1. Introduction
Coronavirus disease 2019 (COVID-19) is a disease caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Ever since it emerged in December 2019, SARS-CoV-2 has caused millions of infection cases and deaths [
1,
2] and the greatest global public health and economic crisis around the world and the United States [
3].
SARS-CoV-2 is a single-stranded RNA-enveloped virus whose genome encodes four major structural proteins, including the spike (S), membrane (M), envelope (E), and nucleocapsid (N) proteins [
4,
5]. Among them, the S protein plays a key role in the receptor recognition (S1 unit) and cell membrane fusion processes (S2 unit), and it determines the infectivity of the virus and its transmissibility in the host. The S1 contains the receptor binding domain (RBD), which is mainly responsible for binding the virus to the angiotensin-converting enzyme 2 (ACE2) receptor. Once the RBD binds to the host ACE2 receptors, the cell surface serine protease TMPRSS2 starts to promote SARS-CoV-2 viral uptake and fusion at the cellular level. The virus enters the host cell and releases the viral RNA into the cytoplasm, after which it expresses and replicates its genomic RNA in order to make full-length copies that are integrated into freshly produced viral particles [
5,
6,
7]. In addition, the S protein contains a site that is recognized and is activated by furin, which is a host cell enzyme expressed in various human organs, such as the liver, the lungs, and the small intestines [
8]. Because of the S protein’s specific structure, it allows the SARS-CoV-2 viruses to bind at least 10 times more tightly to ACE2 receptors than the corresponding S protein of other SARS viruses and can potentially attack several organs at the same time [
9].
The biggest challenge to fighting COVID-19 is the lack of specific anti-SARS-CoV-2 drugs that target different variants. The common strategy of current COVID-19 treatment is “old drug, new use”. Among these drugs are RNA-dependent RNA polymerase inhibitors (e.g., Remdesivir, Favipiravir, Ribavirin and interferons), protease inhibitors (e.g., Lopinavir and Ritonavir), hydroxychloroquine, azithromycin, monoclonal antibody, or convalescent plasma [
10]. Most of these potential drugs are being investigated for their safety and efficacy against COVID-19. Remdesivir has shown to be the most promising and hopeful anti-viral therapeutic, while many other drugs have shown severe side effects, uncertain benefits, or contraindicated conditions [
11,
12,
13]. Recently, the U.S. FDA has authorized Pfizer’s Paxlovid for emergency use within five days of symptom onset but not for pre-exposure or post-exposure prevention of COVID-19. At the same time, the FDA noted that Paxlovid may result in significant drug interactions [
14]. On the other hand, many research laboratories are continuing to study and develop new drugs against COVID-19. Neutralizing antibodies for the prevention and treatment of COVID-19 is one of the fields that focuses on the N-terminal domain (NTD) and receptor-binding domain (RBD) of the S protein. This includes humanized monoclonal antibodies [
15], antibodies cloned from human B cells [
16], and single-chain camelid antibodies [
17]. However, coronaviruses are RNA viruses that continue to mutate, evolve, and hence develop resistance to drugs easily. So far, different mutations have been found in all four structural proteins and other viral proteins that may lead to escape from antibody recognition, resulting in antibodies that have a weaker effect or no effect at all for the new variant types of coronavirus [
18,
19,
20,
21,
22]. For example, the study by Li et al. indicated that SARS-CoV-2 variants that include mutations of A475V, L452R, V483A, F490L, and N234Q became resistant or markedly resistant to some neutralizing antibodies [
23].
More than that, antibody-based vaccines and therapeutics could potentially increase the risk of exacerbation of COVID-19 severity through antibody-dependent enhancement (ADE), which may lead to an increase of unwanted immune reactions, virus infectivity, and virulence [
24,
25]. Although the relevance of in vitro ADE for human coronaviruses remains less clear, several viruses, including human immunodeficiency virus (HIV), Ebola, and influenza have been well documented [
26,
27,
28,
29,
30]. Wan et al. showed that neutralizing monoclonal antibody against the RBD of MERS-COV increased the uptake of virion into macrophages and various cell lines transfected with Fc
RIIa (Fc gamma receptor IIa) [
31]. Thus, to address these limitations, it is urgent and necessary to develop an effective anti-SARS-CoV-2 drug to inhibit viral infection with less side effects.
Aptamers are short, single-stranded RNA or DNA molecules that have a high affinity for specific target molecules. Aptamers are often referred to as chemical antibodies because their interaction with their target is similar to that of an antigen–antibody interaction [
32]. However, aptamers have many advantages over antibodies, such as smaller size, lower immunogenicity, long shelf life and stability, less batch-to-batch variation, ease of modification, cost-effectiveness, and short production time. Moreover, aptamers’ flexible three-dimensional structures allow them to fold around the complex surfaces of their target molecules, facilitating greater flexibility in selecting aptamers for various targets such as peptides, proteins, small organic compounds, toxins, cells, viruses, bacteria, etc. [
33,
34,
35,
36,
37,
38,
39]. Recently, several groups have identified DNA aptamers that recognize the S protein of SARS-CoV-2. The majority of them were focused on developing aptamers that bind the RBD or the S1 domain of S protein [
40,
41,
42,
43,
44,
45].
In this study, we report the development of a series of single-stranded DNA (ssDNA) aptamers targeting the trimer S proteins of Wuhan original strain. Selected aptamers were studied for their binding affinity and inhibitory efficacy. The truncation approach was used to improve the binding capacity of aptamers as well as their inhibitory efficiency to prevent the binding of trimer S protein to the ACE2 receptors. The results show that our aptamers were not only able to bind to the trimer S protein of the Wuhan original strain but also bind multiple variants of trimer S proteins of Delta, Delta plus, Alpha, Lambda, Mu, and Omicron and inhibit their binding to ACE2 receptors in the Vero E6 cell line. To further analyze the inhibitory efficacy of the selected aptamers, we used virus-like particles (VLPs) packaged with Green Fluorescent Protein (GFP) plasmid to mimic the SARS-CoV-2 virus. Our modified aptamers AYA2012004_L and AYA2012004_L-M1 showed up to 70% inhibition of the binding of virus-like particles (VLPs) expressing S protein to the ACE2 receptor expressed in Human Embryonic Kidney 293T (HEK293T) cells that overexpress ACE2 receptors. Overall, the findings suggest that our reported aptamers could be an innovative therapy for the treatment of COVID-19. They hold many advantages over existing therapies due to better efficacy, the ability to identify different variants of SARS-CoV-2 and safety.
2. Materials
Human ACE2 protein, Fc-tag (# AC2-H5257); SARS-CoV-2 spike protein trimer, His-tag Wuhan strain (# SPN-C52H2); SARS-CoV-2 S protein, His Tag, Super stable trimer Wuhan strain (# SPN-C52H9, SPN-C52H7); SARS-CoV-2 S protein (HV69-70del, Y144del, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H), His Tag Alpha strain (# SPN-C52H6); SARS-CoV-2 Spike Trimer (T19R, G142D, EF156-157del, R158G, L452R, T478K, D614G, P681R, D950N), His Tag Delta strain (# SPN-C52He); SARS-CoV-2 Spike Trimer (G75V, T76I, SYLTPGD 247-253 del, L452Q, F490S, D614G, T859N), His Tag Lambda strain (# SPN-C52Hs); SARS-CoV-2 Spike Trimer (T19R, V70F, FR157-158Del, A222V, W258L, K417N, L452R, T478K, D614G, P681R, D950N), His Tag Delta plus strain (# SPN-C52Ht); SARS-CoV-2 Spike Trimer (T95I, Y144S, Y145N, R346K, E484K, N501Y, D614G, P681H, D950N), His Tag Mu strain (# SPN-C52Ha) and SARS-CoV-2 Spike Trimer, His Tag (B.1.1.529/Omicron) strain (# SPN-C52Hz); and anti-SARS-CoV-2 RBD neutralizing antibody, human IgG1(# SAD-S35) were purchased from ACRO Biosystems; DNA Polymerase kit for PCR (# 170-8870) was from Bio-Rad; ssDNA library consisting of 40 random nucleotides that are flanked by two 23 bases of primer sequences (5TAG GGA AGA GAA GGA CAT ATG AT (N40) TTG ACT AGT ACA TGA CCA CTT GA 3, # O-32140-10), forward, reverse and 5biotin reverse primers for PCR (# O-32201, # O-32211 and # O-32212, respectively) were from TriLink Biotechnologies. Phosphate-Buffered Saline (PBS, # 10010-031), Dulbecco’s Phosphate Buffered Saline (DPBS, # 1404-133), HisPur Ni-NTA Magnetic Beads (# 88831), Nunc MaxiSorp Flat-Bottom 96-well Plates (# 44-2404-21), Pierce High-Sensitivity Streptavidin–HRP (# 21130), 1-Step Ultra TMB-ELISA (# 34028), and Pierce™ Nickel Coated Plates, Clear, 8-Well Strip (# 15142) were purchased from ThermoFisher Scientific; High-Capacity Streptavidin Magnetic Beads (# 1497) were from Click Chemistry Tools; DNA Clean & Concentrator-5 (# D4013) was from Zymo Research; HS Next-Generation Sequencing (NGS) Fragment kit (1–6000 bp), 500 (# 5191-6578) was from Agilent Technologies; TruSeq ChiP Library Preparation Kit (# IP-202-1012) was from Illumina; Horseradish Peroxidase-conjugated Goat Anti-Mouse IgG (# 115-035-062) was from Jackson ImmunoResearch Laboratories; SARS-CoV/SARS-CoV-2 (COVID-19) spike antibody (1A9) (# GTX632604) was from GeneTex; plasticware for cell culture were from CellTreat Scientific Products; Pulmonary surfactant (# THP-0147) was from Creative BioMart; ssDNA and biotinylated DNA HPLC purified were from Integrated DNA Technologies; Influenza A H1N1 hemagglutinin protein (# 40005-V08H1) was from Sino Biological; SARS-CoV-2 Nucleocapsid protein (# C5227 and # 230-01104) was from AcroBiosystems and RayBiotech, respectively. Antibodies specific for anti-His Tag (clone: J095G46, # 362605), isotype control mouse IgG2a (clone: MOPC-173, # 400220), and FluoroFix Buffer (# 422101) were purchased from BioLegend. Antibodies specific for anti-human ACE2 (# FAB933A) and isotype control Goat IgG (# IC108A) were purchased from R&D System. Fixable Viability Dye eFluor 450 (# 65-0863-14) was purchased from Invitrogen. COVID-19 S protein/(GFP)-(6His) VLP (# VLP001) were purchased from Gentarget, Inc.; SPRi-Biochip CSe (# 1123299207) was from Horiba; all other reagents were from Sigma-Aldrich. Sequencing of the aptamers were performed on MiSeq (Illumina, San Diego, CA, USA); the surface plasmon resonance experiments were performed using an OpenPlex SPRi system (HORIBA, Loos, France); Navios EX flow cytometer (Beckman Coulter, Indianapolis, IN, USA) was used for flow cytometry experiments and flow cytometry data were analyzed with FlowJo v10 (FlowJo LLC, Ashland, OR, USA.
