

Reactive Molecular Dynamics in Ionic Liquids: A Review of Simulation Techniques and Applications


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
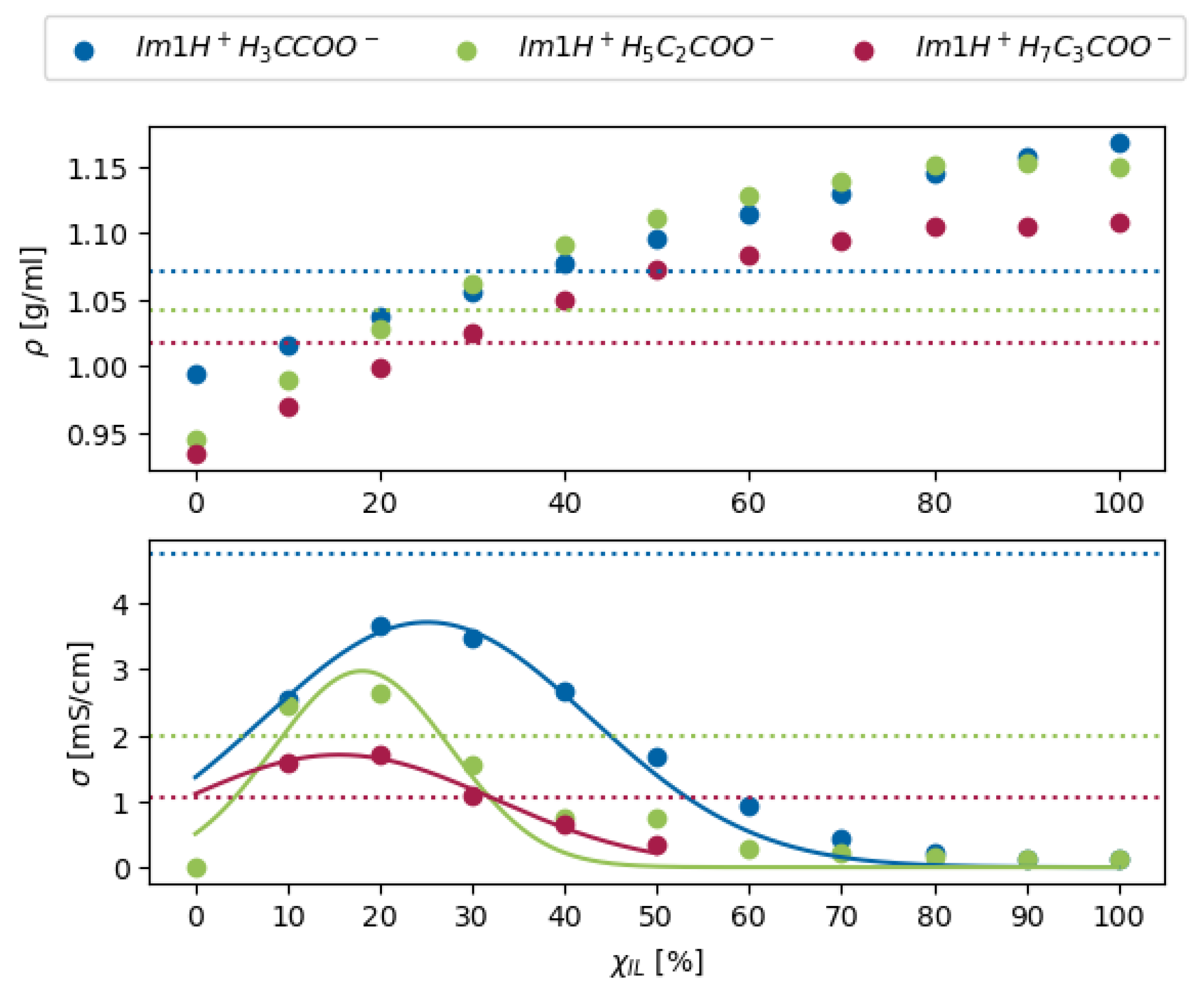
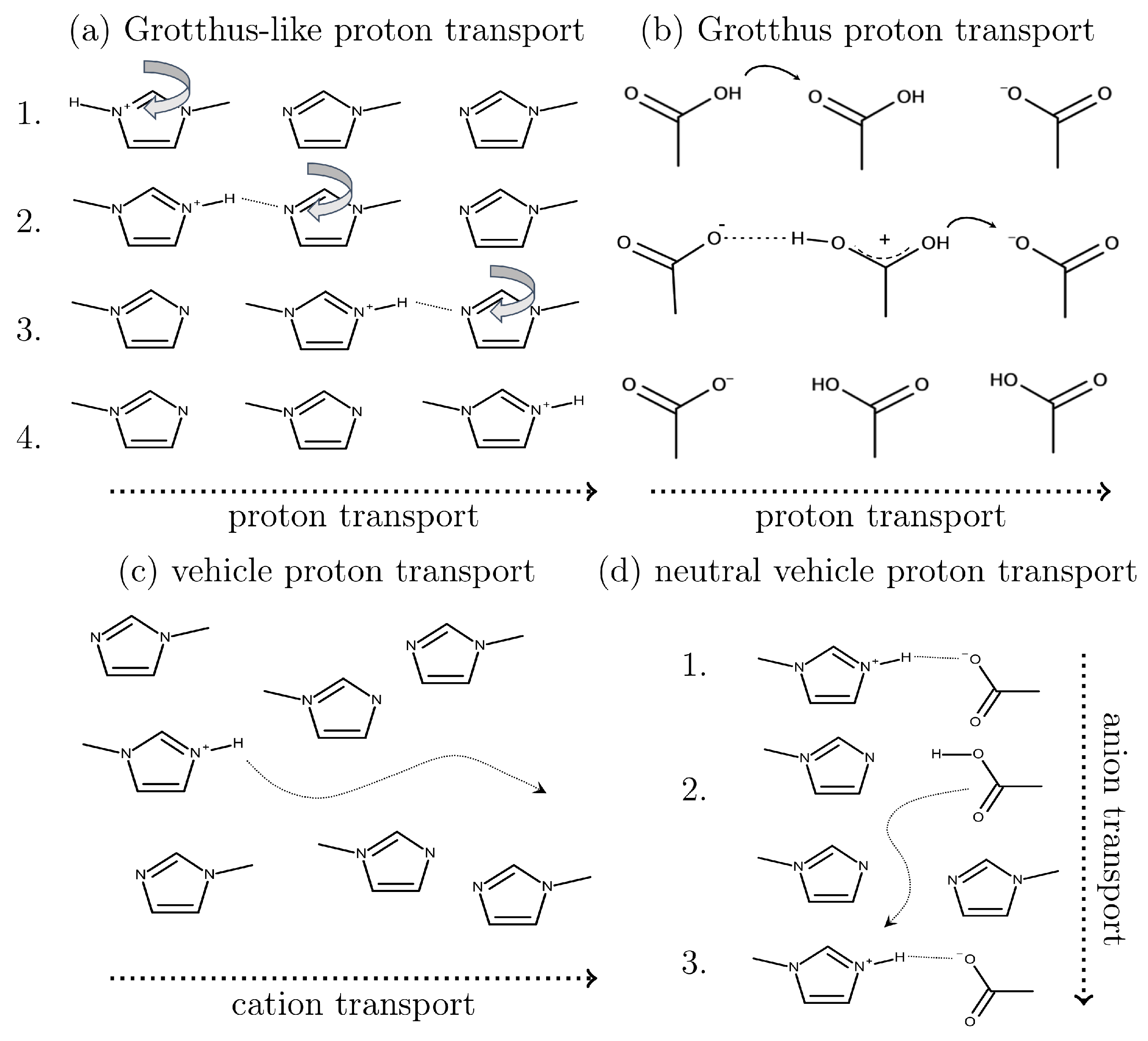
1.1. Proton Transfer Reactions in Ionic Liquids
