

Molecular Dynamics Simulations Suggest SARS-CoV-2 3CLpro Mutations in Beta and Omicron Variants Do Not Alter Binding Affinities for Cleavage Sites of Non-Structural Proteins
 , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Site-Directed Mutation In Silico
2.2. Thermodynamic Integration
3. Results and Discussion
3.1. Molecular and Structural Relationships of the 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron
3.2. Predicted Binding Affinity of Different Substrates Interacting with 3CLproH41A, 3CLproWT, 3CLproBeta, and 3CLproOmicron
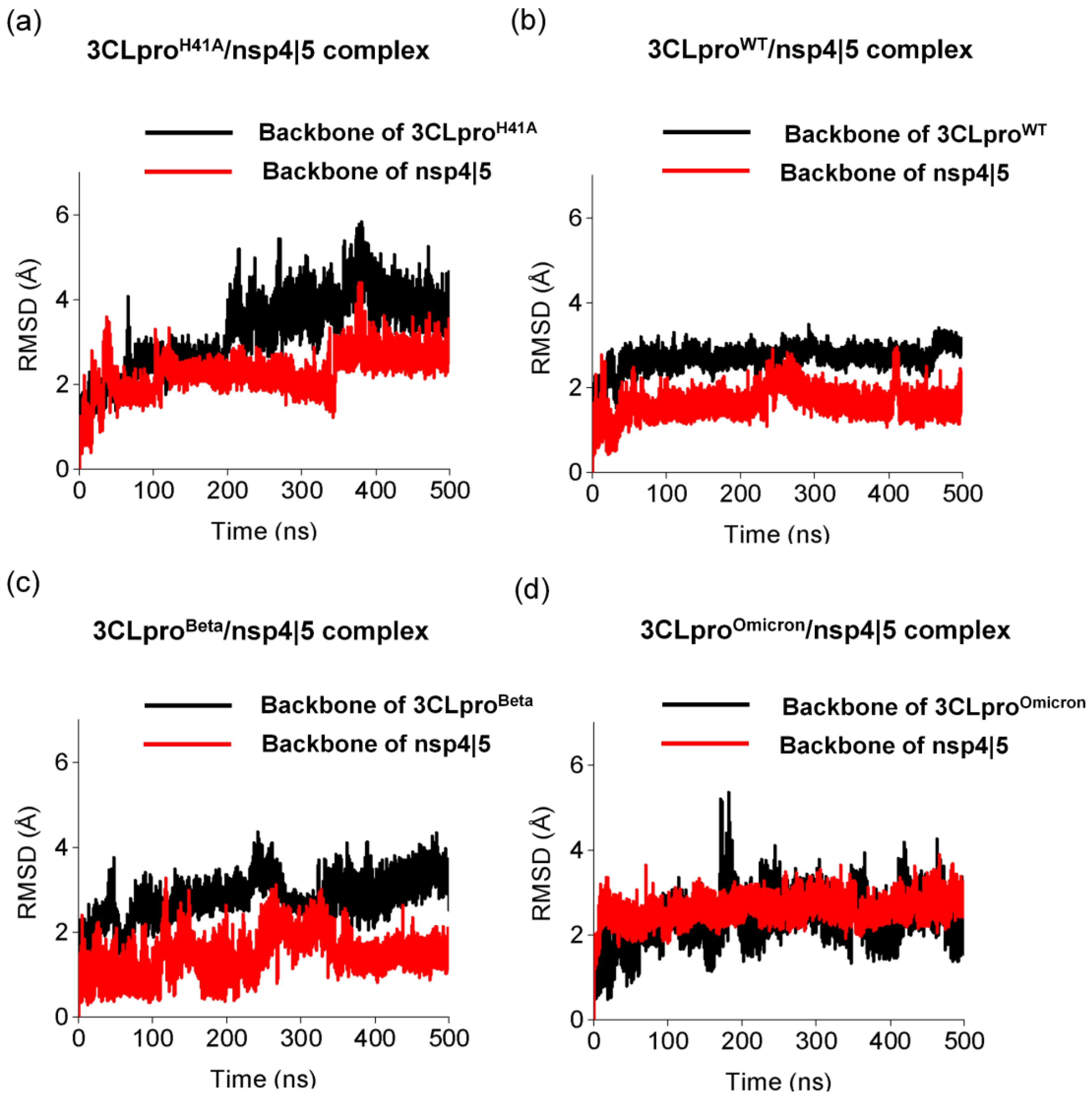
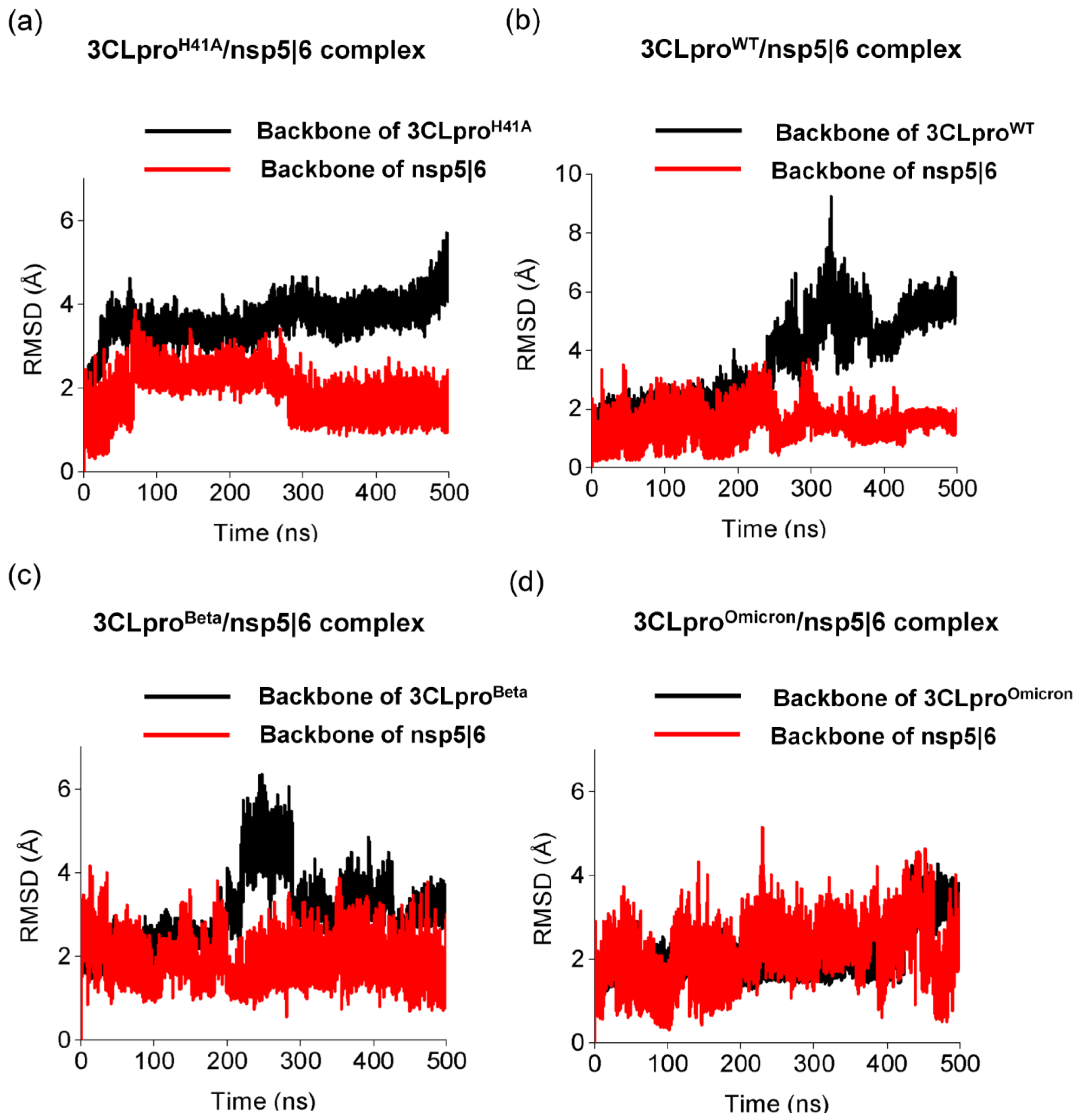
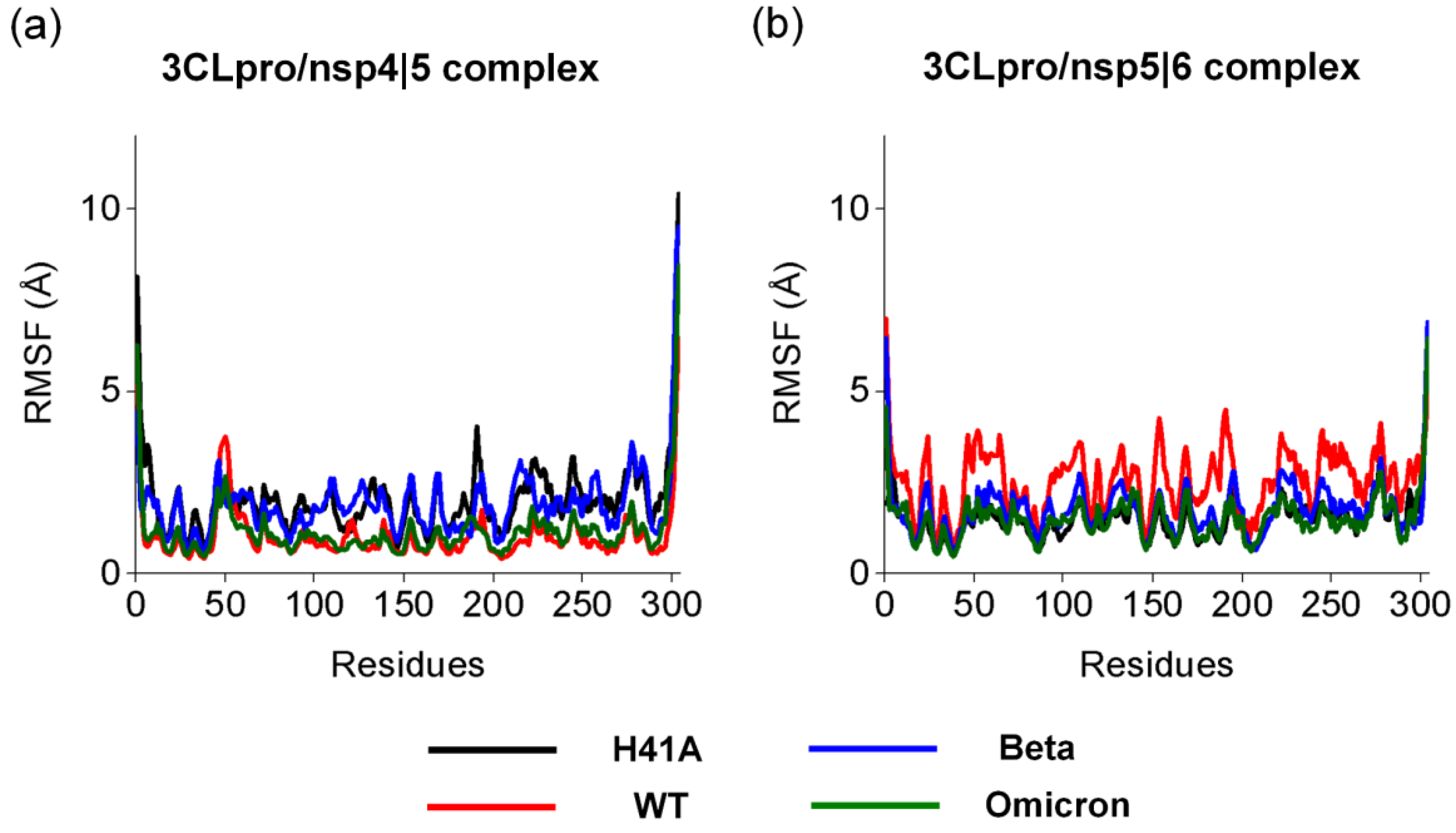
3.3. Pharmacophoric Hypothesis Based on Molecular Dynamics Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Souza, A.S.; de Freitas Amorim, V.M.; Guardia, G.D.A.; Dos Santos, F.F.; Ulrich, H.; Galante, P.A.F.; de Souza, R.F.; Guzzo, C.R. Severe Acute Respiratory Syndrome Coronavirus 2 Variants of Concern: A Perspective for Emerging More Transmissible and Vaccine-Resistant Strains. Viruses 2022, 14, 827. [Google Scholar] [CrossRef]
- WHO. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 15 April 2023).
