 with enhanced in vivo therapeutic potency. Proc. Natl. Acad. Sci. USA 2020, 117, 32370–32379. [Google Scholar] [CrossRef]
- Anderson, B.A.; Freestone, G.C.; Low, A.; De-Hoyos, C.L.; Drury, W.J., III; Østergaard, M.E.; Migawa, M.T.; Fazio, M.; Wan, W.B.; Berdeja, A.; et al. Towards next generation antisense oligonucleotides: Mesylphosphoramidate modification improves therapeutic index and duration of effect of gapmer antisense oligonucleotides. Nucleic Acids Res. 2021, 49, 9026–9041. [Google Scholar] [CrossRef] [PubMed]
- Majlessi, M.; Nelson, N.C.; Becker, M.M. Advantages of 2′-O-methyl oligoribonucleotide probes for detecting RNA targets. Nucleic Acids Res. 1998, 26, 2224–2229. [Google Scholar] [CrossRef]
- Miroshnichenko, S.K.; Amirloo, B.; Bichenkova, E.V.; Vlassov, V.V.; Zenkova, M.A.; Patutina, O.A. 2’OMe modification of antimirna-21 oligonucleotide–peptide conjugate improves its hybridization properties and catalytic activity. Russ. J. Bioorg. Chem. 2019, 45, 803–812. [Google Scholar] [CrossRef]
- Geary, R.S.; Watanabe, T.A.; Truong, L.; Freier, S.; Lesnik, E.A.; Sioufi, N.B.; Sasmor, H.; Manoharan, M.; Levin, A.A. Pharmacokinetic properties of 2′-O-(2-methoxyethyl)-modified oligonucleotide analogs in rats. J. Pharmacol. Exp. Ther. 2001, 296, 890–897. [Google Scholar] [PubMed]
- Koizumi, M.; Takagi-Sato, M.; Okuyama, R.; Araki, K.; Sun, W.; Nakai, D.; Tsutsumi, S.; Kawai, K. Direct comparison of in vivo antisense activity of ENA oligonucleotides targeting ptp1b mRNA with that of 2′-O-(2-methoxy) ethyl-modified oligonucleotides. Oligonucleotides 2006, 16, 253–262. [Google Scholar] [CrossRef]
- Veedu, R.N.; Wengel, J. Locked nucleic acids: Promising nucleic acid analogs for therapeutic applications. Chem. Biodivers. 2010, 7, 536–542. [Google Scholar] [CrossRef]
- Le, B.T.; Adams, A.M.; Fletcher, S.; Wilton, S.D.; Veedu, R.N. Rational design of short locked nucleic acid-modified 2′-O-methyl antisense oligonucleotides for efficient exon-skipping in vitro. Mol. Ther. Nucleic Acids 2017, 9, 155–161. [Google Scholar] [CrossRef]
- Veedu, R.N.; Wengel, J. Locked nucleic acid as a novel class of therapeutic agents. RNA Biol. 2009, 6, 321–323. [Google Scholar] [CrossRef]
- Yu, H.; Zhang, S.; Chaput, J.C. Darwinian evolution of an alternative genetic system provides support for TNA as an RNA progenitor. Nat. Chem. 2012, 4, 183–187. [Google Scholar] [CrossRef]
- Lin, Y.; Qiu, Q.; Gill, S.C.; Jayasena, S.D. Modified RNA sequence pools for in vitro selection. Nucleic Acids Res. 1994, 22, 5229–5234. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Le, B.T.; Chakravarthy, M.; Kosbar, T.R.; Veedu, R.N. Systematic evaluation of 2′-fluoro modified chimeric antisense oligonucleotide-mediated exon skipping in vitro. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Alves Ferreira-Bravo, I.; Cozens, C.; Holliger, P.; DeStefano, J.J. Selection of 2′-deoxy-2′-fluoroarabinonucleotide (FANA) aptamers that bind HIV-1 reverse transcriptase with picomolar affinity. Nucleic Acids Res. 2015, 43, 9587–9599. [Google Scholar] [CrossRef] [PubMed]
- Le, B.T.; Chen, S.; Abramov, M.; Herdewijn, P.; Veedu, R.N. Evaluation of anhydrohexitol nucleic acid, cyclohexenyl nucleic acid and d-altritol nucleic acid-modified 2′-O-methyl RNA mixmer antisense oligonucleotides for exon skipping in vitro. Chem. Commun. 2016, 52, 13467–13470. [Google Scholar] [CrossRef]
- Pallan, P.S.; Allerson, C.R.; Berdeja, A.; Seth, P.P.; Swayze, E.E.; Prakash, T.P.; Egli, M. Structure and nuclease resistance of 2′,4′-constrained 2′-O-methoxyethyl (cMOE) and 2′-O-ethyl (cEt) modified DNAs. Chem. Comm. 2012, 48, 8195–8197. [Google Scholar] [CrossRef] [PubMed]
- Hyrup, B.; Nielsen, P.E. Peptide nucleic acids (PNA): Synthesis, properties and potential applications. Bioorg. Med. Chem. 1996, 4, 5–23. [Google Scholar] [CrossRef]
- Summerton, J.; Weller, D. Morpholino antisense oligomers: Design, preparation, and properties. Antisense Nucleic Acid Drug Dev. 1997, 7, 187–195. [Google Scholar] [CrossRef]
- Le, B.T.; Veedu, R.N.; Fletcher, S.; Wilton, S.D. Antisense oligonucleotide development for the treatment of muscular dystrophies. Expert Opin. Orphan Drugs 2016, 4, 139–152. [Google Scholar]
- Agrawal, S.; Jiang, Z.; Zhao, Q.; Shaw, D.; Cai, Q.; Roskey, A.; Channavajjala, L.; Saxinger, C.; Zhang, R. Mixed-backbone oligonucleotides as second generation antisense oligonucleotides: In vitro and in vivo studies. Proc. Natl. Acad. Sci. USA 1997, 94, 2620–2625. [Google Scholar] [CrossRef]
- Stanton, R.; Sciabola, S.; Salatto, C.; Weng, Y.; Moshinsky, D.; Little, J.; Walters, E.; Kreeger, J.; DiMattia, D.; Chen, T.; et al. Chemical modification study of antisense gapmers. Nucleic Acid Ther. 2012, 22, 344–359. [Google Scholar] [CrossRef]