African Green Monkey Kidney Cell (Vero E6 cells, # ATCC CRL-1586) and Eagle’s Minimum Essential Medium (EMEM) (# 30-2003) were obtained from ATCC; Geneticin (Antibiotic G418, # G271) was from ABM, Opti-MEM I Reduced Serum Medium was purchased from ThermoFisher Scientific, HEK293T cell line human (# 12022001) was from Sigma-Aldrich, Dulbecco’s Modified Eagle’s Medium (DMEM, # 10-013-CV) was purchased from Corning, plasmid ACE2 (NM_021804) Human Tagged ORF Clone (Angiotensin-Converting Enzyme 2) (# RC208442) was purchased from Origene, Lipofectamine 2000 Reagent (# 11668-030), StemPro Accutase (# A11105-01) and Penicillin Streptomycin (# 15140-122) were purchased from ThermoFisher Scientific; Fetal Bovine serum (# SH30088.03) was from HyClone.
3. Methods
3.1. Selection of the Aptamers for SARS-CoV-2 Trimer S Protein
The SELEX (systematic evolution of ligands by exponential enrichment) procedure was performed as previously described [
46] with some modifications (
Figure 1). A library of single-stranded DNA oligonucleotides (10
random unique sequences) flanked by two 23-based primer sequences (5
TAG GGA AGA GAA GGA CAT ATG AT (N40) TTG ACT AGT ACA TGA CCA CTT GA 3
) were used. To obtain the ssDNA in their unique confirmation, 10 nM of ssDNA library in 2 mL of activation buffer (20 mM Na-phosphate, 0.2 M NaCl and 0.5 mM MgCl
, pH∼7.4) were heated at 95 °C for 10 min followed by 20 min incubation on ice. Nickel–nitrilotriacetic acid (Ni-NTA) modified magnetic beads were prepared by equilibrating 480 μL of slurry (6 mg beads) into binding/washing buffer A (20 mM Na-phosphate, 0.2 M NaCl, 0.01% Tween 20 and 0.5 mM MgCl
, pH∼7.4). Activated ssDNA was first pre-cleared by incubation with 6 mg of washed Ni-NTA beads to remove any ssDNA that binds non-specifically to the matrix. Two milliliters of flow-through ss-DNA were collected for the first step of SELEX. Protein-bound Ni-NTA magnetic beads were prepared by incubation of 40 μg of His-tagged SARS-CoV-2 trimer S protein with washed Ni-NTA beads (3 mg) for 1 h with rotation in 1 mL of buffer A. Unbound S-protein was removed by washing the beads 2 times with the same buffer on a magnetic stand. Two milliliters of pre-cleared ssDNA were added to SARS-CoV-2 trimer S protein bound Ni-NTA beads. After 15 min incubation with rotation at room temperature, beads were collected by a magnetic stand and washed three times with 1.5 mL of buffer A. Captured ssDNA were eluted by 10 min incubation with 100 μL of 20 mM NaOH followed by neutralization with 12 μL of 0.2 M NaH
PO
to adjust the pH to ∼7.2. The eluted ssDNA was amplified by PCR using 5
biotinylated reverse primer. The amplification was performed in seven PCR tubes, and the total reaction volume in each of the tubes was 50 μL, which included 10 μL of eluted ssDNA (template); 5 μL of 10× iTaq buffer; 1.5 μL of 50 mM MgCl
; 1 μL of 10 mM dNTP; 0.25 μL iTaq DNA polymerase; 1.5 μL of 10 μM forward primers; 1.5 μL of 10 μM reverse primers; and 29.25 μL of miliQ water. The following PCR program was used: polymerase activation 95 °C for 3 min, amplification at 95 °C for 20 s, 60 °C for 20 s and 72 °C for 20 s repeated 17 cycles, final extension at 72 °C for 1 min. PCR products of each tube were combined, cleaned up and concentrated using Zymo Research kit per the product insert. The collected dsDNA was bound to streptavidin beads (35 μL of slurry, 0.3 mg of beads) prewashed in buffer A in the presence of 1 M NaCl. Single strands were eluted from dsDNA with 100 μL of 20 mM NaOH and neutralized with 12 μL of 0.2 M NaH
PO
to adjust the final pH to ∼7.2. The quality of ssDNA was analyzed using Fragment Analyzer with Agilent Technologies reagents. The eluted ssDNA was activated as described above and represents a new enriched ssDNA library pool that was used for subsequent (rolling) round of SELEX) (
Figure 1A). For all following rounds of selection, the amount of S-protein was decreased to 10 μg bounded to 3 mg of Ni-NTA beads. The stringency of wash buffer was increased by adjusting the salt concentration in buffer A to 0.5 M NaCl with a simultaneous increase in washing time up to 10 min for each wash. To remove ssDNA that non-specifically binds to Ni-NTA matrix, negative depletion for the ssDNA pool was performed after every third round of selection. To enhance the specificity of the aptamers, counter SELEX was performed using pulmonary surfactant, BSA, and human plasma. For counter-selection with pulmonary surfactant, aptamers incubation with immobilized trimer S protein was performed in the presence of 0.1 mg/mL pulmonary surfactant at the 5th and 9th rounds of selection. In addition, BSA and human plasma proteins were used for counter-selections at 7th and 10th rounds, respectively, by incubating the aptamers with immobilized S protein in the presence of 1% BSA (round 7) or human plasma that was diluted three times with wash/binding buffer A (round 10). After six rounds of selection, the eluted ssDNA pool was used to specifically select neutralizing ssDNA aptamers (
Figure 1B). Human ACE-2 receptor was added to trimer S-protein immobilized on the beads in a 3:1 molar ratio to block the receptor binding site on the S-protein. The eluted ssDNA from round six of selection was incubated with this S proteins complex immobilized on the beads. The flow-through, representing aptamers that specifically bind to S-protein site concealed by an ACE-2 receptor, were collected and processed for five more rounds of selection. A total of twelve rounds of selection were performed.
3.2. Next-Generation Sequencing of Aptamers
The ssDNA pool eluted from the 4th, 8th, 9th and 12th rounds of selection were subjected to library preparation for sequencing using a TruSeq ChiP Library Preparation Kit from Illumina. Four pooled paired-end indexed DNA libraries were prepared, using the reagents provided in the Illumina TruSeq ChiP Sample preparation Kit, for subsequent cluster generation and DNA sequencing. Input DNA (50 μL of 200 pg/μL) was blunt-ended and phosphorylated. A single “A” nucleotide was added to the 3
ends of the fragments in preparation for ligation to an adapter that has a single–base “T” overhang. Adapter sequences were added onto the ends of DNA to generate indexed single read or paired-end sequencing libraries. The ligation products were purified and accurately size-selected by agarose gel electrophoresis. Size-selected DNA was purified and PCR-amplified to enrich for fragments that have adapters on both ends. The final product was then quantitated before cluster generation. Sequencing was performed on Illumina MiSeq instrument. Sequence analysis was conducted using FASTAptamer software to identify sequences that showed enrichment across selection pools [
47].
3.3. ELISA-Based Binding Assay
To test the binding of 5-biotinylated aptamers to trimer S protein, 100 μL of trimer S protein per well at a concentration 2 μg/mL in 50 mM Na carbonate–bicarbonate buffer was adsorbed overnight in a MaxiSorp plate. The plate was blocked with 1% BSA in PBS buffer for 1 h at room temperature. Then, 10 nM of biotinylated aptamers was added to each well and incubated with the immobilized trimer S protein in PBS buffer supplemented with 0.05% tween and 1 mM MgCl. The same buffer was used to wash out unbound aptamers. Bound biotinylated aptamers were detected with streptavidin–HRP and 3,3,5,5-tetramethylbenzidine (TMB) as a substrate. After the reaction was stopped with 1 M sulfuric acid, absorbance was measured at 450 nM. To show specific binding of 5-biotinylated aptamers to trimer S protein, competition assays were performed using one-hundred-fold excess of the respective non-biotinylated aptamer. To test the specificity of modified aptamers, the same ELISA-based binding assay was performed with high stringency wash buffer that contains PBS, 0.5 M NaCl, 0.05% Tween 20, and 1 mM MgCl, PH ∼7.4.