- Milman, O.; Yelin, I.; Aharony, N.; Katz, R.; Herzel, E.; Ben-Tov, A.; Kuint, J.; Gazit, S.; Chodick, G.; Patalon, T.; et al. Community-Level Evidence for SARS-CoV-2 Vaccine Protection of Unvaccinated Individuals. Nat. Med. 2021, 27, 1367–1369. [Google Scholar] [CrossRef] [PubMed]
- Giordano, G.; Colaneri, M.; Di Filippo, A.; Blanchini, F.; Bolzern, P.; De Nicolao, G.; Sacchi, P.; Colaneri, P.; Bruno, R. Modeling Vaccination Rollouts, SARS-CoV-2 Variants and the Requirement for Non-Pharmaceutical Interventions in Italy. Nat. Med. 2021, 27, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Wilder-Smith, A.; Mulholland, K. Effectiveness of an Inactivated SARS-CoV-2 Vaccine. N. Eng. J. Med. 2021, 385, 946–948. [Google Scholar] [CrossRef]
- Halley, J.M.; Vokou, D.; Pappas, G.; Sainis, I. SARS-CoV-2 Mutational Cascades and the Risk of Hyper-Exponential Growth. Microb. Pathog. 2021, 161, 105237. [Google Scholar] [CrossRef]
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated Transmissibility and Impact of SARS-CoV-2 Lineage B.1.1.7 in England. Science 2021, 372, eabg3055. [Google Scholar] [CrossRef]
- Choi, J.Y.; Smith, D.M. SARS-CoV-2 Variants of Concern. Yonsei Med. J. 2021, 62, 961–968. [Google Scholar] [CrossRef]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; Corado, A.d.L.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 in Amazonas, Brazil, Was Driven by the Persistence of Endemic Lineages and P.1 Emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef]
- Li, M.; Lou, F.; Fan, H. SARS-CoV-2 Variants of Concern Delta: A Great Challenge to Prevention and Control of COVID-19. Signal Transduct. Target. Ther. 2021, 6, 349. [Google Scholar] [CrossRef] [PubMed]
- Barnard, R.C.; Davies, N.G.; Pearson, C.A.B.; Jit, M.; John Edmunds, W. Projected Epidemiological Consequences of the Omicron SARS-CoV-2 Variant in England, December 2021 to April 2022. medRxiv, 2021; preprint. [Google Scholar]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly. 2021, 3, 1049–1051. [Google Scholar] [CrossRef]
- GISAID—NextStrain. Available online: https://gisaid.org/phylodynamics/global/nextstrain/ (accessed on 30 March 2023).
- De Souza, A.S.; de Souza, R.F.; Guzzo, C.R. Quantitative Structure-Activity Relationships, Molecular Docking and Molecular Dynamics Simulations Reveal Drug Repurposing Candidates as Potent SARS-CoV-2 Main Protease Inhibitors. J. Biomol. Struct. Dyn. 2022, 40, 11339–11356. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.S.; de Freitas Amorim, V.M.; Guardia, G.D.A.; Dos Santos, F.R.C.; Dos Santos, F.F.; de Souza, R.F.; de Araujo Juvenal, G.; Huang, Y.; Ge, P.; Jiang, Y.; et al. Molecular Dynamics Analysis of Fast-Spreading Severe Acute Respiratory Syndrome Coronavirus 2 Variants and Their Effects on the Interaction with Human Angiotensin-Converting Enzyme 2. ACS Omega 2022, 7, 30700–30709. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.S.; Amorim, V.M.d.F.; de Souza, R.F.; Guzzo, C.R. Molecular Dynamics Simulations of the Spike Trimeric Ectodomain of the SARS-CoV-2 Omicron Variant: Structural Relationships with Infectivity, Evasion to Immune System and Transmissibility. J. Biomol. Struct. Dyn. 2022, 1–18. [Google Scholar] [CrossRef]
- Souza, A.S.; Rivera, J.D.; Almeida, V.M.; Ge, P.; de Souza, R.F.; Farah, C.S.; Ulrich, H.; Marana, S.R.; Salinas, R.K.; Guzzo, C.R. Molecular Dynamics Reveals Complex Compensatory Effects of Ionic Strength on the Severe Acute Respiratory Syndrome Coronavirus 2 Spike/Human Angiotensin-Converting Enzyme 2 Interaction. J. Phys. Chem. Lett. 2020, 11, 10446–10453. [Google Scholar] [CrossRef] [PubMed]
- Mody, V.; Ho, J.; Wills, S.; Mawri, A.; Lawson, L.; Ebert, M.C.C.J.C.; Fortin, G.M.; Rayalam, S.; Taval, S. Identification of 3-Chymotrypsin like Protease (3CLPro) Inhibitors as Potential Anti-SARS-CoV-2 Agents. Commun. Biol. 2021, 4, 93. [Google Scholar] [CrossRef]