- Monia, B.P.; Lesnik, E.A.; Gonzalez, C.; Lima, W.F.; McGee, D.; Guinosso, C.J.; Kawasaki, A.M.; Cook, P.D.; Freier, S.M. Evaluation of 2′-modified oligonucleotides containing 2′-deoxy gaps as antisense inhibitors of gene expression. J. Biol. Chem. 1993, 268, 14514–14522. [Google Scholar] [CrossRef]
- Chen, S.; Le, B.T.; Rahimizadeh, K.; Shaikh, K.; Mohal, N.; Veedu, R.N. Synthesis of a morpholino nucleic acid (MNA)-uridine phosphoramidite, and exon skipping using MNA/2′-O-methyl mixmer antisense oligonucleotide. Molecules 2016, 21, 1582. [Google Scholar] [CrossRef] [PubMed]
- Langner, H.K.; Jastrzebska, K.; Caruthers, M.H. Synthesis and characterization of thiophosphoramidate morpholino oligonucleotides and chimeras. J. Am. Chem. Soc. 2020, 142, 16240–16253. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.K.; Corey, D.R. Silencing disease genes in the laboratory and the clinic. J. Pathol. 2012, 226, 365–379. [Google Scholar] [CrossRef] [PubMed]
- Siomi, H.; Siomi, M.C. On the road to reading the RNA-interference code. Nature 2009, 457, 396–404. [Google Scholar] [CrossRef]
- Sajid, M.I.; Moazzam, M.; Kato, S.; Yeseom Cho, K.; Tiwari, R.K. Overcoming barriers for siRNA therapeutics: From bench to bedside. Pharmaceuticals 2020, 13, 294. [Google Scholar] [CrossRef]
- Hu, B.; Zhong, L.; Weng, Y.; Peng, L.; Huang, Y.; Zhao, Y.; Liang, X.-J. Therapeutic siRNA: State of the art. Signal. Transduct. Target. Ther. 2020, 5, 101. [Google Scholar] [CrossRef]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef]
- Jackson, A.L.; Burchard, J.; Leake, D.; Reynolds, A.; Schelter, J.; Guo, J.; Johnson, J.M.; Lim, L.; Karpilow, J.; Nichols, K.; et al. Position-specific chemical modification of siRNAs reduces “off-target” transcript silencing. RNA 2006, 12, 1197–1205. [Google Scholar] [CrossRef]
- Balachandran, A.A.; Larcher, L.M.; Chen, S.; Veedu, R.N. Therapeutically significant microRNAs in primary and metastatic brain malignancies. Cancers 2020, 12, 2534. [Google Scholar] [CrossRef]
- Lennox, K.A.; Behlke, M.A. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011, 18, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Piva, R.; Spandidos, D.A.; Gambari, R. From microRNA functions to microRNA therapeutics: Novel targets and novel drugs in breast cancer research and treatment (review). Int. J. Oncol. 2013, 43, 985–994. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Wang, S.C.; Hsu, C.Y.; Miao, Y.; Martin, M.; Yin, Y.; Wu, C.C.; Wang, Y.T.; Wu, G.; Chien, S.; et al. MicroRNA-92a mediates endothelial dysfunction in CKD. J. Am. Soc. Nephrol. 2017, 28, 3251–3261. [Google Scholar] [CrossRef] [PubMed]
- Hinkel, R.; Penzkofer, D.; Zühlke, S.; Fischer, A.; Husada, W.; Xu, Q.F.; Baloch, E.; van Rooij, E.; Zeiher, A.M.; Kupatt, C.; et al. Inhibition of microRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation 2013, 128, 1066–1075. [Google Scholar] [CrossRef]
- Moreno, J.A.; Hamza, E.; Guerrero-Hue, M.; Rayego-Mateos, S.; García-Caballero, C.; Vallejo-Mudarra, M.; Metzinger, L.; Meuth, V.M.-L. Non-coding RNAs in kidney diseases: The long and short of them. Int. J. Mol. Sci. 2021, 22, 6077. [Google Scholar] [CrossRef]
- Li, N.; Cui, Y.; Yin, M.; Liu, F. Screening potential prognostic biomarkers of long non-coding RNAs for predicting the risk of chronic kidney disease. Braz. J. Med. Biol. Res. 2019, 52, e8333. [Google Scholar] [CrossRef]
- Daniel, C.; Takabatake, Y.; Mizui, M.; Isaka, Y.; Kawashi, H.; Rupprecht, H.; Imai, E.; Hugo, C. Antisense oligonucleotides against thrombospondin-1 inhibit activation of TGF- β in fibrotic renal disease in the rat in vivo. Am. J. Pathol. 2003, 163, 1185–1192. [Google Scholar] [CrossRef]
- Guha, M.; Xu, Z.G.; Tung, D.; Lanting, L.; Natarajan, R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007, 21, 3355–3368. [Google Scholar] [CrossRef]
- Wang, J.H.; Newbury, L.J.; Knisely, A.S.; Monia, B.; Hendry, B.M.; Sharpe, C.C. Antisense knockdown of Kras inhibits fibrosis in a rat model of unilateral ureteric obstruction. Am. J. Pathol. 2012, 180, 82–90. [Google Scholar] [CrossRef]
- Ravichandran, K.; Zafar, I.; He, Z.; Doctor, R.B.; Moldovan, R.; Mullick, A.E.; Edelstein, C.L. An mTOR anti-sense oligonucleotide decreases polycystic kidney disease in mice with a targeted mutation in Pkd2. Hum. Mol. Genet. 2014, 23, 4919–4931. [Google Scholar] [CrossRef]
- Ravichandran, K.; Ozkok, A.; Wang, Q.; Mullick, A.E.; Edelstein, C.L. Antisense-mediated angiotensinogen inhibition slows polycystic kidney disease in mice with a targeted mutation in Pkd2. Am. J. Physiol. Ren. Physiol. 2015, 308, F349–F357. [Google Scholar] [CrossRef]