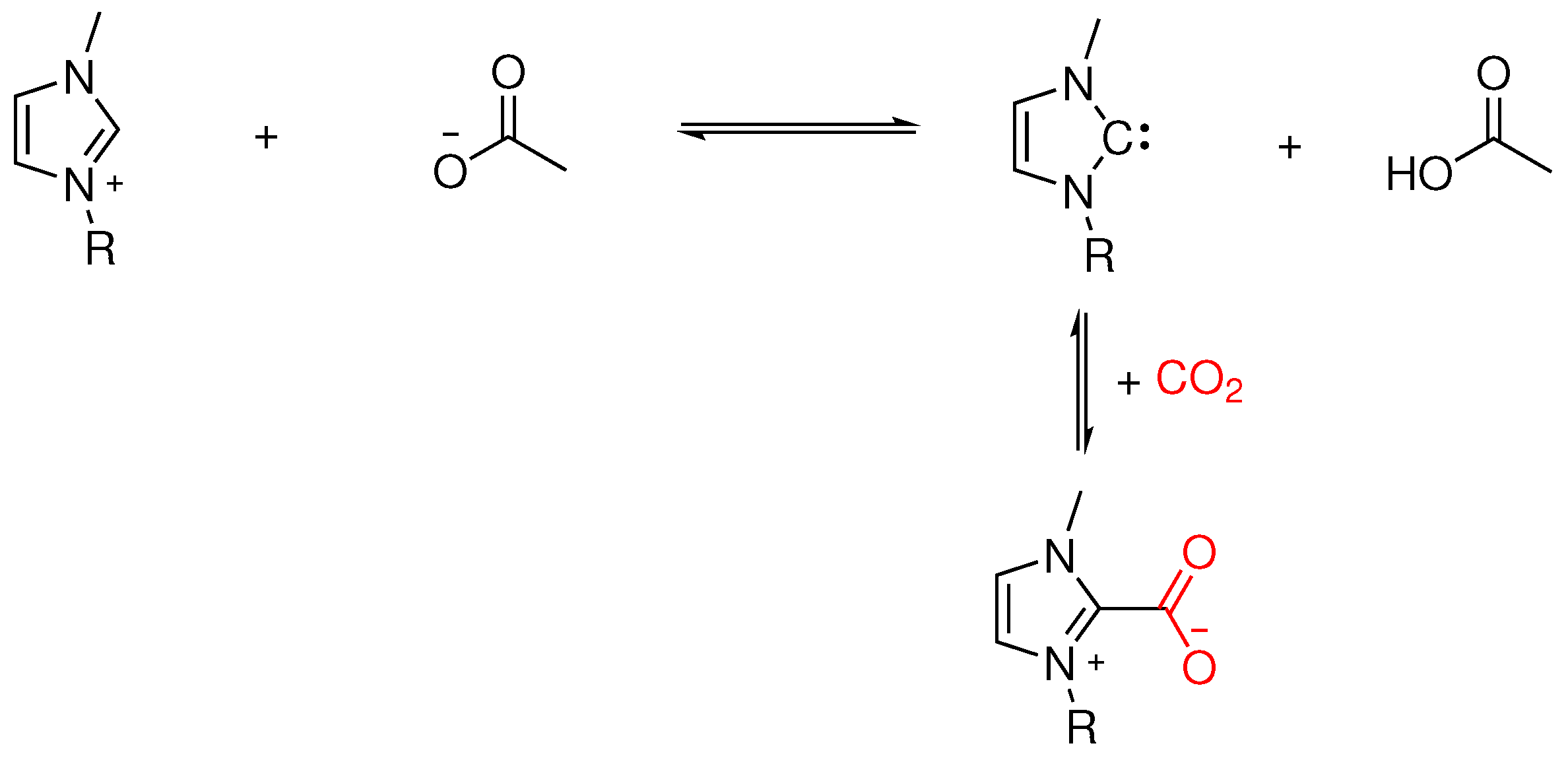
1.2. Chemisorption in Ionic Liquids
1.3. Demand for Computational Studies
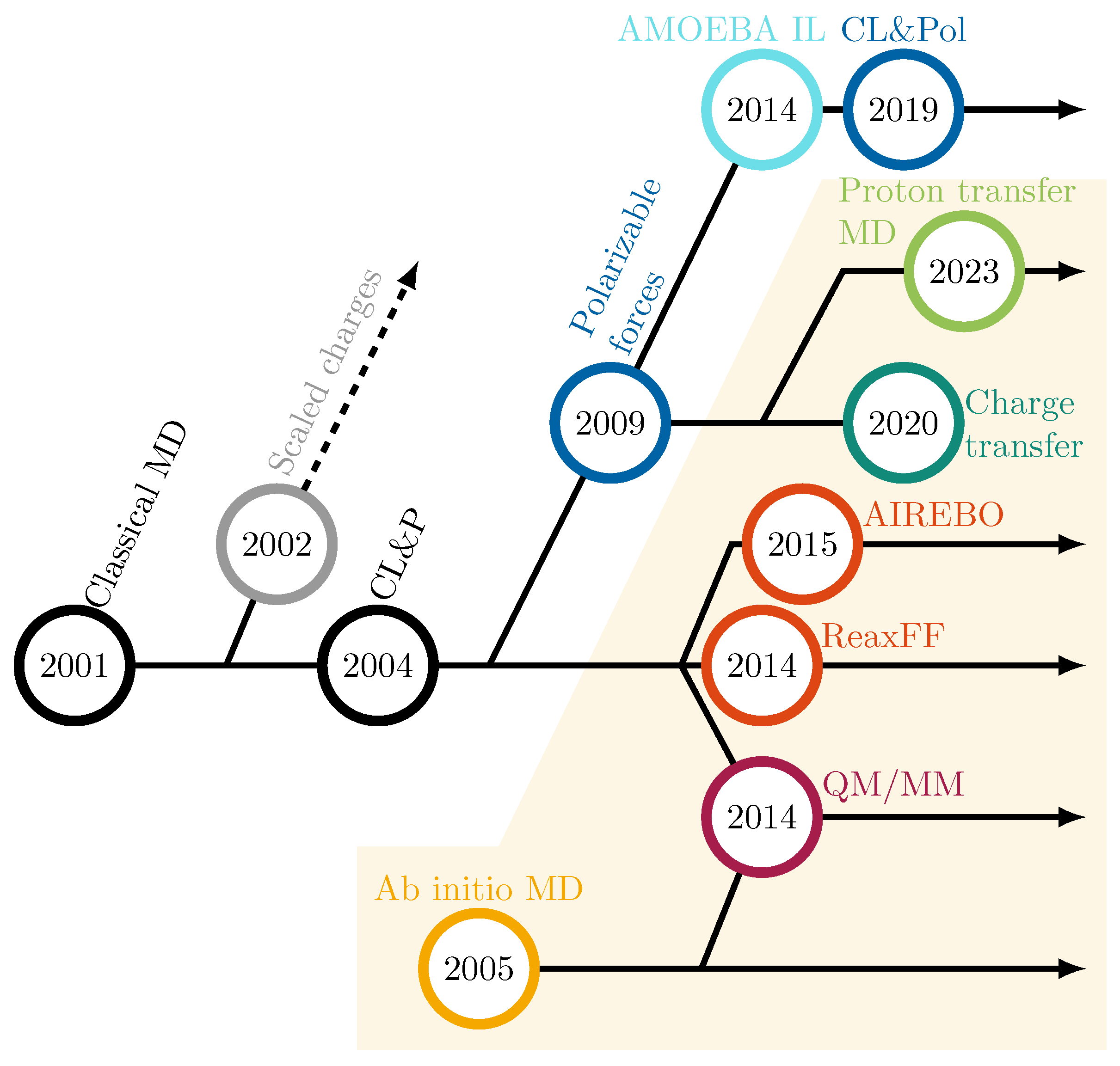
2. Theoretical Investigations
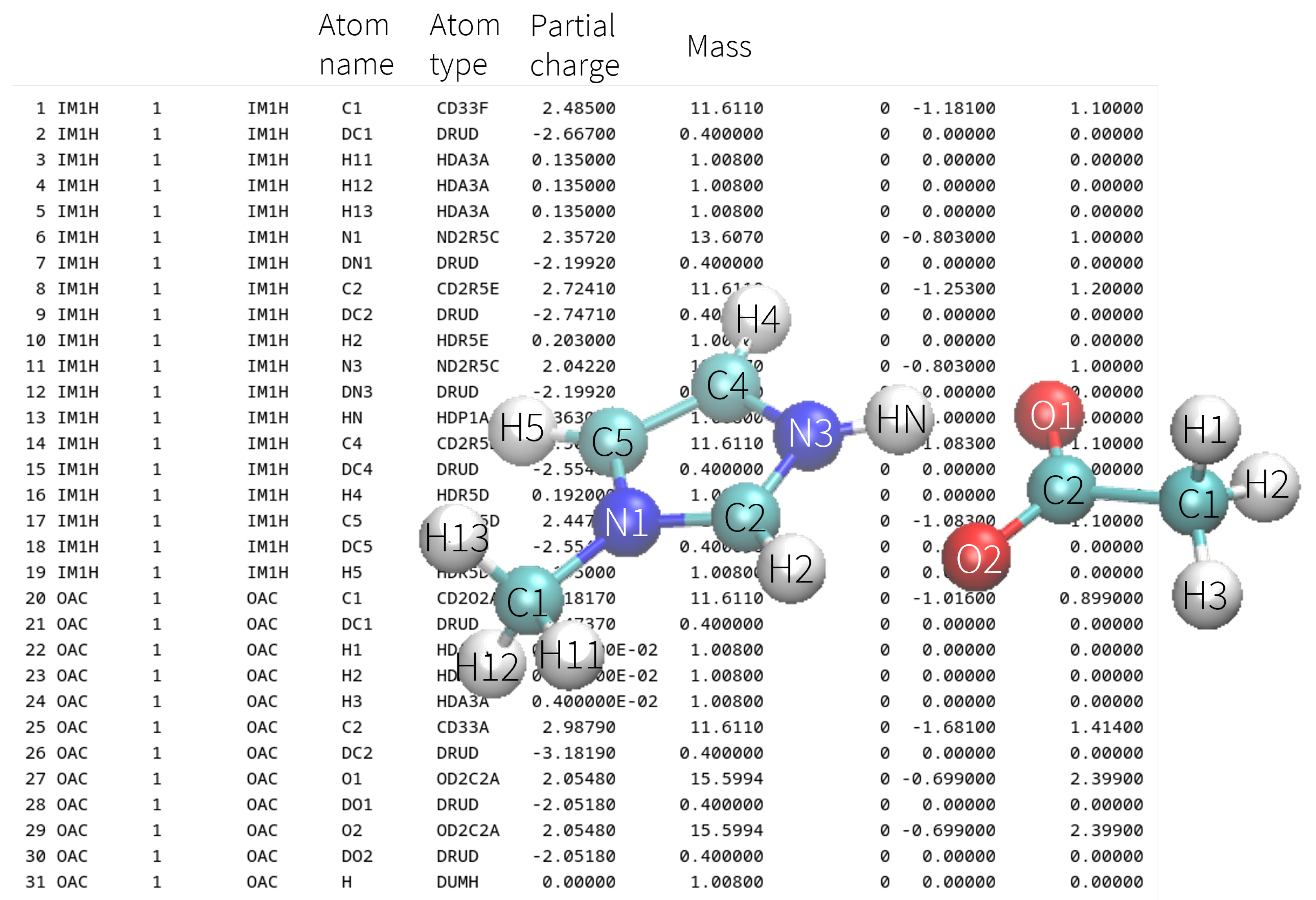
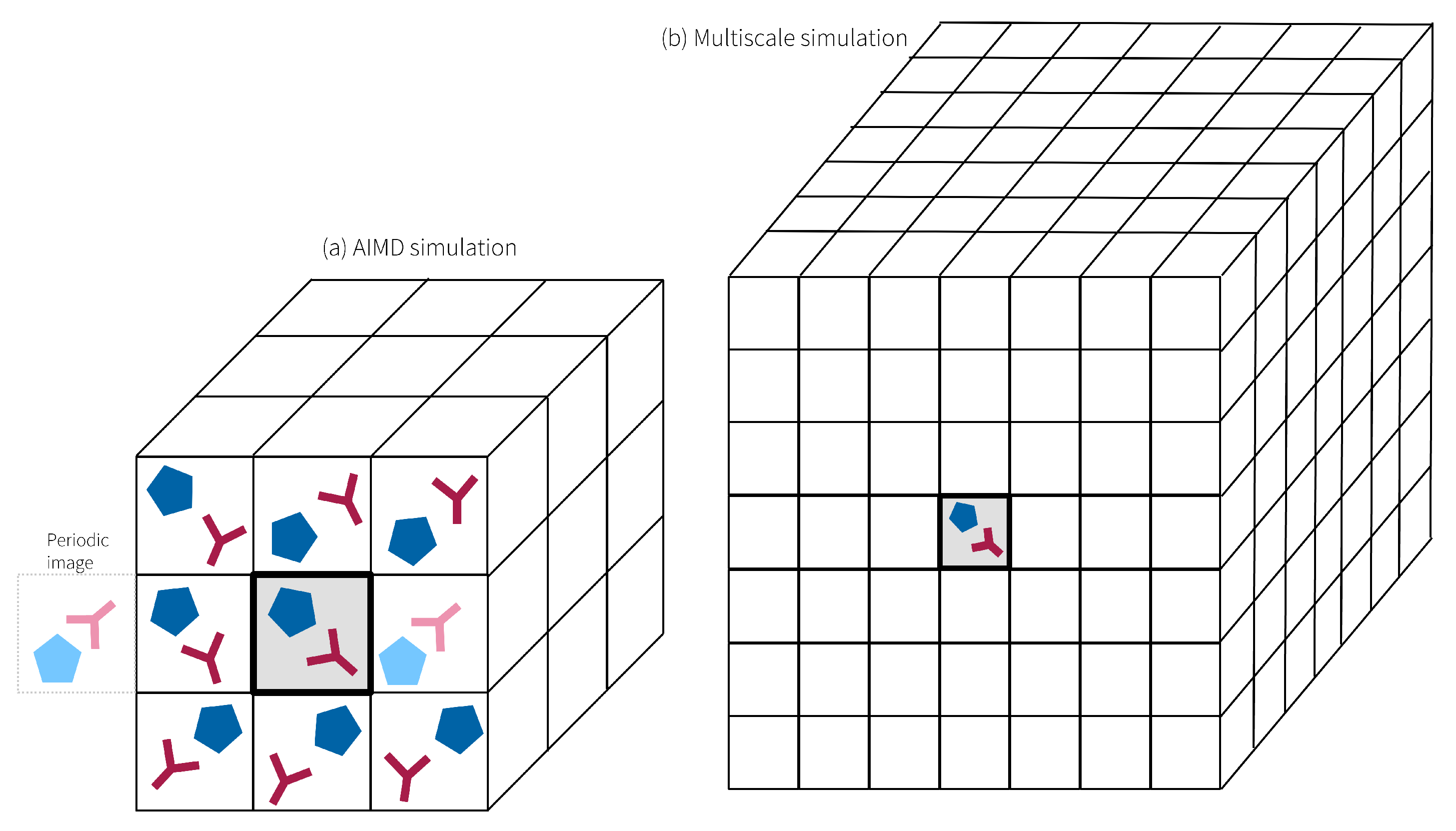