- Yoshino, R.; Yasuo, N.; Sekijima, M. Identification of Key Interactions between SARS-CoV-2 Main Protease and Inhibitor Drug Candidates. Sci. Rep. 2020, 10, 12493. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef]
- Shi, J.; Song, J. The Catalysis of the SARS 3C-like Protease Is under Extensive Regulation by Its Extra Domain. FEBS J. 2006, 273, 1035–1045. [Google Scholar] [CrossRef]
- Anand, K.; Palm, G.J.; Mesters, J.R.; Siddell, S.G.; Ziebuhr, J.; Hilgenfeld, R. Structure of Coronavirus Main Proteinase Reveals Combination of a Chymotrypsin Fold with an Extra Alpha-Helical Domain. EMBO J. 2002, 21, 3213–3224. [Google Scholar] [CrossRef]
- Iketani, S.; Hong, S.J.; Sheng, J.; Bahari, F.; Culbertson, B.; Atanaki, F.F.; Aditham, A.K.; Kratz, A.F.; Luck, M.I.; Tian, R.; et al. Functional Map of SARS-CoV-2 3CL Protease Reveals Tolerant and Immutable Sites. Cell Host Microbe 2022, 30, 1354–1362. [Google Scholar] [CrossRef]
- Kiemer, L.; Lund, O.; Brunak, S.; Blom, N. Coronavirus 3CLpro Proteinase Cleavage Sites: Possible Relevance to SARS Virus Pathology. BMC Bioinform. 2004, 5, 72. [Google Scholar] [CrossRef]
- Grottesi, A.; Bešker, N.; Emerson, A.; Manelfi, C.; Beccari, A.R.; Frigerio, F.; Lindahl, E.; Cerchia, C.; Talarico, C. Computational Studies of SARS-CoV-2 3CLpro: Insights from MD Simulations. Int. J. Mol. Sci. 2020, 21, 5346. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, Y.; Liu, X.; Jin, Z.; Duan, Y.; Zhang, Q.; Wu, C.; Feng, L.; Du, X.; Zhao, J.; et al. Structural Basis for Replicase Polyprotein Cleavage and Substrate Specificity of Main Protease from SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2022, 119, e2117142119. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the Size of the Active Site in Proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Hilgenfeld, R. From SARS to MERS: Crystallographic Studies on Coronaviral Proteases Enable Antiviral Drug Design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ali, S.S.; Khan, M.T.; Saleem, S.; Ali, A.; Suleman, M.; Babar, Z.; Shafiq, A.; Khan, M.; Wei, D.-Q. Combined Drug Repurposing and Virtual Screening Strategies with Molecular Dynamics Simulation Identified Potent Inhibitors for SARS-CoV-2 Main Protease (3CLpro). J. Biomol. Struct. Dyn. 2021, 39, 4659–4670. [Google Scholar] [CrossRef]
- Jade, D.; Ayyamperumal, S.; Tallapaneni, V.; Nanjan, C.M.J.; Barge, S.; Mohan, S.; Nanjan, M.J. Virtual High Throughput Screening: Potential Inhibitors for SARS-CoV-2 PLPRO and 3CLPRO Proteases. Eur. J. Pharmacol. 2021, 901, 174082. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Kim, D.Y.; Kim, M.-S.; Shin, D.H. Flavonoids with Inhibitory Activity against SARS-CoV-2 3CLpro. J. Enzym. Inhib. Med. Chem. 2020, 35, 1539–1544. [Google Scholar] [CrossRef] [PubMed]
- Molavi, Z.; Razi, S.; Mirmotalebisohi, S.A.; Adibi, A.; Sameni, M.; Karami, F.; Niazi, V.; Niknam, Z.; Aliashrafi, M.; Taheri, M.; et al. Identification of FDA Approved Drugs against SARS-CoV-2 RNA Dependent RNA Polymerase (RdRp) and 3-Chymotrypsin-like Protease (3CLpro), Drug Repurposing Approach. Biomed. Pharmacother. 2021, 138, 111544. [Google Scholar] [CrossRef]
- Oostra, M.; Te Lintelo, E.G.; Deijs, M.; Verheije, M.H.; Rottier, P.J.M.; de Haan, C.A.M. Localization and Membrane Topology of Coronavirus Nonstructural Protein 4: Involvement of the Early Secretory Pathway in Replication. J. Virol. 2007, 81, 12323–12336. [Google Scholar] [CrossRef]
- Puhach, O.; Adea, K.; Hulo, N.; Sattonnet, P.; Genecand, C.; Iten, A.; Jacquérioz, F.; Kaiser, L.; Vetter, P.; Eckerle, I.; et al. Infectious Viral Load in Unvaccinated and Vaccinated Individuals Infected with Ancestral, Delta or Omicron SARS-CoV-2. Nat. Med. 2022, 28, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Schrodinger Maestro. Available online: https://www.schrodinger.com/maestro (accessed on 15 April 2023).