- Aghajan, M.; Booten, S.L.; Althage, M.; Hart, C.E.; Ericsson, A.; Maxvall, I.; Ochaba, J.; Menschik-Lundin, A.; Hartleib, J.; Kuntz, S.; et al. Antisense oligonucleotide treatment ameliorates IFN-γ-induced proteinuria in APOL1-transgenic mice. JCI Insight 2019, 4, e126124. [Google Scholar] [CrossRef] [PubMed]
- Bülow, R.D.; Boor, P. Extracellular matrix in kidney fibrosis: More than just a scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Border, W.A.; Noble, N.A. Transforming growth factor-β in tissue fibrosis. N. Engl. J. Med. 1994, 331, 1286–1292. [Google Scholar] [PubMed]
- Okuda, S.; Languino, L.R.; Rouslahti, E.; Border, W.A. Elevated expression of transforming growth factor-β and proteoglycan production in experimental glomerulonephritis. J. Clin. Investig. 1990, 86, 453–462. [Google Scholar] [CrossRef]
- Kopp, J.B.; Factor, V.M.; Mozes, M.; Nagy, P.; Sanderson, N.; Böttinger, E.P.; Klotman, P.E.; Thorgeirsson, S.S. Transgenic mice with increased plasma levels of TGF-β1 develop progressive renal disease. Lab. Investig. 1996, 74, 991–1003. [Google Scholar]
- Sharma, K.; Ziyadeh, F.N. The emerging role of transforming growth factor-β in kidney disease. Am. J. Physiol. 1994, 35, F829–F842. [Google Scholar] [CrossRef]
- Akagi, Y.; Isaka, Y.; Arai, M.; Kaneko, T.; Takenaka, M.; Moriyama, T.; Kaneda, Y.; Ando, A.; Orita, Y.; Kamada, T.; et al. Inhibition of TGF-β 1 expression by antisense oligonucleotides suppressed extracellular matrix accumulation in experimental glomerulonephritis. Kidney Int. 1996, 50, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschmann, T. TGF-β2 knockout mice have multiple developmental defects that are nonoverlapping with other TGF-β knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [CrossRef] [PubMed]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warbuton, D.; Bu, D.; Heitserkamp, N.; Groffen, J. Abnormal lung development and cleft palate in mice lacking TGF-β3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef]
- Schultz-Cherry, S.; Murphy-Ullrich, J.E. Thrombospondin causes activation of latent transforming growth factor-β secreted by endothelial cells by a novel mechanism. J. Cell Biol. 1993, 122, 923–932. [Google Scholar] [CrossRef]
- Tada, H.; Isogai, S. The fibronectin production is increased by thrombospondin via activation of TGF-β in cultured human mesangial cells. Nephron 1998, 79, 38–44. [Google Scholar] [CrossRef]
- Crawford, S.E.; Chen, H.; Mosher, D.; Misenheimer, T.; Krutzsch, H.; Roberts, D.D.; Murphy-Ullrich, J.E. Thrombospondin-1 is a major activator of TGF-β1 in vivo. Cell 1998, 93, 1159–1170. [Google Scholar] [CrossRef]
- Lawler, J.; Sunday, M.; Thibert, V.; Duquette, M.; George, E.L.; Rayburn, H.; Hynes, R.O. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J. Clin. Investig. 1998, 101, 982–992. [Google Scholar] [CrossRef]
- Hugo, C.; Shankland, S.J.; Pichler, R.H.; Couser, W.G.; Johnson, R.J. Thrombospondin 1 precedes and predicts the development of tubulointerstitial fibrosis in glomerular disease in the rat. Kidney Int. 1998, 53, 302–311. [Google Scholar] [CrossRef]
- Hugo, C.; Pichler, R.; Meek, R.; Gordon, K.; Kyriakides, T.; Floege, J.; Bornstein, P.; Couser, W.; Johnson, R.J. Thrombospondin1 is expressed by proliferating mesangial cells in vivo and is up-regulated by PDGF and bFGF. Kidney Int. 1995, 48, 1846–1856. [Google Scholar] [CrossRef]
- Kreisberg, J.I.; Ayo, S.H. The glomerular mesangium in diabetes mellitus. Kidney Int. 1993, 43, 109–113. [Google Scholar] [CrossRef][Green Version]
- Ziyadeh, F.N.; Sharma, K.; Ericksen, M.; Wolf, G. Stimulation of collagen gene expression and protein synthesis in murine mesangial cells by high glucose is mediated by autocrine activation of transforming growth factor-β. J. Clin. Investig. 1994, 93, 536–542. [Google Scholar] [CrossRef]
- Ziyadeh, F.N.; Han, D.C. Involvement of transforming growth factor-beta and its receptors in the pathogenesis of diabetic nephrology. Kidney Int. 1997, 60, S7–S11. [Google Scholar]
- Hoffman, B.B.; Sharma, K.; Zhu, Y.; Ziyadeh, F.N. Transcriptional activation of transforming growth factor-β1 in mesangial cell culture by high glucose concentration. Kidney Int. 1998, 54, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Han, D.C.; Isono, M.; Hoffman, B.B.; Ziyadeh, F.N. High glucose stimulates proliferation and collagen type I synthesis in renal cortical fibroblasts: Mediation by autocrine activation of TGF-beta. J. Am. Soc. Nephrol. 1999, 10, 1891–1899. [Google Scholar] [CrossRef]