3.4. Cell Culture, Transfection, and Stable Cell Line
HEK-293T cells were set up for experiments on day 0 (2 × 10/60-mm dish) and cultured in 5% CO at 37 °C in Medium A (Dulbecco’s modified Eagle’s medium containing 100 units/mL penicillin and 100 μg/mL streptomycin sulfate) supplemented with 10% (v/v) fetal bovine serum. On day 1, the cells were washed and refed with Opti-MEM reduced serum medium and transfected with 2 μg of plasmid ACE2 (Myc-DDK-tagged)-human angiotensin I converting enzyme (peptidyl-dipeptidase A) 2 using existing protocol from Invitrogen (Protocol Pub. No. MAN0007824 Rev1). Briefly, DNA was incubated with 6 μL of Lipofectamine 2000 in Opti-MEM reduced serum medium for 5 min at room temperature and DNA–Lipofectamine 2000 complex was added to the cells. Eight hours after transfection, cells were refed with medium A. On day 3, cells were fed with medium A containing 200 μg/mL of G418 antibiotic. Cells were maintained in this media for one week before decreasing the G418 concentration to 100 μg/mL. African Green Monkey Kidney Cell (Vero E6 cells) were set up for experiments and cultured in 5% CO at 37 °C in Medium B (Eagle’s Minimum Essential Medium (EMEM) containing 100 units/mL penicillin and 100 μg/mL streptomycin sulfate) supplemented with 10% (v/v) fetal bovine serum.
3.5. Cell-Based Inhibition Assay of SARS-CoV-2 Trimer S Protein Binding to ACE2 Receptors on the Surface of Vero E6 Cells
SARS-CoV-2 trimer S protein at 10 nM was pre-incubated with aptamers at different concentrations in 200 μL of PBS buffer containing 1 mM MgCl and 0.5% fish gelatin for 1 h at room temperature. Vero E6 cells were grown in 10 cm plates to nearly 100% of confluency, washed with PBS and detached from the plate with 3 mL of StemPro Accutase by incubation at 37 °C for 20 min. Cells were collected in 15 mL centrifuge tubes and pelleted at 1000× g for 5 min. The pellet of the cells was washed once with PBS containing 1 mM MgCl and 0.1% fish gelatin and resuspended in the same buffer. The suspension of cells was divided for aliquots (5 × 10 cells per condition) in microcentrifuge tubes and pelleted again at 3000× g for 4 min. The supernatant was carefully and completely removed, and 200 μL of each condition of trimer S protein pre-incubated with aptamers was added to each pellet. The protein without aptamers in the same buffer was added as a positive control. Samples were resuspended, and S protein was allowed to bind to cells for 1.5 h with occasional gentle vortexing every 15 min at room temperature. Cells with bound trimer S protein were washed out twice with PBS by centrifugation at 3000× g for 4 min. One hundred microliters of 1% Triton X-100 in PBS were added to each pellet to solubilize cell membranes and proteins bound to them. After 1 h incubation at room temperature with rotation, insoluble cell debris was removed by centrifugation at 23,000× g for 1 h, and 100 μL of supernatant from each sample was added to each well of Nickel-Coated Plate and incubated overnight to allow His-tagged SARS-CoV-2 trimer S protein to bind to the surface of the wells. Bound trimer S protein was detected with mouse monoclonal antibody against SARS-CoV-2 spike protein and Horseradish Peroxidase-conjugated Goat Anti-Mouse IgG. Each incubation with antibodies was performed in PBS buffer containing 0.05% tween, 1 mM MgCl and 0.5% fish gelatin for 1 h at room temperature. After each incubation, the wells were washed with the same buffer without fish gelatin three times. Bound Horseradish Peroxidase-conjugated antibodies were detected with TMB substrate. Reaction was stopped with 2 M sulfuric acid, and absorbance was measured at 450 nM.
3.6. Secondary Structure Prediction
The secondary structure analysis of our aptamers was performed using the mFold webserver [
48]. A temperature of 37 °C and 0.2 M of NaCl were used for the secondary structure simulation. The truncation was performed by removing the big loop (nucleotides from position 38 to 85) from the AYA2012004_L.
3.7. Flow Cytometry Study of SARS-CoV-2 Trimer S Protein Binding to Vero E6 Cells and Its Inhibition by Aptamers
SARS-CoV-2 trimer S protein was pre-incubated with aptamers, control aptamers with random sequences, or neutralizing antibody in PBS buffer containing 1 mM MgCl prior to the addition to Vero E6 cells. Trimer S protein in the same buffer alone was used as a positive control. Vero E6 cells (105 cells per well) were diluted in 200 μL PBS buffer and then centrifuged at 250× g for 3 min. The pre-incubated S protein at different conditions listed above was added to Vero E6 cells for 30 min at room temperature followed by two washes with PBS buffer. Anti-his-tag-APC antibodies (1 μL per cells in 50 μL of PBS), control antibody, and fixable viability dye eFluor 450 (dilution 1:1000) were added to the corresponding wells and incubated for an additional 30 min at 4 °C followed by washing with PBS buffer. Cells were fixed with 200 μL FluoroFix buffer per well. Cells were acquired on Navios-Ex flow cytometry, and data were analyzed by FlowJo v10 software.
3.8. Detection of SARS-CoV-2 Trimer S Protein Binding to Transfected 293T Cell Line Using Flow Cytometry
To confirm the expression of the ACE2 receptor on the surface of stably transfected HEK293T cells, ACE2-transfected cells and mock-transfected HEK293T cells were stained with anti-hACE2-APC, Goat IgG-APC, and fixable viability dye eFluor 450 at 4 °C for 30 min, which was followed by washing with PBS buffer. Cells were fixed with 200 μL per well FluoroFix. Fluoro fixed cells were acquired on Navios-Ex flow cytometry, and data were analyzed by FlowJo v10. To demonstrate that S protein binds to ACE2 receptors on the surface of the cells, His-tagged SARS-CoV-2 trimer S protein was incubated with HEK 293T expressing ACE2 receptors and mock-transfected cells at 4 °C for 30 min. After washing with PBS, cells were incubated with anti-his-tag-APC (1 μL per 10 cells) or isotype control antibodies and the fixable viability dye eFluor 450 (dilution 1:1000) in 50 μL of PBS at 4 °C for 30 min; then, cells were washed and fluoro fixed. Cells were acquired on Navios-Ex flow cytometry, and data were analyzed by FlowJo v10.
3.9. Uptake of SARS-CoV-2 S Protein Expressing Virus-like Particles (VLPs) by HEK293T Cells That Overexpress ACE2 Receptors
Mock and ACE2 receptor overexpressing HEK293T cells were cultured in 48-well plates at 60% confluency (about 10 per well) in medium A supplemented with 10% FBS. VLPs that have the full-length SARS-CoV-2 S protein expressed/presented at the surface of lentiviral particle to mimic the coronavirus were used. The VLPs are packaged with Green Fluorescent Protein plasmid (GFP) as a reporter signal. VLPs ( particles) were diluted in 50 μL PBS buffer in the absence or presence of aptamers and incubated at room temperature for 30 min. Pre-incubated VLPs of each condition were added to cultured HEK293T cells that are mock-transfected or ACE2 receptor stably transfected and incubated for 72 h. Cells were washed once with PBS and stained with viability dye eFluor 450 (dilution 1:1000) in 50 μL of PBS at 4 °C for 30 min. The uptaking of VLPs was assessed by measuring the GFP signal in the cells by flow cytometry on the Navios-EX system, and data were analyzed by FlowJo v10.
3.10. Binding Affinity Measurement Using Surface Plasmon Resonance (SPR)
To determine the binding affinity of aptamers to SARS-CoV-2 trimer S protein, biotinylated aptamers were immobilized as a dot in volume ∼1 μL on a CSe surface coated chip with an extravidin layer (HORIBA France, France) at a concentration of 20 μM overnight. Random biotinylated ssDNA sequences were used as a negative control. The biochip was blocked with 10 μg/mL of biotin in PBS and saturated with 1% BSA in PBS. The analytes, original strain SARS-CoV-2 trimer S protein, Delta variant of SARS-CoV-2 trimer S protein, and Alpha variant of SARS-CoV-2 trimer S protein were diluted with PBS and injected over the flow cell at concentrations of 20 nM, 5 nM, 1 nM and 0.2 nM at a flow rate of 50 μL/min with PBS as a running buffer at a temperature of 25 °C. The complex was allowed to associate and dissociate for 200 s and 360 s, respectively. Influenza A H1N1 hemagglutinin protein, SARS-CoV-2 Nucleocapsid protein, and human ACE2/ACEH protein were injected at 10 nM, and serum from the healthy human donor was injected at 1:100 dilution as negative controls. The surface was regenerated with a 200 sec injection of 1 M NaCl. Triplicate ligand spots and a buffer blank were flowed over the surface. The entire experiment was repeated on two different biochips in duplicates. The data were fitted to a simple 1:1 interaction model using the global data analysis with ScrubberGen software.
4. Statistical Analysis
Significance was determined in Prism 9.0 (GraphPad Software) using the one-way ANOVA test (Dunnett’s multiple comparisons test) or two-way ANOVA test (Tukey’s multiple comparisons test) for comparisons. The p values < 0.05 were considered statistically significant.