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A Package for Molecular Simulation and Trajectory Analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Robertson, M.J.; Tirado-Rives, J.; Jorgensen, W.L. Improved Peptide and Protein Torsional Energetics with the OPLSAA Force Field. J. Chem. Theory Comput. 2015, 11, 3499–3509. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lyngse, F.P.; Mølbak, K.; Skov, R.L.; Christiansen, L.E.; Mortensen, L.H.; Albertsen, M.; Møller, C.H.; Krause, T.G.; Rasmussen, M.; Michaelsen, T.Y.; et al. Increased Transmissibility of SARS-CoV-2 Lineage B.1.1.7 by Age and Viral Load. Nat. Commun. 2021, 12, 7251. [Google Scholar] [CrossRef] [PubMed]
- Luttens, A.; Gullberg, H.; Abdurakhmanov, E.; Vo, D.D.; Akaberi, D.; Talibov, V.O.; Nekhotiaeva, N.; Vangeel, L.; De Jonghe, S.; Jochmans, D.; et al. Ultralarge Virtual Screening Identifies SARS-CoV-2 Main Protease Inhibitors with Broad-Spectrum Activity against Coronaviruses. J. Am. Chem. Soc. 2022, 144, 2905–2920. [Google Scholar] [CrossRef] [PubMed]
- Astuti, I. Ysrafil Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An Overview of Viral Structure and Host Response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef]
- Avti, P.; Chauhan, A.; Shekhar, N.; Prajapat, M.; Sarma, P.; Kaur, H.; Bhattacharyya, A.; Kumar, S.; Prakash, A.; Sharma, S.; et al. Computational Basis of SARS-CoV 2 Main Protease Inhibition: An Insight from Molecular Dynamics Simulation Based Findings. J. Biomol. Struct. Dyn. 2021, 40, 8894–8904. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, N.; Sakthivel, R.; Velmurugan, D.; Gromiha, M.M. Computational Studies of Drug Repurposing and Synergism of Lopinavir, Oseltamivir and Ritonavir Binding with SARS-CoV-2 Protease against COVID-19. J. Biomol. Struct. Dyn. 2021, 39, 2673–2678. [Google Scholar] [CrossRef]
- Zheng, Y.; Deng, J.; Han, L.; Zhuang, M.-W.; Xu, Y.; Zhang, J.; Nan, M.-L.; Xiao, Y.; Zhan, P.; Liu, X.; et al. SARS-CoV-2 NSP5 and N Protein Counteract the RIG-I Signaling Pathway by Suppressing the Formation of Stress Granules. Signal Transduct. Target 2022, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug Repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Barrett, B.S.; Morrison, J.H.; Mickens, K.L.; Vladar, E.K.; Hasenkrug, K.J.; Poeschla, E.M.; Santiago, M.L. Interferon Resistance of Emerging SARS-CoV-2 Variants. Proc. Natl. Acad. Sci. USA 2022, 119, e2203760119. [Google Scholar] [CrossRef]
- Tao, K.; Tzou, P.L.; Nouhin, J.; Gupta, R.K.; de Oliveira, T.; Kosakovsky Pond, S.L.; Fera, D.; Shafer, R.W. The Biological and Clinical Significance of Emerging SARS-CoV-2 Variants. Nat. Rev. Genet. 2021, 22, 757–773. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Flury, P.; Krüger, N.; Su, H.; Schäkel, L.; Barbosa Da Silva, E.; Eppler, O.; Kronenberger, T.; Nie, T.; Luedtke, S.; et al. Small-Molecule Thioesters as SARS-CoV-2 Main Protease Inhibitors: Enzyme Inhibition, Structure-Activity Relationships, Antiviral Activity, and X-Ray Structure Determination. J. Med. Chem. 2022, 65, 9376–9395. [Google Scholar] [CrossRef]
- Johnson, T.O.; Adegboyega, A.E.; Ojo, O.A.; Yusuf, A.J.; Iwaloye, O.; Ugwah-Oguejiofor, C.J.; Asomadu, R.O.; Chukwuma, I.F.; Ejembi, S.A.; Ugwuja, E.I.; et al. A Computational Approach to Elucidate the Interactions of Chemicals From Targeted Toward SARS-CoV-2 Main Protease Inhibition for COVID-19 Treatment. Front. Med. 2022, 9, 907583. [Google Scholar] [CrossRef]