- Reeves, W.B.; Andreoli, T.E. Transforming growth factor beta contributes to progressive diabetic nephropathy. Proc. Natl. Acad. Sci. USA 2000, 97, 7667–7669. [Google Scholar] [CrossRef] [PubMed]
- Riser, B.L.; Denichilo, M.; Cortes, P.; Baker, C.; Grondin, J.M.; Yee, J.; Narins, R.G. Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J. Am. Soc. Nephrol. 2000, 11, 25–38. [Google Scholar] [CrossRef] [PubMed]
- Sakharova, O.V.; Taal, M.W.; Brenner, B.M. Pathogenesis of diabetic nephropathy: Focus on transforming growth factor-beta and connective tissue growth factor. Curr. Opin. Nephrol. Hypertens. 2001, 10, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.; Godson, C.; Cannon, S.; Kato, S.; Mackenzie, H.S.; Martin, F.; Brady, H.R. Suppression subtractive hybridization identifies high glucose levels as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J. Biol. Chem. 1999, 274, 5830–5834. [Google Scholar] [CrossRef]
- Okada, H.; Kikuta, T.; Kobayashi, T.; Inoue, T.; Kanno, Y.; Takigawa, T.; Sugaya, T.; Kopp, J.B.; Suzuki, H. Connective tissue growth factor expressed in tubular epithelium plays a pivotal role in renal fibrogenesis. J. Am. Soc. Nephrol. 2005, 16, 133–143. [Google Scholar] [CrossRef]
- Norman, J.T.; Fine, L.G. Progressive renal disease: Fibroblasts, extracellular matrix, and integrins. Exp. Nephrol. 1999, 7, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Wittinghofer, A.; Scheffzek, K.; Ahmadian, M.R. The interaction of Ras with GTPase-activating proteins. FEBS Lett. 1997, 410, 63–67. [Google Scholar] [CrossRef]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Muller, G.A.; Kalbacher, H.; Salant, D.J.; Muller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grunert, S. Ras and TGFβ cooperatively regulate epithelial cell plasticity and metastasis: Dissection of Ras signaling pathways. J. Cell Biol. 2002, 156, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Clarke, H.C.; Kocher, H.M.; Khwaja, A.; Kloog, Y.; Cook, H.T.; Hendry, B.M. Ras antagonist farnesylthiosalicylic acid (FTS) reduces glomerular cellular proliferation and macrophage number in rat thy-1 nephritis. J. Am. Soc. Nephrol. 2003, 14, 848–854. [Google Scholar] [CrossRef]
- Kocher, H.M.; Moorhead, J.; Sharpe, C.C.; Dockrell, M.E.; Al-Nawab, M.; Hendry, B.M. Expression of Ras GTPases in normal kidney and in glomerulonephritis. Nephrol. Dial. Transplant. 2003, 18, 2284–2292. [Google Scholar] [CrossRef]
- Martinez-Salgado, C.; Fuentes-Calvo, I.; Garcia-Cenador, B.; Santos, E.; Lopez-Novoa, J.M. Involvement of H- and N-Ras isoforms in transforming growth factor-β1-induced proliferation and in collagen and fibronectin synthesis. Exp. Cell Res. 2006, 312, 2093–2106. [Google Scholar] [CrossRef]
- Rodriguez-Pena, A.B.; Grande, M.T.; Eleno, N.; Arevalo, M.; Guerrero, C.; Santos, E.; Lopez-Novoa, J.M. Activation of Erk1/2 and Akt following unilateral ureteral obstruction. Kidney Int. 2008, 74, 196–209. [Google Scholar] [CrossRef]
- Dockrell, M.E.; Phanish, M.K.; Hendry, B.M. Tgf-β auto-induction and connective tissue growth factor expression in human renal tubule epithelial cells requires N-ras. Nephron Exp. Nephrol. 2009, 112, e71–e79. [Google Scholar] [CrossRef]
- Lahsnig, C.; Mikula, M.; Petz, M.; Zulehner, G.; Schneller, D.; van Zijl, F.; Huber, H.; Csiszar, A.; Beug, H.; Mikulits, W. ILEI requires oncogenic Ras for the epithelial to mesenchymal transition of hepatocytes and liver carcinoma progression. Oncogene 2009, 28, 638–650. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; Fuentes-Calvo, I.; Arevalo, M.; Heredia, F.; Santos, E.; Martinez-Salgado, C.; Rodriguez-Puyol, D.; Nieto, M.A.; Lopez-Novoa, J.M. Deletion of H-Ras decreases renal fibrosis and myofibroblast activation following ureteral obstruction in mice. Kidney Int. 2010, 77, 509–518. [Google Scholar] [CrossRef]
- Sharpe, C.C.; Dockrell, M.E.; Noor, M.I.; Monia, B.P.; Hendry, B.M. Role of Ras isoforms in the stimulated proliferation of human renal fibroblasts in primary culture. J. Am. Soc. Nephrol. 2000, 11, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Sharpe, C.C.; Dockrell, M.E.; Scott, R.; Noor, M.I.; Cowsert, L.M.; Monia, B.P.; Hendry, B.M. Evidence of a role for Ki-Ras in the stimulated proliferation of renal fibroblasts. J. Am. Soc. Nephrol. 1999, 10, 1186–1192. [Google Scholar] [CrossRef]
- Klahr, S.; Morrissey, J. Obstructive nephropathy and renal fibrosis. Am. J. Physiol. Ren. Physiol. 2002, 283, F861–F875. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.J.; Revenko, A.S.; Hanson, L.L.; Ellston, R.; Staniszewska, A.; Whalley, N.; Pandey, S.K.; Revill, M.; Rooney, C.; Buckett, L.K.; et al. Targeting KRAS-dependent tumors with AZD4785, a high-affinity therapeutic antisense oligonucleotide inhibitor of KRAS. Sci. Transl. Med. 2017, 9, eaal5253. [Google Scholar] [CrossRef]