5. Results
5.1. Developing High-Affinity Neutralizing DNA Apatmers against SARS-CoV-2 Trimer S Protein
To identify single-stranded DNA aptamers that bind to the SARS-CoV-2 trimer S protein and inhibit its binding to ACE2 receptors, we employed the SELEX procedure as described [
46] with some modifications. The SARS-CoV-2 trimer S protein that carries a polyhistidine tag at the C-terminus was immobilized on Ni-NTA magnetic beads. A library of single-stranded DNA oligonucleotides (10
random unique sequences) that are flanked by two 23-based of primer sequences was used for the selection of specific aptamers. To allow ssDNA to fold into their unique conformation, 10 nM of ssDNA library was heated at 95 °C followed by incubation on ice. Bare Ni-NTA magnetic beads were introduced to the activated ssDNA library to prevent the enrichment of aptamers that recognize the beads only. The SELEX procedure was performed using trimer S protein-coated magnetic beads as a target (
Figure 1A). The counter-selection steps against Ni-NTA magnetic beads were performed after every third round of selection. The selection stringency was increased by decreasing the concentration of trimer S protein and increasing the stringency of wash buffer with a simultaneous increase in washing times. After six rounds of conventional selection, the human ACE2 receptor was added to trimer S protein immobilized on the beads to block the receptor binding site on trimer S protein. This complex was used to select aptamers that specifically bind to the receptor-binding site on trimer S protein (
Figure 1B). The eluted ssDNA from round six of selection was incubated with the proteins complex (S and ACE2 proteins) immobilized on the beads. The flowthough, representing aptamers that specifically bind to trimer S protein sites concealed by ACE2 receptors were collected and proceeded for five more rounds of selection. To enhance the specificity of the aptamers, counter-selection was performed using pulmonary surfactant, human plasma, and BSA. The ssDNA pools eluted from the 4th, 8th, 9th, and 12th rounds of selection were chosen for high-throughput sequencing using the MiSeq (Illumina) platform. Sequence analysis was made according to the FASTAptamer to identify sequences that showed enrichment across selection pools [
47]. The ten most enriched sequences after twelve rounds of selection are presented in
Table 1.
5.2. Aptamer Binding to SARS-CoV-2 Trimer S Protein
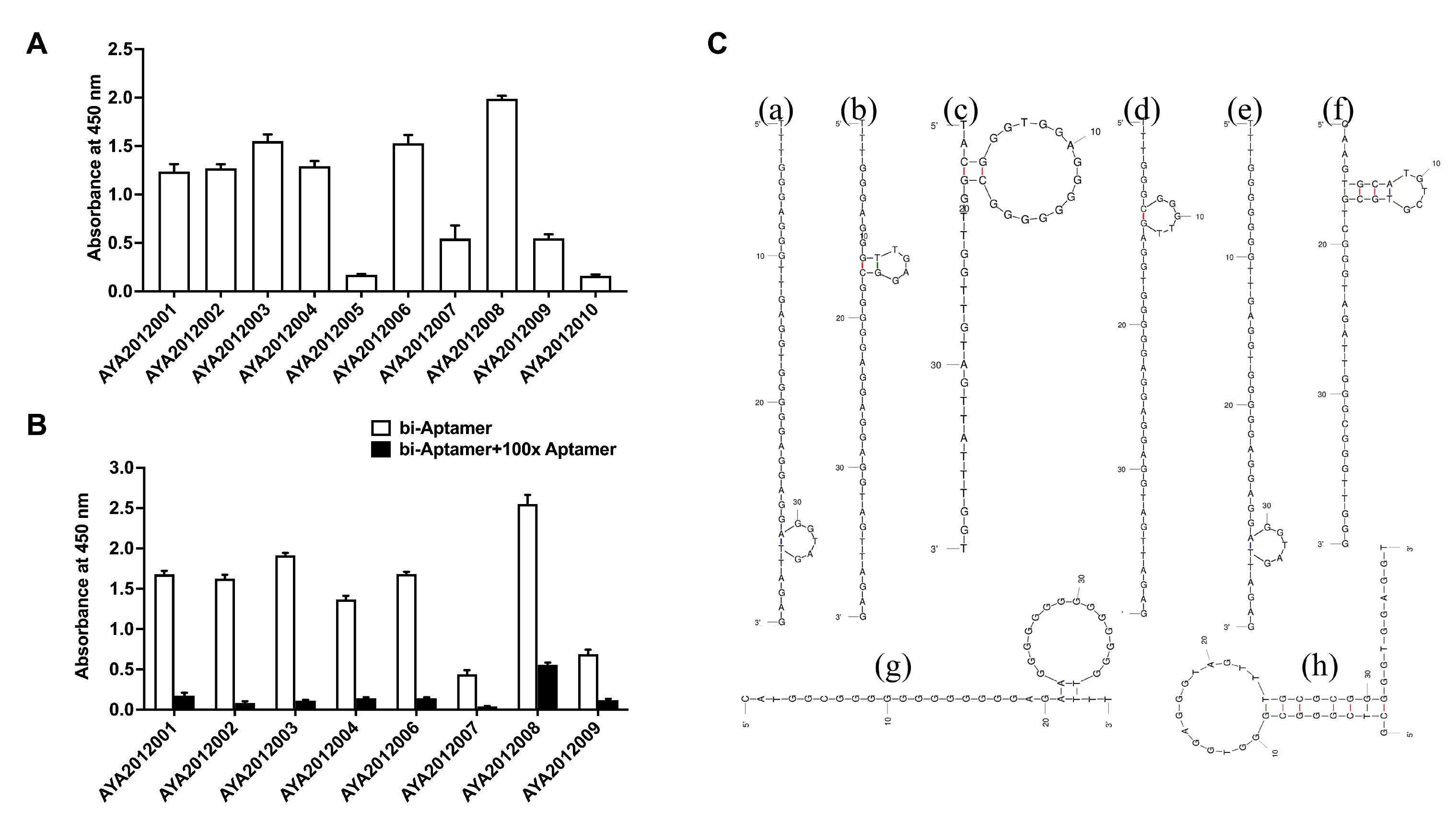
The ten most enriched aptamer sequences were evaluated for their binding to SARS-CoV-2 trimer S protein using an ELISA-based binding assay. The recombinant SARS-CoV-2 trimer S protein was immobilized on a MaxiSorp plate, and biotinylated aptamers were incubated with immobilized protein. Bound aptamers were detected using streptavidin–HRP. All aptamers exhibited binding ability except AYA2012005 and AYA2012010 (
Figure 2A). The specificity of the aptamers with good binding affinity was determined using competition ELISA-based binding assay (
Figure 2B). For this purpose, one-hundred-fold excess of the non-biotinylated aptamers was added to the wells with respective biotinylated aptamers. If binding is specific, non-biotinylated aptamers compete out biotinylated aptamers, and absorbance at 450 nM will significantly decrease. As shown in
Figure 2B, all tested aptamers were competed out with one-hundred-fold excess of the non-biotinylated respective aptamers and demonstrated specific binding to the SARS-CoV-2 trimer S protein. The predicted secondary structures of selected aptamers are shown in
Figure 2C. All selected aptamers have a hairpin structure with the unpaired loop size ranging from 4 nucleotides (AYA2012002) to 14 nucleotides (AYA2012003, AYA2012008, and AYA2012009). AYA2012001 and AYA2012006 share an almost identical predicted secondary structure, as the only difference is the A/G change at site 7. The increased base pairs in AYA2012007 and AYA2012009 as compared to others could result in enhanced stability. It is worth noting that the selected aptamers are rich in G nucleotides, indicating that the formation of the G quadruplex, which has been identified as a common motif in many DNA aptamers, may improve the stability of these aptamers [
49].
5.3. Aptamers’ Inhibition of the Binding of SARS-CoV-2 Trimer S Protein to ACE2 Receptors Expressed on the Surface of Vero E6 Cells
Angiotensin-converting enzyme 2 (ACE2) serves as the cell surface receptor to bind the SARS-CoV-2 trimer S protein and facilitates the entry of these coronaviruses into the cell [
50]. The African green monkey kidney cell line, Vero E6, are commonly used to isolate, propagate and study SARS-CoV-like viruses as they support viral entry and replication to high titers [
51]. To investigate the inhibitory effect of our aptamers on the SARS-CoV-2 trimer S protein binding to ACE2 receptors, the Vero E6 cells line was used. His-tagged SARS-CoV-2 trimer S protein was incubated with Vero E6 cells in the absence or presence of the indicated aptamers. Random 40-nucleotide ssDNA was used as a control. Unbound S-protein was washed out and the cell pellet, containing the bound His-tagged SARS-CoV-2 trimer S protein, was solubilized with 1% Triton X100. Insoluble cell debris was removed by centrifugation and supernatant was added to wells of nickel coated plate. The His-tagged S protein bound to nickel was detected using mouse monoclonal antibodies against S protein and horseradish peroxidase-conjugated goat anti-mouse antibodies (
Figure 3A). Our results show that the majority of the aptamers inhibit the binding of SARS-CoV-2 trimer S protein to ACE2 receptors. Aptamers AYA2012001, AYA2012004 and AYA2012006 had the best inhibition efficiency up to 70%. The affinity of those aptamers was verified by surface plasmon resonance (SPR) (
Figure 3B). Aptamers AYA2012001, AYA2012004 and AYA2012006 showed a high affinity for the SARS-CoV-2 trimer S-protein with Kd 4.9 nM, 3.3 nM and 6.2 nM, respectively.