2.1. Fundamentals of Conventional, Fixed-Charge Molecular Dynamics Simulations
2.2. Molecular Dynamics in Ionic Liquids
2.3. How to Include Chemical Reactions?
2.3.1. Quantum-Mechanical-Based Methods
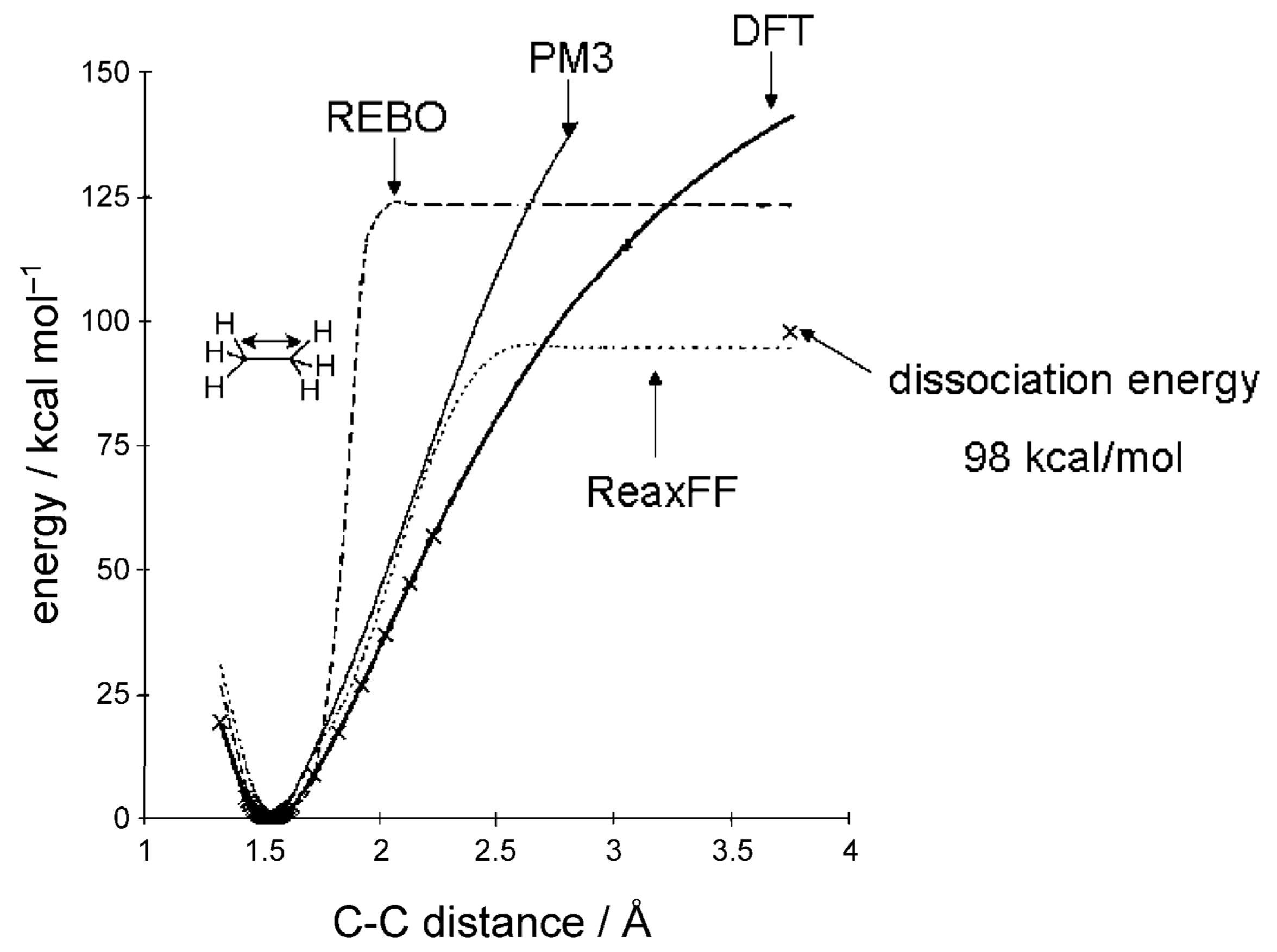