- Grantham, J.J. Clinical practice. Autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2008, 359, 1477–1485. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, C.L. Mammalian target of rapamycin and caspase inhibitors in polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2008, 3, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Shillingford, J.M.; Murcia, N.S.; Larson, C.H.; Low, S.H.; Hedgepeth, R.; Brown, N.; Flask, C.A.; Novick, A.C.; Goldfarb, D.A.; Kramer-Zucker, A.; et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. USA 2006, 103, 5466–5471. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.G.; Weimbs, T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J. Am. Soc. Nephrol. 2010, 21, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Wahl, P.R.; Serra, A.L.; Le Hir, M.; Molle, K.D.; Hall, M.N.; Wuthrich, R.P. Inhibition of mTOR with sirolimus slows disease progression in Han:SPRD rats with autosomal dominant polycystic kidney disease (ADPKD). Nephrol. Dial. Transplant. 2006, 21, 598–604. [Google Scholar] [CrossRef]
- Fischer, D.C.; Jacoby, U.; Pape, L.; Ward, C.J.; Kuwertz-Broeking, E.; Renken, C.; Nizze, H.; Querfeld, U.; Rudolph, B.; Mueller-Wiefel, D.E.; et al. Activation of the AKT/mTOR pathway in autosomal recessive polycystic kidney disease (ARPKD). Nephrol. Dial. Transplant. 2009, 24, 1819–1827. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef]
- Rodrik-Outmezguine, V.S.; Chandarlapaty, S.; Pagano, N.C.; Poulikakos, P.I.; Scaltriti, M.; Moskatel, E.; Baselga, J.; Guichard, S.; Rosen, N. mTOR kinase inhibition causes feedback-dependent biphasic regulation of AKT signaling. Cancer Discov. 2011, 1, 248–259. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Belibi, F.; Ravichandran, K.; Zafar, I.; He, Z.; Edelstein, C.L. mTORC1/2 and rapamycin in female Han:SPRD rats with polycystic kidney disease. Am. J. Physiol. 2011, 300, F236–F244. [Google Scholar] [CrossRef]
- Natoli, T.A.; Smith, L.A.; Rogers, K.A.; Wang, B.; Komarnitsky, S.; Budman, Y.; Belenky, A.; Bukanov, N.O.; Dackowski, W.R.; Husson, H.; et al. Inhibition of glucosylceramide accumulation results in effective blockade of polycystic kidney disease in mouse models. Nat. Med. 2010, 16, 788–792. [Google Scholar] [CrossRef]
- Zafar, I.; Ravichandran, K.; Belibi, F.; Doctor, R.B.; Edelstein, C.L. Sirolimus attenuates disease progression in an orthologous mouse model of human autosomal dominant polycystic kidney disease. Kidney Int. 2010, 78, 754–761. [Google Scholar] [CrossRef]
- Serra, A.L.; Poster, D.; Kistler, A.D.; Krauer, F.; Raina, S.; Young, J.; Rentsch, K.M.; Spanaus, K.S.; Senn, O.; Kristanto, P.; et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2010, 363, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Shor, B.; Gibbons, J.J.; Abraham, R.T.; Yu, K. Targeting mTOR globally in cancer; thinking beyond rapamycin. Cell Cycle 2009, 8, 3831–3837. [Google Scholar] [CrossRef] [PubMed]
- Dazert, E.; Hall, M.N. mTOR signaling in disease. Curr. Opin. Cell Biol. 2011, 23, 744–755. [Google Scholar] [CrossRef]
- Wilson, P.D. Polycystic kidney disease. N. Engl. J. Med. 2004, 350, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Raizada, V.; Skipper, B.; Luo, W.; Griffith, J. Intracardiac and intrarenal renin-angiotensin systems: Mechanisms of cardiovascular and renal effects. J. Investig. Med. 2007, 55, 341–359. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Ruperez, M.; Esteban, V.; Rodriguez-Vita, J.; Sanchez-Lopez, E.; Carvajal, G.; Egido, J. Angiotensin II: A key factor in the inflammatory and fibrotic response in kidney diseases. Nephrol. Dial. Transplant. 2006, 21, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W. Renal volume, renin-angiotensin-aldosterone system, hypertension, and left ventricular hypertrophy in patients with autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2009, 20, 1888–1893. [Google Scholar] [CrossRef]
- Wu, C.; Lu, H.; Cassis, L.A.; Daugherty, A. Molecular and pathophysiological features of angiotensinogen: A mini review. Am. J. Med. Sci. 2011, 4, 183–190. [Google Scholar] [CrossRef]
- Ronco, C.; Di Lullo, L. Cardiorenal syndrome. Heart Fail. Clin. 2014, 10, 251–280. [Google Scholar] [CrossRef]
- Long, D.A.; Price, K.L.; Herrera-Acosta, J.; Johnson, R.J. How does angiotensin II cause renal injury? Hypertension 2004, 43, 722–723. [Google Scholar] [CrossRef]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Classical renin-angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201–1228. [Google Scholar]