5.4. Binding of Selected Aptamers to Variants of SARS-CoV-2 Trimer S Protein
Since the pandemic started, the SARS-CoV-2 virus has mutated frequently [
52], and mutations in the SARS-CoV-2 S proteins have conferred an advantage to the virus. For example, the N501Y mutation in B.1.1.7 (Alpha), T487K, P681R, and L452R mutations in B.1.617.2 (Delta), K417N mutation in AY.1/AY.2 (Delta plus), D614G, P681H, and D950N mutations in B.1.621 (Mu), G75V, T76I, Δ246-252, L452Q, F490S, D614G, and T859N mutations in C.37 (Lambda), T478K, Q498R, and H655Y mutations in B.1.1.529 (Omicron) have been shown to majorly contribute to the virus’s ability to become more infectious, bind more tightly to human cells ACE2 receptors, and evade vaccines and some neutralizing antibodies [
53,
54,
55,
56,
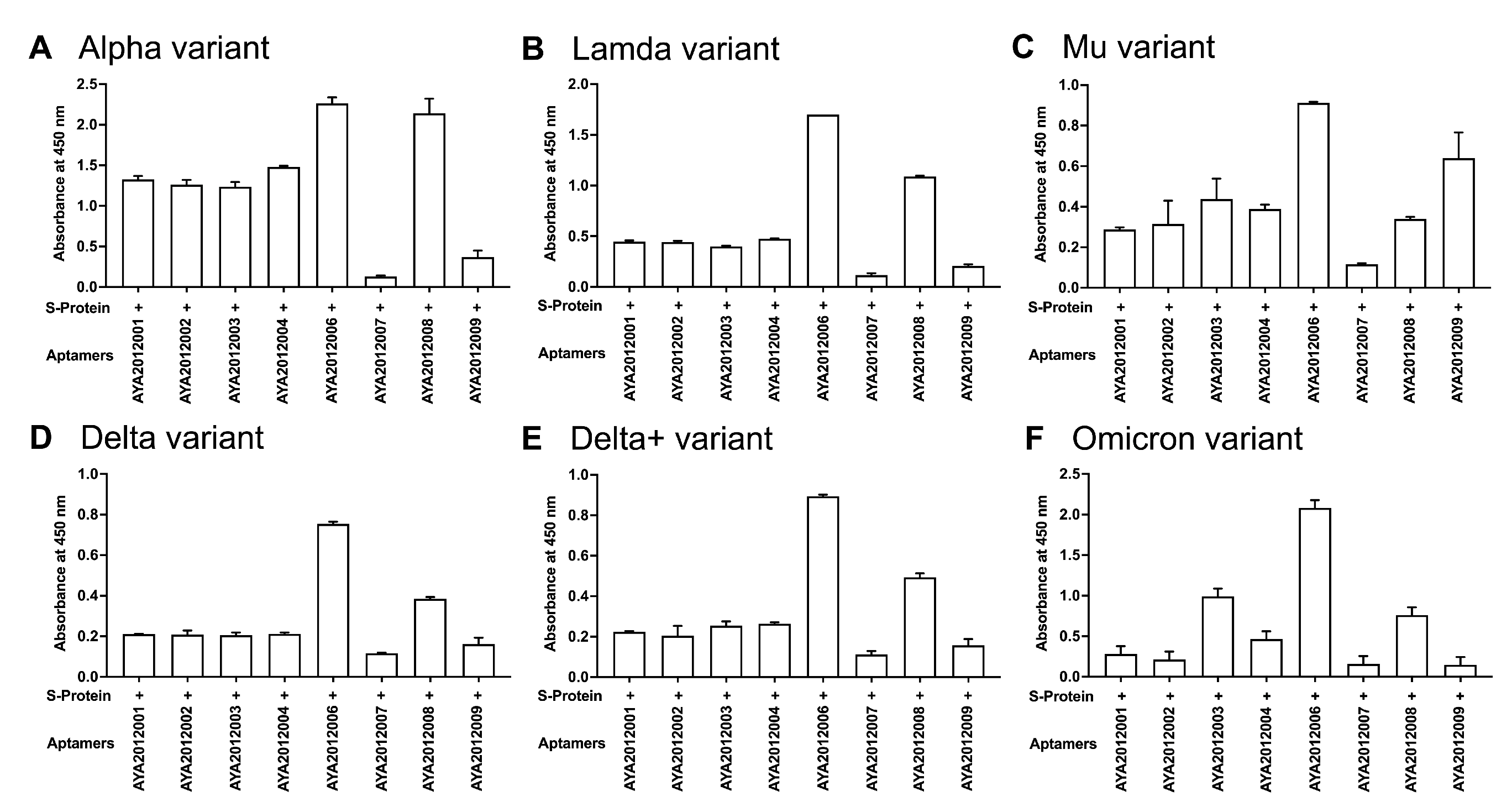
57]. In this study, ELISA-based binding assay was performed to determine if selected aptamers could recognize the mutated S proteins of the COVID-19 variants (
Figure 4). Recombinant SARS-CoV-2 trimer S proteins from (A) Alpha, (B) Lambda, (C) Mu, (D) Delta, (E) Delta plus, and (F) Omicron variants were immobilized on a MaxiSorp plate. Biotinylated aptamers were added, and bound aptamers were detected using streptavidin–HRP. As seen in
Figure 4, our aptamers demonstrated different binding affinities to the SARS-CoV-2 trimer S protein variants with AYA2012006 and AYA20122008 showing the best binding to all variants and aptamer AYA2012009 showing the best binding to Mu as compared to other tested variants. This presents the advantage of potentially developing combined therapy consisting of aptamers that can target different variants of the S protein.
5.5. Long Aptamers Bind to SARS-CoV-2 Trimer S Protein and Inhibit the S Protein Binding to Vero E6 Cells
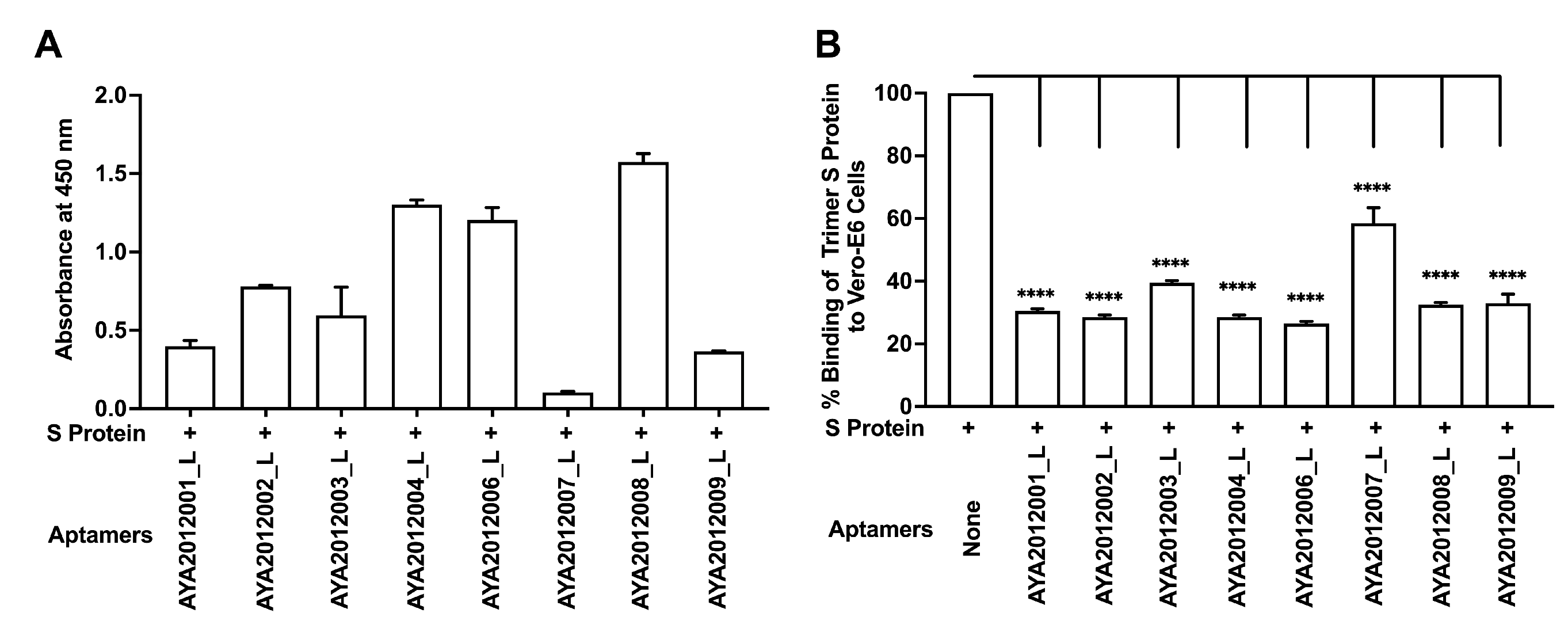
Candidate aptamers were modified by reinserting the primer sequences that were part of the original library (long aptamers) and were tested for their binding ability to SARS-CoV-2 trimer S protein and inhibitory effect on SARS-CoV-2 trimer S protein binding to ACE2 receptors on Vero E6 cells (
Figure 5). The experiments were performed as previously described. All long aptamers except AYA2012007_L demonstrated good binding ability as seen in
Figure 5A. Among the long aptamers, AYA212001_L, AYA 212002_L, AYA2012004_L, and AYA2012006_L showed better inhibitory activity (
Figure 5). It is valuable to note that aptamer AYA2012004_L provided the best balance between binding ability and inhibition efficiency. The predicted secondary structures of long aptamers are shown in
Supplementary Figure S1. Based on the predicted structures, the incorporation of the two conserved primer sequences changed the secondary structures of all aptamers. For example, an additional hairpin structure with 36 unpaired nucleotides along with 4 base pairs of stems appears in AYA2012004_L. Among the 8 selected aptamers, AYA2012001_L, AYA2012004_L, AYA2012006_L, AYA2012007_L and AYA2012009_L appeared to have a divalent structure (two hairpin motifs). The additional pairs in the stems of these long aptamers are expected to enhance their stability.