2.3.2. Continuous Force Fields
2.3.3. Fractional Force Fields
3. Modelling the Reaction
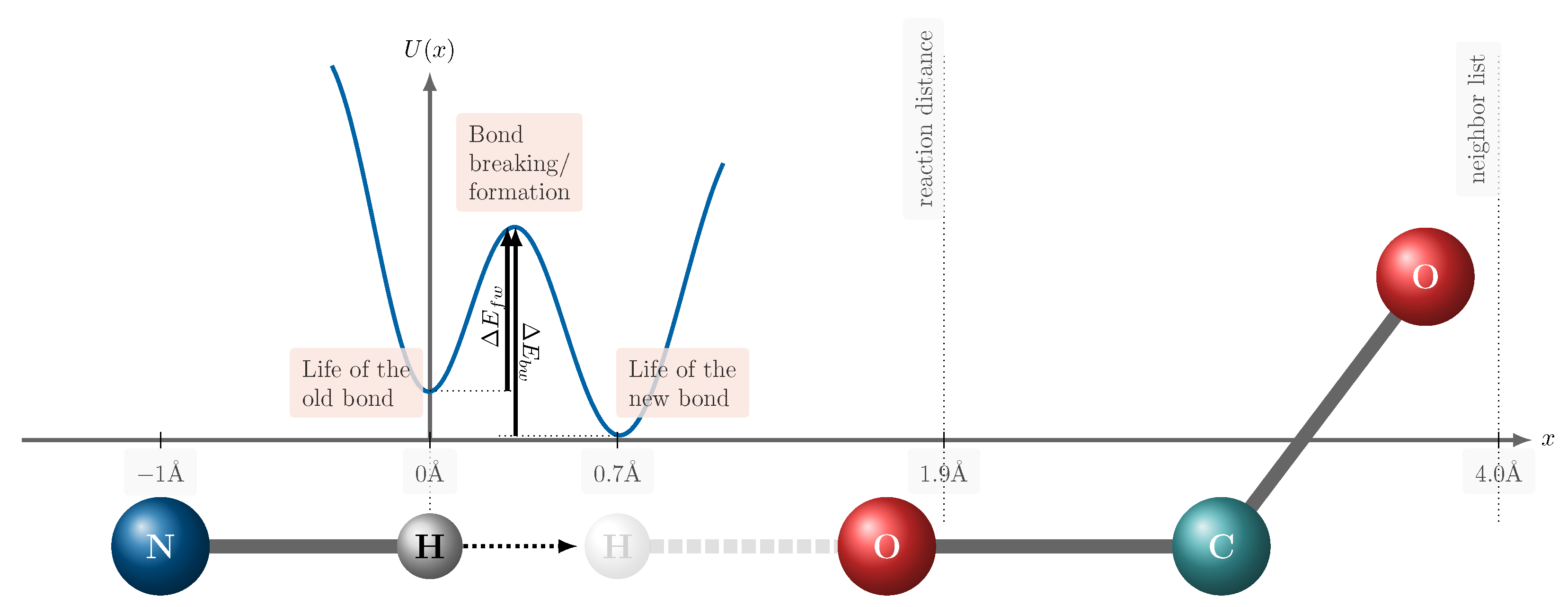
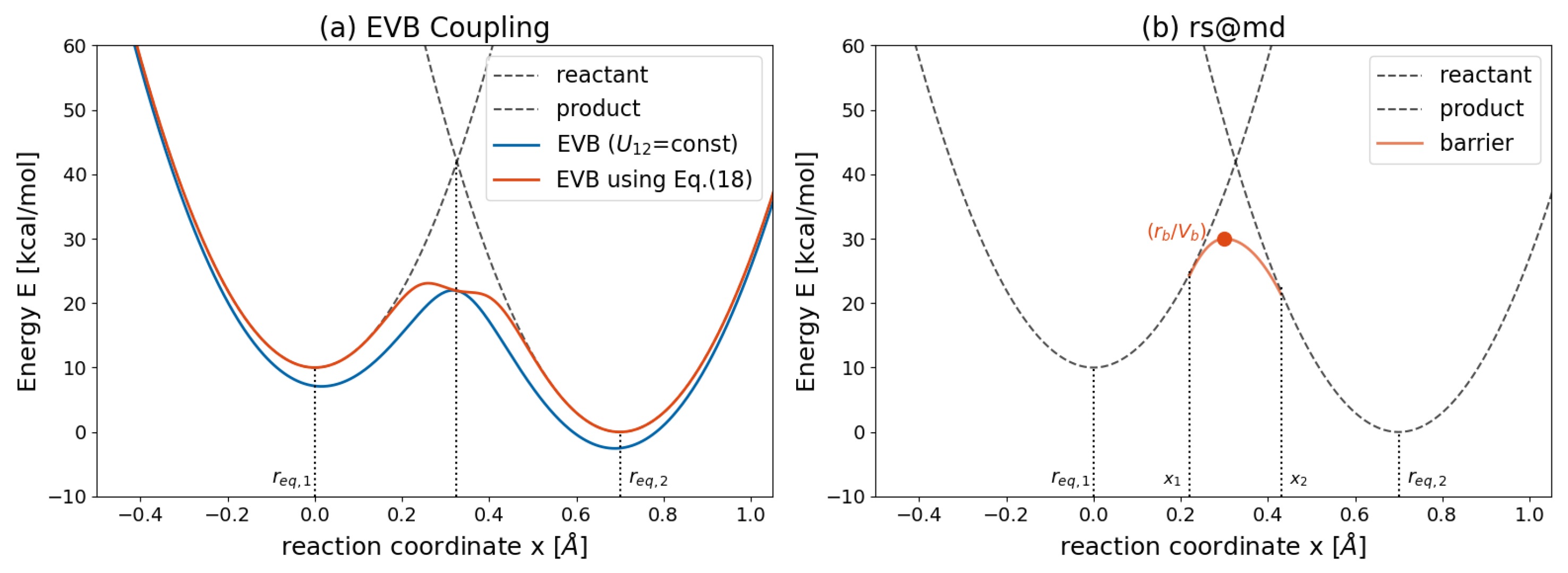
3.1. Life of the Old Bond
3.2. Bond Breaking and Bond Formation
3.3. Life of the New Bond