- Ramos-Guzmán, C.A.; Ruiz-Pernía, J.J.; Tuñón, I. Unraveling the SARS-CoV-2 Main Protease Mechanism Using Multiscale Methods. ACS Catal. 2020, 10, 12544–12554. [Google Scholar] [CrossRef]
- Alamri, M.A.; Tahir Ul Qamar, M.; Mirza, M.U.; Bhadane, R.; Alqahtani, S.M.; Muneer, I.; Froeyen, M.; Salo-Ahen, O.M.H. Pharmacoinformatics and Molecular Dynamics Simulation Studies Reveal Potential Covalent and FDA-Approved Inhibitors of SARS-CoV-2 Main Protease 3CL. J. Biomol. Struct. Dyn. 2021, 39, 4936–4948. [Google Scholar] [CrossRef]
- Khan, A.; Heng, W.; Wang, Y.; Qiu, J.; Wei, X.; Peng, S.; Saleem, S.; Khan, M.; Ali, S.S.; Wei, D.-Q. In Silico and in Vitro Evaluation of Kaempferol as a Potential Inhibitor of the SARS-CoV-2 Main Protease (3CLpro). Phytother. Res. 2021, 35, 2841–2845. [Google Scholar] [CrossRef]
- Sk, M.F.; Roy, R.; Jonniya, N.A.; Poddar, S.; Kar, P. Elucidating Biophysical Basis of Binding of Inhibitors to SARS-CoV-2 Main Protease by Using Molecular Dynamics Simulations and Free Energy Calculations. J. Biomol. Struct. Dyn. 2021, 39, 3649–3661. [Google Scholar] [CrossRef]
- Pathak, N.; Chen, Y.-T.; Hsu, Y.-C.; Hsu, N.-Y.; Kuo, C.-J.; Tsai, H.P.; Kang, J.-J.; Huang, C.-H.; Chang, S.-Y.; Chang, Y.-H.; et al. Uncovering Flexible Active Site Conformations of SARS-CoV-2 3CL Proteases through Protease Pharmacophore Clusters and COVID-19 Drug Repurposing. ACS Nano 2021, 15, 857–872. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, Y.; Wu, Y.; Yu, H.; Wang, Z.; Ma, Y. Pharmacophore-Based Virtual Screening of Potential SARS-CoV-2 Main Protease Inhibitors from Library of Natural Products. Nat. Prod. Commun. 2022, 17, 1934578X2211436. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the Sensitivity of Progressive Multiple Sequence Alignment through Sequence Weighting, Position-Specific Gap Penalties and Weight Matrix Choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Sawant, S.; Patil, R.; Khawate, M.; Zambre, V.; Shilimkar, V.; Jagtap, S. Computational Assessment of Select Antiviral Phytochemicals as Potential SARS-Cov-2 Main Protease Inhibitors: Molecular Dynamics Guided Ensemble Docking and Extended Molecular Dynamics. In Silico Pharmacol. 2021, 9, 44. [Google Scholar] [CrossRef]
- Hamed, M.I.A.; Darwish, K.M.; Soltane, R.; Chrouda, A.; Mostafa, A.; Abo Shama, N.M.; Elhady, S.S.; Abulkhair, H.S.; Khodir, A.E.; Elmaaty, A.A.; et al. β-Blockers Bearing Hydroxyethylamine and Hydroxyethylene as Potential SARS-CoV-2 Mpro Inhibitors: Rational Based Design and SAR Studies for Lead Optimization. RSC Adv. 2021, 11, 35536–35558. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ΔG (kJ·mol−1) | ||||
|---|---|---|---|---|
| 3CLproH41A | 3CLproWT | 3CLproBeta | 3CLproOmicron | |
| nsp 4|5 | 3037.6 ± 18.3 | 3017.4 ± 21.2 | 3016.6 ± 22.3 | 3026.0 ± 18.6 |
| nsp 5|6 | 3428.1 ± 13.7 | 3418.7 ± 19.7 | 3402.9 ± 11.0 | 3412.3 ± 19.6 |
| nsp 14|15 | 3290.3 ± 15.2 | 3276.8 ± 13.3 | 3293.0 ± 14.5 | 3266.0 ± 14.1 |
| KD (μM) | |||||
|---|---|---|---|---|---|
| Experimental | Prediction | ||||
| 3CLproH41A | 3CLproH41A | 3CLproWT | 3CLproBeta | 3CLproOmicron | |
| nsp 4|5 | 28 ± 3 [26] | 35.6 | 27.8 | 27.5 | 30.9 |
| nsp 5|6 | 2730 ± 900 [26] | 4216.2 | 3756.1 | 3098.6 | 3473.0 |