5.6. Characterization of Modified Aptamers
Since aptamer AYA2012004_L demonstrated the best balance between binding ability and inhibitory efficiency, we decided to further modify and enhance its efficacy. To assess the importance of different motifs in AYA2012004_L, we modified the aptamer further by truncating its secondary structure as shown in
Figure 6A. Aptamer AYA2012004_L-M1 was modified by removing the big loop (nucleotides from position 38 to 85) from AYA2012004_L. The inhibitory efficiencies of AYA2012004_L and AYA2012004_L-M1 were then tested (
Figure 6B). Interestingly, AYA2012004_L-M1 remains almost as effective as AYA2012004_L with respect to binding and inhibition. The specificity of selected aptamers was challenged in an ELISA-based binding assay with high stringency wash buffer (PBS, 0.5 M NaCl, 0.05% Tween 20 and 1 mM MgCl
, PH∼7.4). Notably, this experiment demonstrated that AYA2012004_L and AYA2012004_L-M1 show good binding to trimer S protein, unlike the control aptamer and the primer sequences, which flank the modified and control aptamer alone (
Supplementary Figure S2). The sequences of modified and control aptamers can be found in
Supplementary Table S2. Concentration dependence of inhibition of the trimer S protein binding to Vero E6 cells was obtained for both modified aptamers, and the results are shown in
Supplementary Figure S3. Modified aptamer AYA2012004_L-M1 demonstrated up to 70% inhibition of trimer S protein binding to Vero E6 cells even at concentrations as low as 0.05 μM.
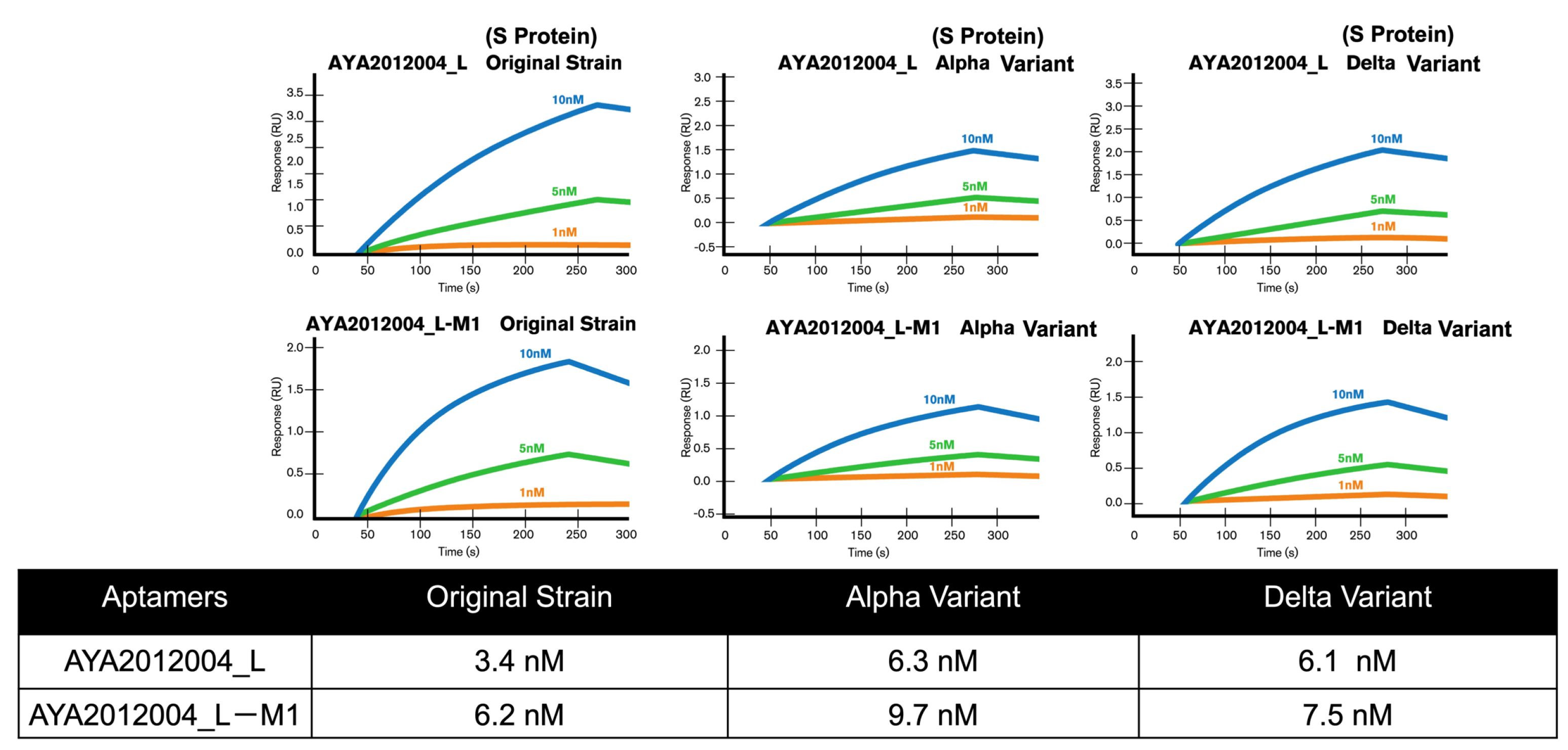
5.7. Binding Affinity and Specificity Determination of Modified Aptamers to Different Variants of S Protein with Surface Plasmon Resonance (SPR)
The selected aptamers AYA2012004_L and AYA2012004_L-M1 were further characterized for their binding affinity to the SARS-CoV-2 trimer S protein of original strain, Delta, and Alpha variants with surface plasmon resonance. Interestingly, both selected aptamers showed a high binding affinity for these variants’ trimer S proteins in the nM range. The Kd numbers were determined and are presented in the table (
Figure 7). The specificity of the selected aptamers was evaluated by measuring their binding to the following four proteins: Influenza H1N1 hemagglutinin proteins, SARS-CoV-2 nucleocapsid protein, Human ACE2/ACEH protein, and serum from a healthy human donor.
Supplementary Figure S4 shows SPR data for all three aptamers. The data clearly indicated that none of the four control proteins showed detectable binding to either aptamer. The result showed that all three aptamers recognized the SARS-CoV-2 trimer S protein with high affinity and specificity.
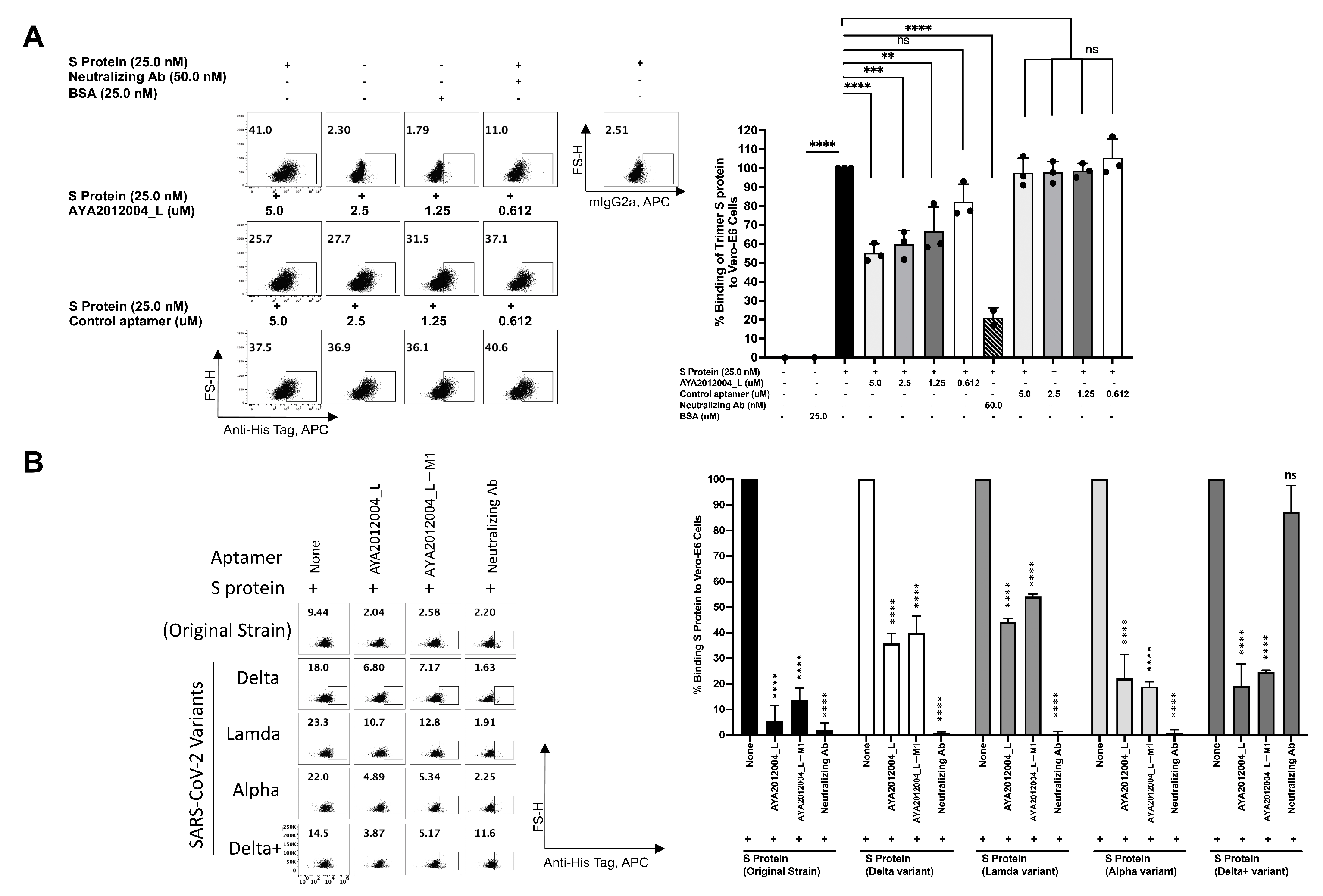
5.8. Flow Cytometry-Based Assay to Assess the Inhibitory Effect of Modified Aptamers on the Binding of Variants of Trimer S Protein to ACE2 Receptors on Vero E6 Cell
A flow cytometry approach was used to confirm SARS-CoV-2 trimer S protein binding to ACE2 receptors on the surface of Vero E6 cells. The binding of the trimer S protein to Vero E6 cells was determined at various concentrations of S protein as indicated in
Supplementary Figure S5. The specificity of the trimer S protein binding was confirmed by the neutralizing antibodies that inhibit the trimer S protein binding to ACE2 receptors at two fold excess. The trimer S protein at 50 nM, 25 nM and 12.5 nM concentrations bind by approximately 39%, 27% and 22%, respectively. The binding percentage was also confirmed on the gMFI index that shows concentration-dependent binding. To test the inhibitory efficiency of AYA2012004_L aptamer, SARS-CoV-2 trimer S protein was pre-incubated with different concentrations of the aptamer before addition to Vero E6 cells. The concentration-dependent inhibition of the trimer S protein binding to Vero E6 cells by aptamer AYA2012004_L is demonstrated in
Figure 8A. The aptamer concentration 5 μM and trimer S protein concentration 12.5 nM were thus chosen for further inhibition experiments for trimer S protein from different strains binding to Vero E6 cells. Aptamers AYA2012004_L and AYA2012004_L-M1 were selected to determine their neutralizing effect on various SARS-CoV-2 trimer S proteins binding to ACE2 expressed Vero E6 cells. Trimer S proteins binding to ACE2 receptors on the surface of Vero E6 cells was significantly neutralized by aptamers AYA2012004_L and AYA2012004_L-M1 at a concentration of 5 μM as indicated (
Figure 8B). Notably, anti-SARS-CoV-2 RBD-neutralizing Ab inhibits the interaction between the S protein of the Wuhan origin strain, Delta, Lambda, and Alpha and ACE2 receptors; however, anti-SARS-CoV-2 RBD neutralizing Ab was not able to inhibit the interaction between the S protein of Delta plus variant and ACE2 receptors. Taken together, these results show that aptamers AYA2012004_L and AYA2012004_L-M1 bind to the trimer S protein of various strains of SARS-CoV-2 and inhibit the binding of these trimer S proteins to ACE2 receptor-expressed Vero E6 cells.