- Theuer, J.; Dechend, R.; Muller, D.N.; Park, J.K.; Fiebeler, A.; Barta, P.; Ganten, D.; Haller, H.; Dietz, R.; Luft, F.C. Angiotensin II induced inflammation in the kidney and in the heart of double transgenic rats. BMC Cardiovasc. Disord. 2002, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.E.; Malik, H.S. The apolipoprotein L family of programmed cell death and immunity genes rapidly evolved in primates at discrete sites of host-pathogen interactions. Genome Res. 2009, 19, 850–858. [Google Scholar] [CrossRef]
- Monajemi, H.; Fontijn, R.D.; Pannekoek, H.; Horrevoets, A.J. The apolipoprotein L gene cluster has emerged recently in evolution and is expressed in human vascular tissue. Genomics 2002, 79, 539–546. [Google Scholar] [CrossRef]
- Shukha, K.; Mueller, J.L.; Chung, R.T.; Curry, M.P.; Friedman, D.J.; Pollak, M.R.; Berg, A.H. Most ApoL1 is secreted by the liver. J. Am. Soc. Nephrol. 2017, 28, 1079–1083. [Google Scholar] [CrossRef]
- Vanhamme, L.; Paturiaux-Hanocq, F.; Poelvoorde, P.; Nolan, D.P.; Lins, L.; Van Den Abbeele, J.; Pays, A.; Tebabi, P.; Van Xong, H.; Jacquet, A.; et al. Apolipoprotein L-I is the trypanosome lytic factor of human serum. Nature 2003, 422, 83–87. [Google Scholar] [CrossRef]
- Pérez-Morga, D.; Vanhollebeke, B.; Paturiaux-Hanocq, F.; Nolan, D.P.; Lins, L.; Homblé, F.; Vanhamme, L.; Tebabi, P.; Pays, A.; Poelvoorde, P.; et al. Apolipoprotein L-I promotes trypanosome lysis by forming pores in lysosomal membranes. Science 2005, 309, 469–472. [Google Scholar] [CrossRef]
- Genovese, G.; Friedman, D.J.; Ross, M.D.; Lecordier, L.; Uzureau, P.; Freedman, B.I.; Bowden, D.W.; Langefeld, C.D.; Oleksyk, T.K.; Uscinski Knob, A.L.; et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 2010, 329, 841–845. [Google Scholar] [CrossRef]
- Nichols, B.; Jog, P.; Lee, J.H.; Blackler, D.; Wilmot, M.; D’Agati, V.; Markowitz, G.; Kopp, J.B.; Alper, S.L.; Pollak, M.R.; et al. Innate immunity pathways regulate the nephropathy gene Apolipoprotein L1. Kidney Int. 2015, 87, 332–342. [Google Scholar] [CrossRef] [PubMed]
- Beckerman, P.; Bi-Karchin, J.; Park, A.S.; Qiu, C.; Dummer, P.D.; Soomro, I.; Boustany-Kari, C.M.; Pullen, S.S.; Miner, J.H.; Hu, C.A.; et al. Transgenic expression of human APOL1 risk variants in podocytes induces kidney disease in mice. Nat. Med. 2017, 23, 429–438. [Google Scholar] [CrossRef]
- Tzur, S.; Rosset, S.; Shemer, R.; Yudkovsky, G.; Selig, S.; Tarekegn, A.; Bekele, E.; Bradman, N.; Wasser, W.G.; Behar, D.M.; et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum. Genet. 2010, 128, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Kopp, J.B.; Nelson, G.W.; Sampath, K.; Johnson, R.C.; Genovese, G.; An, P.; Friedman, D.; Briggs, W.; Dart, R.; Korbet, S.; et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J. Am. Soc. Nephrol. 2011, 22, 2129–2137. [Google Scholar] [CrossRef]
- Saran, R.; Robinson, B.; Abbott, K.C.; Agodoa, L.Y.; Albertus, P.; Ayanian, J.; Balkrishnan, R.; Bragg-Gresham, J.; Cao, J.; Chen, J.L.; et al. US renal data system 2016 annual data report: Epidemiology of kidney disease in the United States. Am. J. Kidney Dis. 2017, 69, A7–A8. [Google Scholar] [CrossRef]
- Parsa, A.; Kao, W.H.; Xie, D.; Astor, B.C.; Li, M.; Hsu, C.Y.; Feldman, H.I.; Parekh, R.S.; Kusek, J.W.; Greene, T.H.; et al. APOL1 risk variants, race, and progression of chronic kidney disease. N. Engl. J. Med. 2013, 369, 2183–2196. [Google Scholar] [CrossRef]
- Johnstone, D.B.; Shegokar, V.; Nihalani, D.; Rathore, Y.S.; Mallik, L.; Ashish; Zare, V.; Ikizler, H.O.; Powar, R.; Holzman, L.B. APOL1 null alleles from a rural village in India do not correlate with glomerulosclerosis. PLoS ONE 2012, 7, e51546. [Google Scholar]
- Joshi, P.P.; Shegokar, V.R.; Powar, R.M.; Herder, S.; Katti, R.; Salkar, H.R.; Dani, V.S.; Bhargava, A.; Jannin, J.; Truc, P. Human trypanosomiasis caused by Trypanosoma evansi in India: The first case report. Am. J. Trop. Med. Hyg. 2005, 73, 491–495. [Google Scholar] [CrossRef]
- Vanhollebeke, B.; Truc, P.; Poelvoorde, P.; Pays, A.; Joshi, P.P.; Katti, R.; Jannin, J.G.; Pays, E. Human Trypanosoma evansi infection linked to a lack of apolipoprotein L-I. N. Engl. J. Med. 2006, 355, 2752–2756. [Google Scholar] [CrossRef] [PubMed]
- Juliano, R.L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. [Google Scholar] [CrossRef] [PubMed]
- Asami, Y.; Yoshioka, K.; Nishina, K.; Nagata, T.; Yokota, T. Drug delivery system of therapeutic oligonucleotides. Drug Discov. Ther. 2016, 10, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Yang, Z.; Tang, X. Chemical modifications of nucleic acid drugs and their delivery systems for gene-based therapy. Med. Res. Rev. 2018, 38, 829–869. [Google Scholar] [CrossRef]