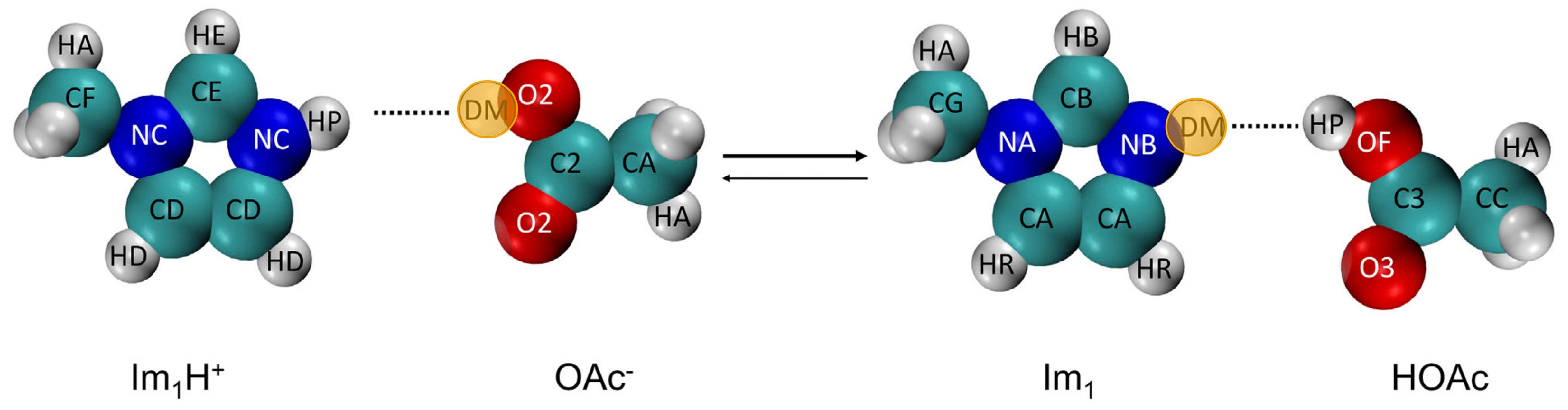
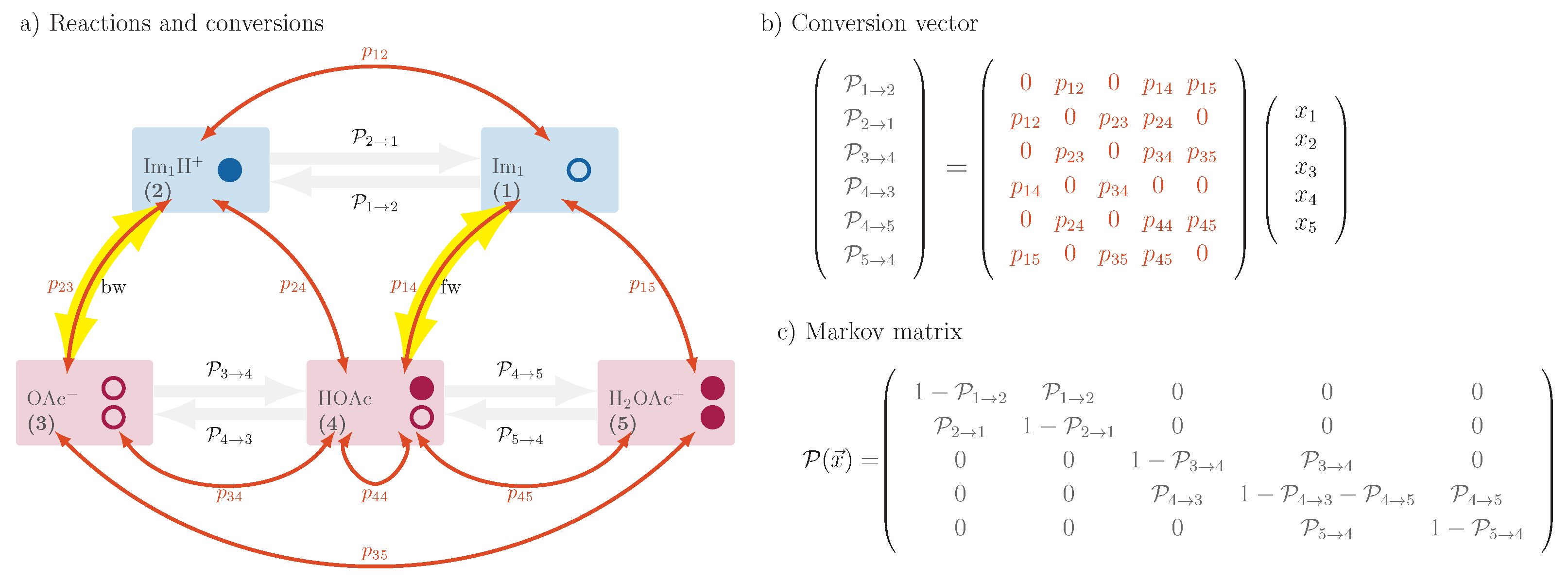
- The reaction with another imidazolium (2) with the probability ;
- The reaction with acetic acid HOAc (4) with probability ;
- The reaction with protonated acetic acid H2OAc+ (5) with probability .