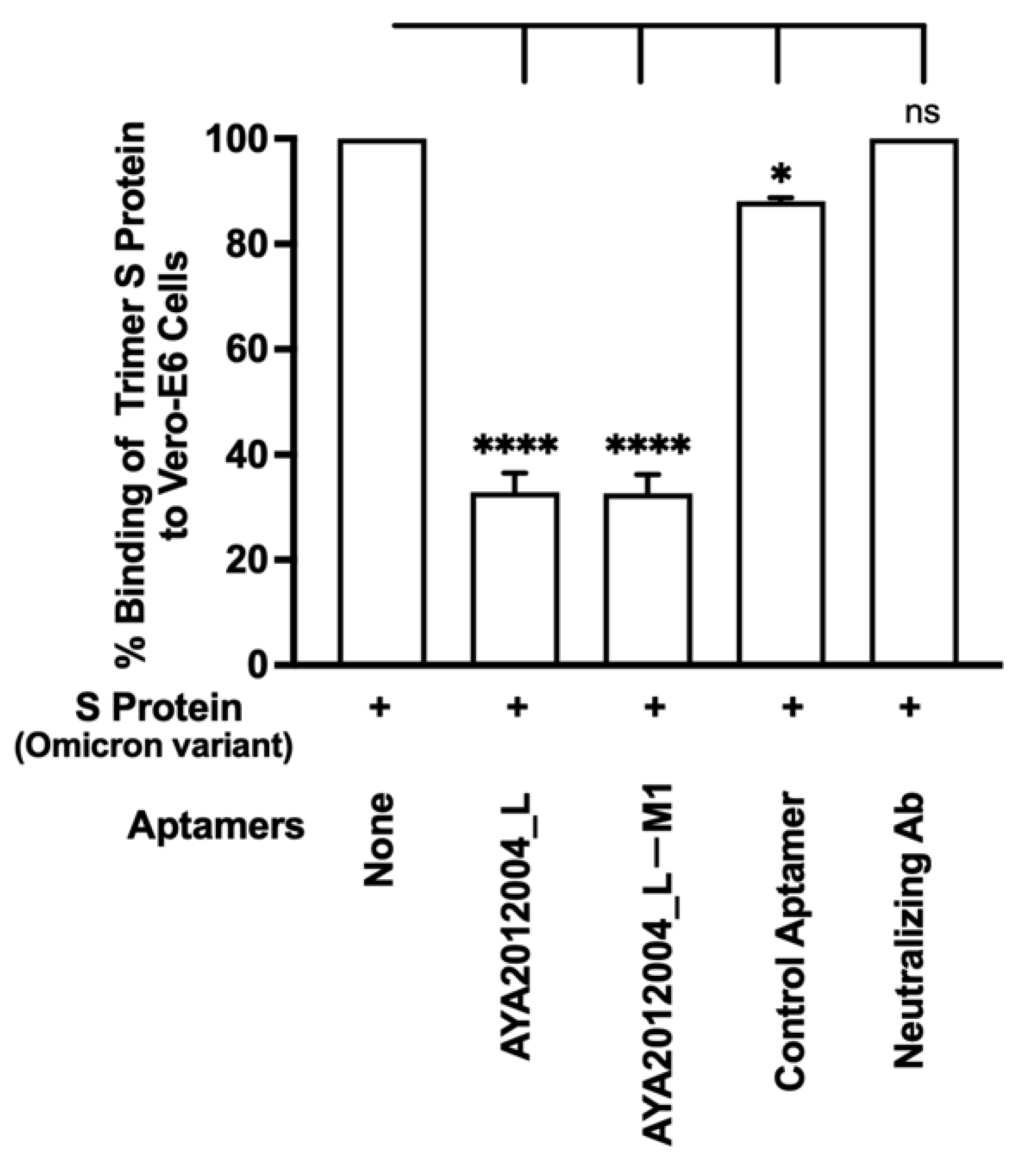
5.9. Selected Aptamers and Omicron
The Omicron variant of SARS-CoV-2 has recently emerged and gradually become the major variant spreading through communities. The Omicron variant has a total of 60 mutations compared to the ancestral variant, 32 of which affect the S protein, and 15 of those that are located in the receptor-binding domain. This resulted in several antibodies that target the S protein RBD to become less effective or ineffective [
23], which created a new concern for the public health system and medical care [
58]. Our aptamers AYA2012004_L and AYA2012004_L-M1 were tested for their inhibitory efficiency for SARS-CoV-2 Omicron trimer S protein binding to ACE2 receptors on the surface of Vero E6 cells (
Figure 9). Our results show that our modified aptamers’ inhibition of the binding of Omicron trimer S protein to ACE2 receptors is similar to other S protein variants. Notably, the anti-SARS-CoV-2 RBD-neutralizing antibody was not able to inhibit the binding of SARS-CoV-2 trimer S protein to ACE2 receptors when tested at 50 nM concentration.
5.10. Neutralizing Aptamers Prevent Entry of S Protein Virus-like Particles (VLPs) into HEK293T Cells Expressing ACE2 Receptor
Stably transfected Human Embryonic Kidney 293 T cells (HEK-293T cells) expressing the ACE2 receptor were obtained using standard protocol from Invitrogen. ACE2 expression on the surface of transfected HEK293T cells was confirmed with flow cytometry (
Supplementary Figure S6A). ACE2 receptor HEK293T cells were stained with anti-hACE2-APC. We observed a shift of fluorescent peak for transfected cells as compared to mock-transfected HEK293T cells (
Supplementary Figure S6A), confirming the expression of the ACE2 receptor on the surface of the transfected cells. To demonstrate that the SARS-CoV-2 trimer S protein binds to ACE2 receptors on the surface of the cells, the His-tagged SARS-CoV-2 trimer S protein was incubated with HEK 293T cells expressing the ACE2 receptor and mock-transfected cells, which was followed by washing unbound trimer S protein. Bound trimer S protein was detected with anti-His-tag-APC antibodies (
Supplementary Figure S6B). The result demonstrated that SARS-CoV-2 trimer S protein specifically binds to ACE2 receptor overexpressing cells.