- Geary, R.S.; Norris, D.; Yu, R.; Bennett, C.F. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug Deliv. Rev. 2015, 87, 46–51. [Google Scholar] [CrossRef]
- Kaczmarek, J.C.; Kowalski, P.S.; Anderson, D.G. Advances in the delivery of RNA therapeutics: From concept to clinical reality. Genome Med. 2017, 9, 60. [Google Scholar] [CrossRef] [PubMed]
- Phillips, M.I.; Kimura, B. Gene therapy for hypertension: Antisense inhibition of the renin-angiotensin system. Methods Mol. Med. 2005, 108, 363–379. [Google Scholar] [PubMed]
- Fiorentino, M.; Grandaliano, G.; Gesualdo, L.; Castellano, G. Acute kidney injury to chronic kidney disease transition. Contrib. Nephrol. 2018, 193, 45–54. [Google Scholar] [PubMed]
- Ko, G.J.; Grigoryev, D.N.; Linfert, D.; Jang, H.R.; Watkins, T.; Cheadle, C.; Racusen, L.; Rabb, H. Transcriptional analysis of kidneys during repair from AKI reveals possible roles for NGAL and KIM-1 as biomarkers of AKI-to-CKD transition. Am. J. Physiol. Ren. Physiol. 2010, 298, F1472–F1483. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | ASO Drug | Approval Year | Indication | Mechanism | Ref. |
|---|---|---|---|---|---|
| 1 | Fomivirsen (Vitravene®) | 1998 | Cytomegalovirus (CMV) retinitis | Downregulate the gene encoding CMV immediate-early 2 protein | [9,10] |
| 2 | Mipomersen (Kynamro®) | 2013 | Familial hypercholesterolemia (FH) | Downregulate the gene APOB encoding apolipoprotein B | [11,12] |
| 3 | Eteplirsen (Exondys 51®) | 2016 | Duchenne muscular dystrophy (DMD) | Rescue the expression of dystrophin through exon-51 skipping of the mRNA of DMD gene | [13,14,15,16] |
| 4 | Nusinersen (Spinraza®) | 2016 | Spinal muscular atrophy (SMA) | Increase the production of the survival motor neuron (SMN) protein by exon-7 inclusion of the mRNA of SMN2 gene | [17,18,19,20] |
| 5 | Inotersen (Tegsedi®) | 2018 | Hereditary transthyretin (TTR) amyloidosis | Downregulate the gene TTR encoding transthyretin | [21,22] |
| 6 | Golodirsen (Vyondys 53®) | 2019 | DMD | Rescue the expression of dystrophin through exon-53 skipping of the mRNA of DMD gene | [23] |
| 7 | Viltolarsen (Viltepso®) | 2020 | DMD | Rescue the expression of dystrophin through exon-53 skipping of the mRNA of DMD gene | [24] |
| 8 | Casimersen (Amondys 45®) | 2021 | DMD | Rescue the expression of dystrophin through exon-45 skipping of the mRNA of DMD gene | [25] |
| In Vitro Study (Initial Screen of ASO Candidates) | Best-Performing ASO Candidates | |||
|---|---|---|---|---|
| Target Gene | Chemistry | Cellular Target | ASO Sequences | Ref. |
| TSP-1 | Eleven 14–25 mer DNAPS ASOs | Mesangial cell | 5′-T*T*C*T*C*C*G*T*T*G*T*G*A*T*T*G*A*A-3′ 5′-C*A*C*C*T*C*C*A*A*T*G*A*G*T*T-3′ | [113] |
| CTGF | 20 mer 4-12-4 MOEPO-DNAPS-MOEPO and MOEPS-DNAPS-MOEPS ASOs | Rat mesangial cell line | 5′-CCACA*A*G*C*T*G*T*C*C*A*G*T*CTAA-3′ 5′-C*C*A*C*A*A*G*C*T*G*T*C*C*A*G*T*C*T*A*A-3′ | [114] |
| KRAS | 20 mer 5-10-5 MOEPS-DNAPS-MOEPS ASOs | Rat renal fibroblast (NRK-49F) | 5′-A*T*T*C*A*C*A*T*G*A*C*T*A*T*A*C*A*C*C*T-3′ 5′-C*A*C*A*C*T*T*A*T*T*C*C*C*T*A*C*T*A*G*G-3′ | [115] |
| MTOR | ~150 20 mer 5-10-5 MOEPS-DNAPS-MOEPS ASOs | Primary murine hepatocytes (for screening), type 1 Madin-Darby Canine Kidney cells (for other in vitro experiments) | 5′-T*C*C*A*C*T*T*T*T*C*A*C*A*G*C*A*C*T*G*C-3′ | [116] |
| AGT | ~150 20 mer 5-10-5 MOEPS-DNAPS-MOEPS ASOs | Primary murine hepatocytes | 5′-T*C*T*T*C*C*A*C*C*C*T*G*T*C*A*C*A*G*C*C-3′ | [117] |
| APOL1 | Over 4000 16 mer MOEPS-DNAPS-MOEPS or 2′-4′ constrained ethyl (cEt)PS-DNAPS-cEtPS ASOs | A-431 cell line | N/A | [118] |
| Target Gene | In Vivo Studies of ASO Mediated Gene Silencing | Ref. | |
|---|---|---|---|
| TSP-1 | Type of CKD model | Animal model | [113] |
| Induced experimental mesangial proliferative glomerulonephritis (the anti-Thy1 model) | Sprague-Dawley rats (150–200 g) | ||
| Therapeutic regimen of ASO | |||
| ASOs were transferred into renal glomeruli via left renal artery perfusion. Five days after the administration, kidneys were isolated for analysis. | |||
| Renal function and/or renal damage markers | |||
| Inhibited glomerular extracellular matrix accumulation determined by significantly reduced collagen IV positive glomerular area (%): TSP-1 ASO-treated group (~16%), scrambled ASO-treated group (~31%), p < 0.01. Markedly reduced mesangial cell activation determined by significantly reduced smooth-muscle-actin positive glomerular area (%): TSP-1 ASO-treated group (~15%), scrambled ASO-treated group (~39%), p < 0.01. | |||