4. Current Limitations in Reactive Molecular Dynamics
5. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reichardt, C.; Welton, T. Solvents and Solvent Effects in Organic Chemistry, 4th ed.; Wiley-VCH: Weinheim, Germany, 2011. [Google Scholar]
- Hallett, J.P.; Welton, T. Room-Temperature Ionic Liquids: Solvents for Synthesis and Catalysis. 2. Chem. Rev. 2011, 111, 3508–3576. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, C.D.; Illner, P.; van Eldik, R. Understanding Chemical Reaction Mechanisms in Ionic Liquids: Successes and Challenges. Chem. Soc. Rev. 2011, 40, 272–290. [Google Scholar] [CrossRef]
- Yoshizawa, M.; Xu, W.; Angell, C.A. Ionic Liquids by Proton Transfer: Vapor Pressure, Conductivity, and the Relevance of ΔpKa from Aqueous Solutions. J. Am. Chem. Soc. 2003, 125, 15411–15419. [Google Scholar] [CrossRef] [PubMed]
- Belieres, J.P.; Angell, C.A. Protic Ionic Liquids: Preparation, Characterization, and Proton Free Energy Level Representation. J. Phys. Chem. B 2007, 111, 4926–4937. [Google Scholar] [CrossRef] [PubMed]
- Greaves, T.L.; Drummond, C.J. Protic Ionic Liquids: Properties and Applications. Chem. Rev. 2008, 108, 206–237. [Google Scholar] [CrossRef]
- Bailey, J.; Byrne, E.L.; Goodrich, P.; Kavanagh, P.; Swadźba-Kwaśny, M. Protic ionic liquids for sustainable uses. Green Chem. 2024, 26, 1092–1131. [Google Scholar] [CrossRef]
- Fumino, K.; Wulf, A.; Ludwig, R. Hydrogen Bonding in Protic Ionic Liquids: Reminiscent of Water. Angew. Chem. Int. Ed. 2009, 48, 3184–3186. [Google Scholar] [CrossRef]
- Fumino, K.; Fossog, V.; Stange, P.; Paschek, D.; Hempelmann, R.; Ludwig, R. Controlling the Subtle Energy Balance in Protic Ionic Liquids: Dispersion Forces Compete with Hydrogen Bonds. Angew. Chem. Int. Ed. 2015, 54, 2792–2795. [Google Scholar] [CrossRef]
- Doi, H.; Song, X.; Minofar, B.; Kanzaki, R.; Takamuku, T.; Umebayashi, Y. A New Proton Conductive Liquid with No Ions: Pseudo-Protic Ionic Liquids. Chem. Eur. J. 2013, 19, 11522–11526. [Google Scholar] [CrossRef]
- Stoimenovski, J.; Izgorodina, E.I.; MacFarlane, D.R. Ionicity and proton transfer in protic ionic liquids. Phys. Chem. Chem. Phys. 2010, 12, 10341–10347. [Google Scholar] [CrossRef]
- MacFarlane, D.R.; Pringle, J.M.; Johansson, K.M.; Forsyth, S.A.; Forsyth, M. Lewis base ionic liquids. Chem. Commun. 2006, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.Y.; Huang, Y.R.; Wang, S.Z.; Bai, B.F. Preparation and Physicochemical Properties of Imidazolium Acetates and the Conductivities of Their Aqueous and Ethanol Solutions. Acta Phys. Chim. Sin. 2011, 27, 2512–2520. [Google Scholar]
- Chen, J.; Chen, L.; Lu, Y.; Xu, Y. Physicochemical properties of aqueous solution of 1-methylimidazolium acetate ionic liquid at several temperatures. J. Mol. Liq. 2014, 197, 374–380. [Google Scholar] [CrossRef]
- Thawarkar, S.; Khupse, N.D.; Shinde, D.R.; Kumar, A. Understanding the behavior of mixtures of protic-aprotic and protic-protic ionic liquids: Conductivity, viscosity, diffusion coefficient and ionicity. J. Mol. Liq. 2019, 276, 986–994. [Google Scholar] [CrossRef]
- Joerg, F.; Schröder, C. Polarizable molecular dynamics simulations on the conductivity of pure 1-methylimidazolium acetate systems. Phys. Chem. Chem. Phys. 2022, 24, 15245. [Google Scholar] [CrossRef]
- Joerg, F.; Sutter, J.; van Dam, L.; Kanellopoulos, K.; Hunger, J.; Schröder, C. Comparative analysis of dielectric spectra in protic ionic liquids: Experimental findings and computational molecular decomposition. J. Mol. Liq. 2024, 396, 123834. [Google Scholar] [CrossRef]
- Overbeck, V.; Ludwig, R. NMR Studies of Protic Ionic Liquids. In Annual Reports on NMR Spectroscopy; Elsevier: Amsterdam, The Netherlands, 2018; Volume 95, pp. 147–190. [Google Scholar]
- Chen, K.; Wang, Y.; Yao, J.; Li, H. Equilibrium in Protic Ionic Liquids: The Degree of Proton Transfer and Thermodynamic Properties. J. Phys. Chem. B 2018, 122, 309–315. [Google Scholar] [CrossRef]
- Hasani, M.; Amin, S.A.; Yarger, J.L.; Davidowski, S.K.; Angell, C.A. Proton Transfer and Ionicity: An 15N NMR Study of Pyridine Base Protonation. J. Phys. Chem. B 2019, 123, 1815–1821. [Google Scholar] [CrossRef]
- Calandra, P.; Caputo, P.; Rossi, C.O.; Kozak, M.; Taube, M.; Pochylski, M.; Gapinski, J. From proton transfer to ionic liquids: How isolated domains of ion pairs grow to form extended network in diphenyl phosphate-bis(2-ethylhexyl) amine mixtures. J. Mol. Liq. 2024, 399, 124402. [Google Scholar] [CrossRef]
- Hoarfrost, M.L.; Tyagi, M.; Segalman, R.A.; Reimer, J.A. Proton Hopping and Long-Range Transport in the Protic Ionic Liquid [Im][TFSI], Probed by Pulsed-Field Gradient NMR and Quasi-Elastic Neutron Scattering. J. Phys. Chem. B 2012, 116, 8201–8209. [Google Scholar] [CrossRef]
- Kobrak, M.N.; Nykypanchuk, D.; Jain, A.; Louz, E.; Jarzecki, A.A. Proton Transfer Equilibrium in Pseudoprotic Ionic Liquids: Inferences on Ionic Populations. J. Phys. Chem. B 2025, 129, 1376–1386. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Arai, N.; Han, J.; Kawana, Y.; Tsuzuki, S.; Umebayashi, Y. Tools for studying ion solvation and ion pair formation in ionic liquids: Isotopic substitution Raman spectroscopy. Anal. Sci. 2022, 38, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Cao, B.; Zhou, X.; Liu, S.; Zhu, X.; Fu, H. Theoretical and experimental studies on proton transfer in acetate-based protic ionic liquids. J. Mol. Liq. 2016, 221, 254–261. [Google Scholar] [CrossRef]
- Mann, S.K.; Brown, S.P.; MacFarlane, D.R. Structure Effects on the Ionicity of Protic Ionic Liquids. ChemPhysChem 2020, 21, 1444–1454. [Google Scholar] [CrossRef]
- Kanzaki, R.; Uchida, K.; Song, X.; Umebayashi, Y.; Ishiguro, S.I. Acidity and Basicity of Aqueous Mixtures of a Protic Ionic Liquid, Ethylammonium Nitrate. Anal. Sci. 2008, 24, 1347–1349. [Google Scholar] [CrossRef]
- Kanzaki, R.; Song, X.; Umebayashi, Y.; Ishiguro, S. Thermodynamic Study of the Solvation States of Acid and Base in a Protic Ionic Liquid, Ethylammonium Nitrate, and Its Aqueous Mixtures. Chem. Lett. 2010, 39, 578–579. [Google Scholar] [CrossRef]
- Song, X.; Kanzaki, R.; Ishiguro, S.; Umebayashi, Y. Physicochemical and Acid-base Properties of a Series of 2-Hydroxyethylammonium-based Protic Ionic Liquids. Anal. Sci. 2012, 28, 469–474. [Google Scholar] [CrossRef]
- Qian, W.; Xu, Y.; Zhu, H.; Yu, C. Properties of pure 1-methylimidazolium acetate ionic liquid and its binary mixtures with alcohols. J. Chem. Thermodyn. 2012, 49, 87–94. [Google Scholar] [CrossRef]
- Ingenmey, J.; Gehrke, S.; Kirchner, B. How to Harvest Grotthuss Diffusion in Protic Ionic Liquid Electrolyte Systems. ChemSusChem 2018, 11, 1900–1910. [Google Scholar] [CrossRef]
- Marx, D. Proton Transfer 200 Years after von Grotthuss: Insights from Ab Initio Simulations. ChemPhysChem 2006, 7, 1848–1870. [Google Scholar] [CrossRef]