| nsp 14|15 | 1530 ± 350 [26] | 781.6 | 663.1 | 807.6 | 580.6 |
| Cleavage Rate (Cut of 4.5 Å) | |||
|---|---|---|---|
| 3CLproWT | 3CLproBeta | 3CLproOmicron | |
| nsp 4|5 | 154 | 9941 | 555 |
| nsp 5|6 | 1025 | 70 | 445 |
| Amino Acid Sequence of 3CLpro Variants | ||||
|---|---|---|---|---|
| nsp 4|5 | H41A | WT | Beta | Omicron |
| MCAla3 | SCGln189HBD (31.6) SCGln189HBA (24.9) | MCThr190HBA (71.0) SCPro168HBD (22.0) | MCThr190HBA (26.8) | - |
| MCVal4 | MCGlu166HBA (62.1) MCGlu166HBD (54.4) | MCGlu166HBA (41.9) MCGlu166HBD (75.6) SCGln189HBA (20.2) | MCGlu166HBA (38.6) MCGlu166HBD (41.9) | MCGlu166HBA (56.3) MCGlu166HBD (49.0) |
| MCLeu5 | - | SCGln189HBA (50.6) | SCGln189HBA (28.7) | - |
| SCGln6 | SCGlu166HBA (72.7) MCLeu141HBA (21.9) | SCGlu166HBA (20.4) | SCGlu166HBA (26.3) | SCHis163HBA (70.3) MCPhe140HBA (47.9) |
| MCGln6 | - | MCHis164HBA (50.6) SCCys145HBD (23.4) MCGly143HBD (68.2) | - | SCCys145HBD (37.4) MCGly143HBD (81.6) |
| SCSer7 | - | SCHis41HBA (29.0) | SCThr25HBD (21.7) | - |
| MCGly8 | MCThr26HBA (30.0) SCAsn142HBD (20.8) | - | SCAsn142HBD (36.2) | SCAsn142HBD (34.4) |
| Amino Acid Sequence of 3CLpro Variants | ||||
|---|---|---|---|---|
| nsp 5|6 | H41A | WT | Beta | Omicron |
| MCVal3 | MCThr190HBA (30.4) SCGln189HBD (20.9) | SCGln189HBD (26.3) | SCGln189HBD (23.4) | SCGln189HBD (27.3) |
| MCThr4 | MCGlu166HBA (58.3) MCGlu166HBD (55.9) | MCGlu166HBA (59.6) MCGlu166HBD (74.9) | MCGlu166HBA (52.5) MCGlu166HBD (70.5) | MCGlu166HBA (59.0) MCGlu166HBD (62.6) |
| SCGln6 | MCGly143HBD (66.3) MCSer144HBD (22.4) | SCGlu166HBD (36.4) | SCGlu166HBD (49.9) | SCGlu166HBA (42.5) |
| MCGln6 | - | MCGly143HBD (31.6) | MCGly143HBD (36.6) | MCGly143HBD (42.9) SCCys145HBD (25.4) mCHis164HBA (22.7) |
| SCSer7 | MCAla41HBA (30.1) | SCHis41HBA (22.2) | SCHis41HBA (27.1) | SCHis41HBA (22.2) |
| MCSer7 | MCTHR26HBA (30.9) | - | MCTHR26HBD (53.1) | - |
| MCAla8 | - | - | - | SCAsn142HBA (21.3) |
| MCVal9 | MCSER46HBA (30.1) | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Freitas Amorim, V.M.; de Souza, R.F.; Guzzo, C.R.; de Souza, A.S. Molecular Dynamics Simulations Suggest SARS-CoV-2 3CLpro Mutations in Beta and Omicron Variants Do Not Alter Binding Affinities for Cleavage Sites of Non-Structural Proteins. COVID 2023, 3, 622-636. https://doi.org/10.3390/covid3040044
de Freitas Amorim VM, de Souza RF, Guzzo CR, de Souza AS. Molecular Dynamics Simulations Suggest SARS-CoV-2 3CLpro Mutations in Beta and Omicron Variants Do Not Alter Binding Affinities for Cleavage Sites of Non-Structural Proteins. COVID. 2023; 3(4):622-636. https://doi.org/10.3390/covid3040044
Chicago/Turabian Stylede Freitas Amorim, Vitor Martins, Robson Francisco de Souza, Cristiane Rodrigues Guzzo, and Anacleto Silva de Souza. 2023. "Molecular Dynamics Simulations Suggest SARS-CoV-2 3CLpro Mutations in Beta and Omicron Variants Do Not Alter Binding Affinities for Cleavage Sites of Non-Structural Proteins" COVID 3, no. 4: 622-636. https://doi.org/10.3390/covid3040044
APA Stylede Freitas Amorim, V. M., de Souza, R. F., Guzzo, C. R., & de Souza, A. S. (2023). Molecular Dynamics Simulations Suggest SARS-CoV-2 3CLpro Mutations in Beta and Omicron Variants Do Not Alter Binding Affinities for Cleavage Sites of Non-Structural Proteins. COVID, 3(4), 622-636. https://doi.org/10.3390/covid3040044