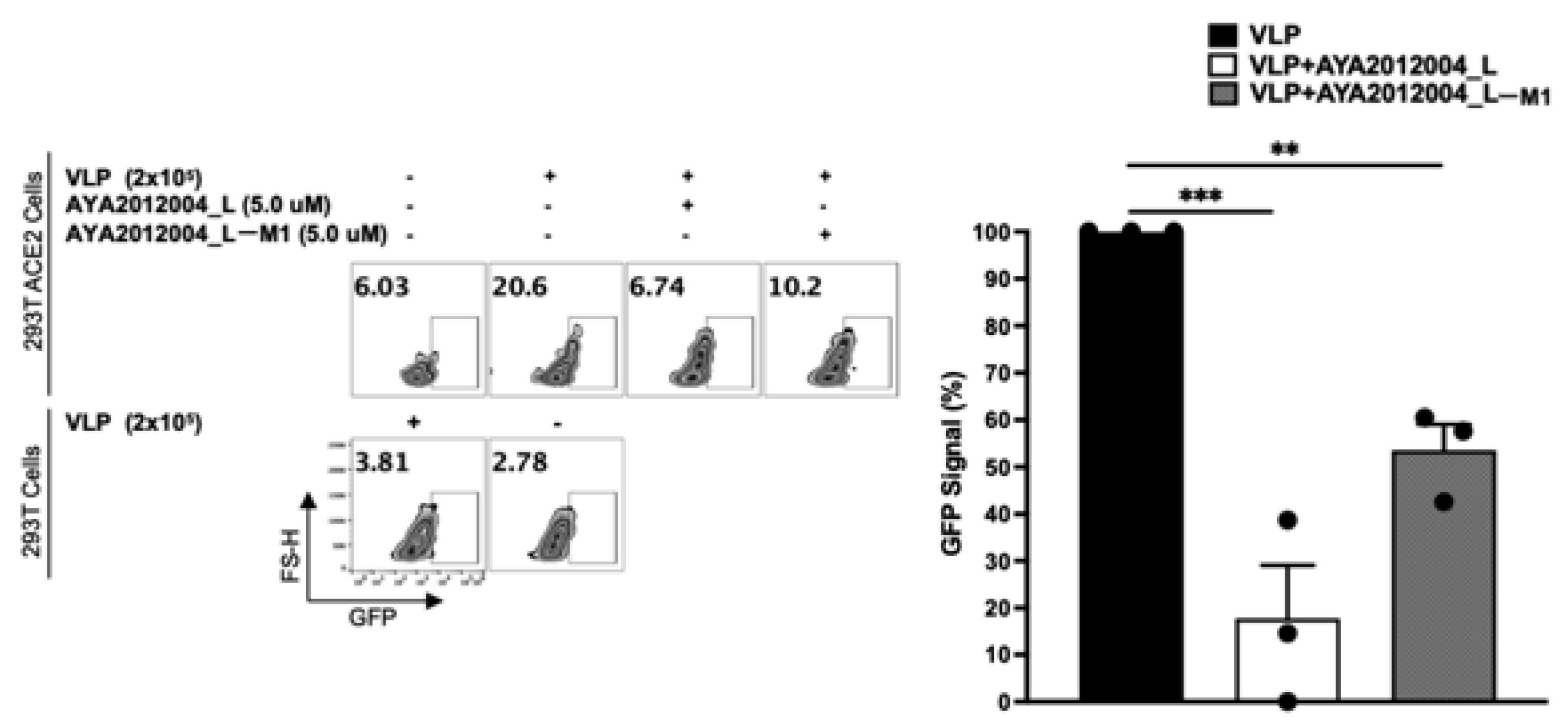
The above cell culture model was used to study the inhibitory activity of our selected aptamers on the uptake of VLPs. In this study, we used VLPs that have the full-length SARS-CoV-2 S protein expressed/presented on the surface of lentiviral particles to mimic the coronavirus. The VLPs are packaged with Green Fluorescent Protein (GFP) plasmid as a reporter signal. When VLPs bind to the cells that express ACE2 receptors, GFP plasmid can enter the cell’s cytoplasm and the signal can be detected after GFP protein is expressed by HEK293T cells.
Figure 10 shows that the GFP signal is detected in 20% of the ACE2 receptor-expressing stable cell line as compared to 3% of mock-transfected cells that show background signal. In other words, our VLPs were taken up by 20% of the ACE2 receptor-expressing cells though binding to S protein. Additionally, all VLPs expressing the S protein significantly decreased the binding and subsequent uptake of VLP by approximately 70% in the presence of aptamer AYA2012004_L and 50% in the presence of AYA2012004_L-M1 (
Figure 10). Our data suggest that the modified aptamers, through their binding to the S protein expressed on the VLP surface, inhibit its interaction with the ACE2 receptors expressed on the cell surface and prevent viral entry.
6. Discussion
Using the SELEX process, we identified a series of neutralizing DNA aptamers that bind with high specificity and affinity to the trimer S protein, which binds to the ACE2 receptor expressed on the surface of cells. The specificity and binding affinity of aptamers are highly dependent on the three-dimensional structure of their target molecule, which is affected by the conditions implemented during the SELEX process. Unlike other studies that developed aptamers utilizing the RBD of the S protein as a target, making it difficult to develop neutralizing aptamers that recognize the ACE2 receptor’s binding site, we performed the SELEX process against the trimer S protein, which is the closest to the native conformation of the target as it is expressed on the envelope of the SARS-CoV-2 virus [
59]. The trimer S protein of SARS-CoV-2 binds to ACE2 receptors, the host cell surface receptor, and mediates the subsequent viral entry via membrane fusion. The selected aptamers were identified after 12 rounds of selection that included stringent wash conditions as well as a series of counter-selections. Counter-selection against blocked beads eliminated non-specifically bound aptamers, whereas counter-selection against the trimer S protein–ACE2 complex enriched for aptamers that bind to the RBD that is concealed by ACE2 (neutralizing aptamers). The first step of viral infection is the entry of the virus to the host cell. In the case of COVID-19 disease, this is initiated by the binding of the S protein that is expressed on the envelope of the SARS-CoV-2 virus to the ACE2 receptor expressed on lung alveolar epithelial cells [
50].
Since aptamers used during the SELEX process consist of 40 variable nucleotides that are flanked by the 23-base conserved sequences on each side, we wanted to test if the additional primer sequences could affect the secondary structure and therefore the binding and subsequent inhibitory properties of our aptamers. Interestingly, adding the primer sequences to aptamers AYA2012004 increased the binding and inhibitory activity of the aptamer.
Aptamers are more stable when they are shorter in length [
60]. The truncation of AYA2012004_L was performed using a similar method implemented by a study conducted by Li et al. [
45] where the hairpin structures and the unpaired nucleic acids sequences are truncated out sequentially from the entire structure. Among the truncated aptamers, AYA2012004_L-M1 appeared to have comparable affinity and inhibition to Wuhan original strain S protein as compared to AYA2012004_L while having only 37 nucleic acids as compared to 85 of AYA2012004_L.
Our study led to the development of a series of neutralizing aptamers that bind to the target trimer S protein in the nM range. Our aptamers depicted different binding affinities to the variants tested. Developing a drug consisting of a combination of aptamers may confer an advantage since it can target different variants of SARS-CoV-2 S protein, resulting in a universal drug for all COVID variants that would emerge.
Our aptamers are specific for the SARS-CoV-2 S protein, since they did not display any binding to influenza H1N1 hemagglutinin proteins, SARS-CoV-2 nucleocapsid protein, or plasma proteins. Modified aptamers AYA2012004_L and AYA2012004_L-M1 blocked the binding of the trimer S protein to ACE2 receptors expressed on the surface of VeroE6 cells as determined by the ELISA-based assay as well as flow cytometry using anti-S protein antibodies.
Recently, the Omicron variant was identified and included 60 mutations, many of which are new to this variant and are in the RBD of S protein. As variants of concern for SARS-CoV-2 virus continue to emerge, the concern about the efficacy of available therapies continues to rise [
61]. Our neutralizing aptamers hold the potential to substitute the antibody used in COVID-19 therapy not only due to cheaper production cost, scalability, very low immunogenicity, no lot-to-lot variation, and higher shelf life stability but also because they can bind to the Delta, Mu, Alpha, Lambda, Delta plus, and Omicron variants of the trimer S protein and inhibit the binding of the different S protein variants to the ACE2 receptors. In fact, we show that the anti-SARS-CoV-2 RBD neutralizing antibody failed to block the interaction between the Delta trimer S protein (
Figure 8) and Omicrom trimer S protein (
Figure 9) and ACE2 receptor, whereas aptamers AYA2012004_L and AYA2012004_L-M1 displayed more than 70% inhibition. Since the strong binding is retained in all of the S protein variants in this study, the attached aptamers prevent the interactions between the ACE2 receptor and S protein, which results in the good inhibition observed in this work.
A number of studies have reported and chosen DNA aptamers to bind RBD [
62,
63], S1 protein [
44], or trimerized S protein [
42] aimed at developing COVID therapies or diagnostic tools. In these studies, their DNA aptamers were found to have high affinity and inhibitory activity. To confirm the uniqueness of our aptamers, the similarity analysis was conducted comparing our selected aptamers and 77 other aptamers from 10 different publications. As shown in the heatmap plots in
Supplementary Figures S7–S16, all similarity scores are below 0.75, indicating our aptamers are unique as compared to the published ones (
Supplementary Table S1). Moreover, our aptamers are rich in G, indicating the stability of these aptamers may be further enhanced with the formation of G quadruplex, which has been reported as a common motif in many DNA aptamers.
Our aptamers can be a potential therapy for COVID-19 due to their stability, ability to recognize different variants of S protein, and ability to prevent viral uptake by inhibiting the binding of S protein to ACE2 receptors. In future studies, the animal model will determine the efficacy of our aptamers in preventing viral uptake in vivo.
7. Conclusions
To address the limitation of current COVID-19 treatment therapy due to the continuous emergence of new SARS-CoV-2 variants of concern, we developed a series of single-stranded DNA (ssDNA) aptamers that were not only able to bind to the trimer S protein of the Wuhan original strain but also bind multiple variants of trimer S proteins of Delta, Delta plus, Alpha, Lambda, Mu, and Omicron. Our selected aptamers inhibited the binding of variants of trimer S protein to ACE2 receptors in the Vero E6 cell line. Furthermore, our modified aptamers AYA2012004_L and AYA2012004_L-M1 showed up to 70% inhibition of the binding and uptake of virus-like particles (VLPs) expressing S protein to ACE2 receptor expressed in Human Embryonic Kidney 293T (HEK293T) cells that overexpress ACE2 receptors. Overall, the findings suggest that our reported aptamers could be an innovative therapy for the treatment of COVID-19. They hold many advantages over existing therapies due to better efficacy, the ability to identify different variants of SARS-CoV-2, and safety.



 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}