- Hayes, R.; Warr, G.G.; Atkin, R. Structure and Nanostructure in Ionic Liquids. Chem. Rev. 2015, 115, 6357–6426. [Google Scholar] [PubMed]
- Anouti, M.; Jacquemin, J.; Porion, P. Transport Properties Investigation of Aqueous Protic Ionic Liquid Solutions through Conductivity, Viscosity, and NMR Self-Diffusion Measurements. J. Phys. Chem. B 2012, 116, 4228–4238. [Google Scholar] [CrossRef] [PubMed]
- Bodo, E. Perspectives in the Computational Modeling of New Generation, Biocompatible Ionic Liquids. J. Phys. Chem. B 2022, 126, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, R.; Kodamatani, H.; Tomiyasu, T.; Watanabe, H.; Umebayashi, Y. A pH Scale for the Protic Ionic Liquid Ethylammonium Nitrate. Angew. Chem. Int. Ed. 2016, 55, 6266–6269. [Google Scholar] [CrossRef]
- Abbott, A.P.; Alabdullah, S.S.M.; Al-Murshedia, A.Y.M.; Ryder, K.S. Brønsted acidity in deep eutectic solvents and ionic liquids. Faraday Discuss. 2018, 206, 365–377. [Google Scholar] [CrossRef]
- Wang, C.; Luo, X.; Zhu, X.; Cui, G.; Jiang, D.E.; Deng, D.; Li, H.; Dai, S. The strategies for improving carbon dioxide chemisorption by functionalized ionic liquids. RSC Adv. 2013, 3, 15518–15527. [Google Scholar] [CrossRef]
- Sheridan, Q.R.; Schneider, W.F.; Maginn, E.J. Role of Molecular Modeling in the Development of CO2-Reactive Ionic Liquids. Chem. Rev. 2018, 118, 5242–5260. [Google Scholar] [CrossRef]
- Elmobarak, W.F.; Almomani, F.; Tawalbeh, M.; Al-Othman, A.; Martis, R.; Rasool, K. Current status of CO2 capture with ionic liquids: Development and progress. Fuel 2023, 344, 128102. [Google Scholar] [CrossRef]
- Bates, E.D.; Mayton, R.D.; Ntai, I.; Davis, J. CO2 capture by a task-specific ionic liquid. J. Am. Chem. Soc. 2002, 124, 926–927. [Google Scholar] [CrossRef]
- Gurkan, B.; Goodrich, B.F.; Mindrup, E.M.; Ficke, L.E.; Massel, M.; Seo, S.; Senftle, T.P.; Wu, H.; Glaser, M.F.; Shah, J.K.; et al. Molecular Design of High Capacity, Low Viscosity, Chemically Tunable Ionic Liquids for CO2 Capture. J. Phys. Chem. Lett. 2010, 1, 3494–3499. [Google Scholar] [CrossRef]
- Lepre, L.F.; Szala-Bilnik, J.; Pison, L.; Traïkia, M.; Padua, A.A.H.; Ando, R.A.; Costa Gomes, M.F. Can the tricyanomethanide anion improve CO2 absorption by acetate-based ionic liquids? Phys. Chem. Chem. Phys. 2017, 19, 12431–12440. [Google Scholar] [CrossRef] [PubMed]
- Gurau, G.; Rodríguez, H.; Kelley, S.P.; Janiczek, P.; Kalb, R.S.; Rogers, R.D. Demonstration of Chemisorption of Carbon Dioxide in 1,3-Dialkylimidazolium Acetate Ionic Liquids. Angew. Chem. Int. Ed. 2011, 50, 12024–12026. [Google Scholar] [CrossRef] [PubMed]
- Blath, J.; Deubler, N.; Hirth, T.; Schiestel, T. Chemisorption of carbon dioxide in imidazolium based ionic liquids with carboxylic anions. Chem. Eng. J. 2012, 181–182, 152–158. [Google Scholar] [CrossRef]
- Karadas, F.; Köz, B.; Jacquemin, J.; Deniz, E.; Rooney, D.; Thompson, J.; Yavuz, C.T.; Khraisheh, M.; Aparicio, S.; Atihan, M. High-pressure CO2 absorption studies on imidazolium-based ionic liquids: Experimental and simulation approaches. Fluid Phase Equil. 2013, 351, 74–86. [Google Scholar] [CrossRef]
- Cao, B.; Yan, W.; Wang, J.; Ding, H.; Yu, Y. Absorption of CO2 with supported imidazolium-based ionic liquid membranes. J. Chem. Technol. Biotechnol. 2015, 90, 1537–1544. [Google Scholar] [CrossRef]
- Wang, C.; Luo, H.; Luo, X.; Li, H.; Dai, S. Equimolar CO2 capture by imidazolium-based ionic liquids and superbase systems. Green Chem. 2010, 12, 2019–2023. [Google Scholar] [CrossRef]
- Peng, Y.; Szeto, K.C.; Santini, C.; Daniele, S. Study of the Parameters Impacting the Photocatalytic Reduction of Carbon Dioxide in Ionic Liquids. ChemPhotoChem 2021, 5, 721–726. [Google Scholar] [CrossRef]
- Peng, Y.; Szeto, K.C.; Santini, C.; Daniele, S. Ionic Liquids as homogeneous photocatalyst for CO2 reduction in protic solvents. Chem. Eng. J. Adv. 2022, 12, 100379. [Google Scholar] [CrossRef]
- Suo, X.; Fu, Y.; Do-Thanh, C.L.; Qiu, L.Q.; Jiang, D.; Mahurin, S.M.; Yang, Z.; Dai, S. CO2 Chemisorption Behavior in Conjugated Carbanion-Derived Ionic Liquids via Carboxylic Acid Formation. J. Am. Chem. Soc. 2022, 144, 21658–21663. [Google Scholar] [CrossRef]
- Arduengo, A.J., III; Harlow, R.L.; Kline, M. A Stable Crystalline Carbene. J. Am. Chem. Soc. 1991, 113, 361–363. [Google Scholar] [CrossRef]
- Cabacço, M.I.; Besnard, M.; Danten, Y.; Coutinho, J.A.P. Carbon Dioxide in 1-Butyl-3-methylimidazolium Acetate. I. UnusualSolubility Investigated by Raman Spectroscopy and DFT Calculations. J. Phys. Chem. A 2012, 116, 1605–1620. [Google Scholar] [CrossRef] [PubMed]
- Canongia Lopes, J.N.A.; Pádua, A.A.H. Nanostructural Organization in Ionic Liquids. J. Phys. Chem. B 2006, 110, 3330–3335. [Google Scholar] [CrossRef] [PubMed]
- Farah, K.; Müller-Plathe, F.; Böhm, M.C. Classical Reactive Molecular Dynamics Implementations: State of the Art. ChemPhysChem 2012, 13, 1127–1151. [Google Scholar] [CrossRef]
- Bedrov, D.; Piquemal, J.P.; Borodin, O.; MacKerell, A.; Roux, B.; Schröder, C. Molecular Dynamics Simulations of Ionic Liquids and Electrolytes Using Polarizable Force Fields. Chem. Rev. 2019, 119, 7940–7995. [Google Scholar] [CrossRef]
- van Gunsteren, W.F.; Oostenbrink, C. Methods for Classical-Mechanical Molecular Simulation in Chemistry: Achievements, Limitations, Perspectives. J. Chem. Inf. Model. 2024, 64, 6281–6304. [Google Scholar] [CrossRef]
- Hunt, P.A. The simulation of imidazolium-based ionic liquids. Mol. Sim. 2006, 32, 1–10. [Google Scholar] [CrossRef]
- Maginn, E.J. Molecular simulation of ionic liquids: Current status and future opportunities. J. Phys. Condens. Matter 2009, 21, 373101. [Google Scholar] [CrossRef]
- Liu, T.; Panwar, P.; Khajeh, A.; Rahman, M.H.; Menezes, P.L.; Martini, A. Review of Molecular Dynamics Simulations of Phosphonium Ionic Liquid Lubricants. Tribol. Lett. 2022, 70, 44. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Dotson, D.L.; Domanski, J.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A python package for the rapid analysis of molecular dynamics simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; Benthall, S., Rostrup, S., Eds.; Los Alamos National Laboratory: Los Alamos, NM, USA, 2016. [Google Scholar]
- McCann, B.W.; Acevedo, O. Pairwise Alternatives to Ewald Summation for Calculating Long-Range Electrostatics in Ionic Liquids. J. Chem. Theory Comput. 2013, 9, 944–950. [Google Scholar] [CrossRef]
- Hünenberger, P.H.; McCammon, J.A. Ewald artifacts in computer simulations of ionic solvation and ion–ion interaction: A continuum electrostatics study. J. Chem. Phys. 1999, 110, 1856–1872. [Google Scholar] [CrossRef]
- Wells, B.A.; Chaffee, A.L. Ewald Summation for Molecular Simulations. J. Chem. Theory Comput. 2015, 11, 3684–3695. [Google Scholar] [CrossRef] [PubMed]