| CTGF | Type of CKD model | Animal model | [114] |
| Mice received streptozotocin (STZ) to develop an experimental model of type 1 diabetes induced diabetic nephropathy, and db/db mice with naturally developed diabetic nephropathy | C57BL/6 mice | ||
| Therapeutic regimen of ASO | |||
| Mice with type 1 diabetes: 20 mg/kg (twice a week) for 16 weeks. db/db mice: 5, 10, 20 mg/kg (twice a week) for 8 weeks | |||
| Renal function and/or renal damage markers | |||
| Mice with type 1 diabetes: Reduced kidney hypertrophy determined by reduced ratio (kidney weight/body weight): CTGF ASO-treated group (1.4%), vehicle-treated group (1.9%), p < 0.02. Attenuated mesangial matrix expansion (a.u.): CTGF ASO-treated group (~1.8), vehicle-treated group (~3.2), p < 0.05. Significantly reduced urinary albumin determined by reduced 24 h urinary albumin excretion (urinary albumin/urinary creatinine, ug/mg): CTGF ASO-treated group (~1.5), vehicle-treated group (~4.0), p < 0.05. db/db mice: Matrix expansion (%): 10 mg/kg CTGF ASO-treated group (~60%), vehicle-treated group (~100%), p < 0.05. Urinary albumin/urinary creatinine (ug/mg): 20 mg/kg CTGF ASO-treated group (~1.2), vehicle-treated group (~2.4), p < 0.05. | |||
| KRAS | Type of CKD model | Animal model | [115] |
| Unilateral ureteric obstruction (UUO) model | Male Wistar rats | ||
| Therapeutic regimen of ASO | |||
| 12.5 mg/kg for six days (administration was performed on alternate days) | |||
| Renal function and/or renal damage markers | |||
| Significantly reduced fibrosis determined by reduced fibrosis score (%): KRAS ASO-1-treated group (17%), scrambled ASO-1 (~40%), p < 0.001; KRAS ASO-2-treated group (20.3%), scrambled ASO-2 (~36%), p < 0.01. | |||
| MTOR | Type of CKD model | Animal model | [116] |
| An orthologous model of human autosomal dominant polycystic kidney disease (ADPKD) caused by a mutation in the Pkd2 gene | C57BL/6 Pkd2WS25/− mice | ||
| Therapeutic regimen of ASO | |||
| Intraperitoneal injection at 100 mg/kg/week for the first 4 weeks and 50 mg/kg/week for the remaining 8 weeks | |||
| Renal function and/or renal damage markers | |||
| Improved kidney function determined by reduced ratio (kidney weight/body weight): MTOR ASO-treated group (1.5%), scrambled ASO-treated group (2.4%), p < 0.001; and cyst volume density: MTOR ASO-treated group (15.1%), scrambled ASO-treated group (34.1%), p < 0.01. | |||
| AGT | Type of CKD model | Animal model | [117] |
| An orthologous model of human ADPKD caused by a mutation in the Pkd2 gene | C57BL/6 Pkd2WS25/− mice | ||
| Therapeutic regimen of ASO | |||
| Intraperitoneal injection at 100 mg/kg/week for the first 4 weeks and 50 mg/kg/week for the remaining 8 weeks | |||
| Renal function and/or renal damage markers | |||
| Improved kidney function determined by reduced ratio (kidney weight/body weight): AGT ASO-treated group (1.5%), scrambled ASO-treated group (2.4%), p < 0.01; and cyst volume density: AGT ASO-treated group (22%), scrambled ASO-treated group (34.1%), p < 0.05. | |||
| APOL1 | Type of CKD model | Animal model | [118] |
| Human APOL1-transgenic mice with induced proteinuria by IFN-γ challenge | Human APOL1-transgenic C57BL/6 mice | ||
| Therapeutic regimen of ASO | |||
| Intraperitoneal injection at 50 mg/kg/week for four weeks | |||
| Renal function and/or renal damage markers | |||
| Prevention of IFN-γ induced proteinuria determined by urinary albumin-to-creatinine ratio (ACR) (ug Alb/mg Cre): APOL1 ASO-treated group (0), control ASO-treated group (~1000), p < 0.001. | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Tan, Y.; Zhang, R.; Wang, T.; Na, N.; Zheng, T.; Veedu, R.N.; Chen, S. Antisense Oligonucleotide: A Potential Therapeutic Intervention for Chronic Kidney Disease. Kidney Dial. 2022, 2, 16-37. https://doi.org/10.3390/kidneydial2010004
Li Y, Tan Y, Zhang R, Wang T, Na N, Zheng T, Veedu RN, Chen S. Antisense Oligonucleotide: A Potential Therapeutic Intervention for Chronic Kidney Disease. Kidney and Dialysis. 2022; 2(1):16-37. https://doi.org/10.3390/kidneydial2010004
Chicago/Turabian StyleLi, Yalin, Yuqin Tan, Rui Zhang, Tao Wang, Ning Na, Tong Zheng, Rakesh N. Veedu, and Suxiang Chen. 2022. "Antisense Oligonucleotide: A Potential Therapeutic Intervention for Chronic Kidney Disease" Kidney and Dialysis 2, no. 1: 16-37. https://doi.org/10.3390/kidneydial2010004
APA StyleLi, Y., Tan, Y., Zhang, R., Wang, T., Na, N., Zheng, T., Veedu, R. N., & Chen, S. (2022). Antisense Oligonucleotide: A Potential Therapeutic Intervention for Chronic Kidney Disease. Kidney and Dialysis, 2(1), 16-37. https://doi.org/10.3390/kidneydial2010004