- Hammonds, K.D.; Heyes, D.M. Optimization of the Ewald method for calculating Coulomb interactions in molecular simulations. J. Chem. Phys. 2022, 157, 074108. [Google Scholar] [CrossRef] [PubMed]
- Aguado, A.; Madden, P.A. Ewald summation of electrostatic multipole interactions up to the quadrupolar level. J. Chem. Phys. 2003, 119, 7471–7483. [Google Scholar] [CrossRef]
- Lamichhane, M.; Newman, K.E.; Gezelter, J.D. Real space electrostatics for multipoles. II. Comparisons with the Ewald sum. J. Chem. Phys. 2014, 141, 134110. [Google Scholar] [CrossRef]
- Hanke, C.G.; Price, S.L.; Lynden-Bell, R.M. termolecular potentials for simulations of liquid imidazolium salts. Mol. Phys. 2001, 99, 801–809. [Google Scholar] [CrossRef]
- Del Pópolo, M.G.; Kohanoff, J.; Lynden-Bell, R.M. Solvation structure and transport of acidic protons in ionic liquids: A first-principles simulation study. J. Phys. Chem. B 2006, 110, 8798–8803. [Google Scholar] [CrossRef]
- Canongia Lopes, J.N.; Deschamps, J.; Pádua, A.A.H. Modeling Ionic Liquids Using a Systematic All-Atom Force Field. J. Phys. Chem. B 2004, 108, 2038–2047. [Google Scholar] [CrossRef]
- Schröder, C. Comparing reduced partial charge models with polarizable simulations of ionic liquids. Phys. Chem. Chem. Phys. 2012, 14, 3089–3102. [Google Scholar] [CrossRef]
- Morrow, T.I.; Maginn, E.J. Molecular Dynamics Study of the Ionic Liquid 1-n-Butyl-3-methylimidazolium Hexafluorophosphat. J. Phys. Chem. 2002, 106, 12807–12813. [Google Scholar] [CrossRef]
- Sappl, M.; Gross, S.; Hugger, P.T.; D’Alessio, P.; Palka, M.; Felix Fritz, S.; Spange, S.; Schröder, C. Decomposition, interpretation and prediction of various ionic liquid solvation parameters: Kamlet-Taft, Catalán and Reichardt’s ETN. J. Mol. Liq. 2024, 417, 126646. [Google Scholar] [CrossRef]
- Dommert, F.; Wendler, K.; Berger, R.; Delle Site, L.; Holm, C. Force Fields for Studying the Structure and Dynamics of Ionic Liquids: A Critical Review of Recent Developments. ChemPhysChem 2012, 13, 1625–1637. [Google Scholar] [CrossRef] [PubMed]
- McDaniel, J.G.; Yethiraj, A. The Influence of Electronic Polarization on the Structure of Ionic Liquids. J. Phys. Chem. Lett. 2018, 9, 4765–4770. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Morales, T.; Carrete, J.; Cabeza, Ó; Russina, O.; Triolo, A.; Gallego, L.J.; Varela, L.M. Solvation of Lithium Salts in Protic Ionic Liquids: A Molecular Dynamics Study. J. Phys. Chem. B 2014, 118, 761–770. [Google Scholar] [CrossRef]
- Paschek, D.; Golub, B.; Ludwig, R. Hydrogen bonding in a mixture of protic ionic liquids: A molecular dynamics simulation study. Phys. Chem. Chem. Phys. 2015, 17, 8431–8440. [Google Scholar] [CrossRef]
- Gontrani, L.; Bodo, E.; Triolo, A.; Leonelli, F.; D’Angelo, P.; Migliorati, V.; Caminiti, R. The Interpretation of Diffraction Patterns of Two Prototypical ProticIonic Liquids: A Challenging Task for Classical Molecular DynamicsSimulations. J. Phys. Chem. B 2012, 116, 13024–13032. [Google Scholar] [CrossRef]
- Docampo-Álvarez, B.; Gómez-González, V.; Méndez-Morales, T.; Rodríguez, J.R.; Cabeza, O.; Turmine, M.; Gallegoa, L.J.; Varela, L.M. The effect of alkyl chain length on the structure and thermodynamics of protic–aprotic ionic liquid mixtures: A molecular dynamics study. Phys. Chem. Chem. Phys. 2018, 20, 9939–9949. [Google Scholar] [CrossRef]
- Gómez-González, V.; Docampo-Álvarez, B.; Otero-Mato, J.M.; Cabeza, O.; Gallego, L.J.; Varela, L.M. Molecular dynamics simulations of the structure of mixtures of protic ionic liquids and monovalent and divalent salts at the electrochemical interface. Phys. Chem. Chem. Phys. 2018, 20, 12767–12776. [Google Scholar] [CrossRef]
- Borodin, O. Polarizable Force Field Development and Molecular Dynamics Simulations of Ionic Liquids. J. Phys. Chem. B 2009, 113, 11463–11478. [Google Scholar] [CrossRef]
- Goloviznina, K.; Gong, Z.; Costa Gomes, M.F.; Pádua, A.A.H. Extension of the CL&Pol Polarizable Force Field to Electrolytes, Protic Ionic Liquids, and Deep Eutectic Solvents. J. Chem. Theory Comput. 2021, 17, 1606–1617. [Google Scholar]
- Goloviznina, K.; Gong, Z.; Pádua, A.A.H. The CL&Pol polarizable force field for the simulation of ionic liquids and eutectic solvents. WIREs Comput. Mol. Sci. 2021, 12, e1572. [Google Scholar]
- Nasrabadi, A.T.; Gelb, L.D. How proton tranfer equilibria influence ionic liquid properties: Molecular simulations of alkylammonium acetates. J. Phys. Chem. B 2018, 122, 5961–5971. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, R.; Joerg, F.; Schröder, C. Emulating proton transfer reactions in the pseudo-protic ionic liquid 1-methylimidazolium acetate. Phys. Chem. Chem. Phys. 2022, 24, 9277–9285. [Google Scholar] [CrossRef] [PubMed]
- Ponder, J.W.; Wu, C.; Ren, P.; Pande, V.S.; Chodera, J.D.; Schnieders, M.J.; Haque, I.; Mobley, D.L.; Lambrecht, D.S.; DiStasio, R.A., Jr.; et al. Current Status of the AMOEBA Polarizable Force Field. J. Phys. Chem. B 2010, 114, 2549–2564. [Google Scholar] [CrossRef]
- Starovoytov, O.N.; Torabifard, H.; Cisneros, G.A. Development of AMOEBA Force Field for 1,3-Dimethylimidazolium Based Ionic Liquids. J. Phys. Chem. B 2014, 118, 7156–7166. [Google Scholar] [CrossRef]
- Vázquez-Montelongo, E.A.; Vázquez-Cervantes, J.E.; Cisneros, G.A. Current Status of AMOEBA–IL: A Multipolar/Polarizable Force Field for Ionic Liquids. Int. J. Mol. Sci. 2020, 21, 697. [Google Scholar] [CrossRef]
- Szabadi, A.; Schröder, C. Recent developments in polarizable molecular dynamics simulations of electrolyte solutions. J. Comput. Biophys. Chem. 2021, 21, 415–429. [Google Scholar] [CrossRef]
- Bernardes, C.E.S.; Shimizu, K.; Lopes, J.N.C.; Marquetand, P.; Heid, E.; Steinhauser, O.; Schröder, C. Additive polarizabilities in ionic liquids. Phys. Chem. Chem. Phys. 2016, 18, 1665–1670. [Google Scholar] [CrossRef]
- Philippi, F.; Quinten, A.; Rauber, D.; Springborg, M.; Hempelmann, R. Density Functional Theory Descriptors for Ionic Liquids and theIntroduction of a Coulomb Correction. J. Phys. Chem. A 2019, 123, 4188–4200. [Google Scholar] [CrossRef]
- Philippi, F.; Goloviznina, K.; Gong, Z.; Gehrke, S.; Kirchner, B.; Pádua, A.A.H.; Hunt, P.A. Charge transfer and polarisability in ionic liquids: A case study. Phys. Chem. Chem. Phys. 2022, 24, 3144–3162. [Google Scholar] [CrossRef]
- Olano, L.R.; Rick, S.W. Fluctuating Charge Normal Modes: An Algorithm forImplementing Molecular Dynamics Simulations withPolarizable Potentials. J. Comput. Chem. 2005, 26, 699–707. [Google Scholar] [CrossRef]
- Patel, S.; Brooks, C.L., III. Fluctuating Charge Force Fields: Recent Developments and Applications From Small Molecules to Macromolecular Biological Systems. Mol. Simul. 2006, 32, 231–249. [Google Scholar] [CrossRef]
- Wendler, K.; Dommert, F.; Zhao, Y.Y.; Berger, R.; Holm, C.; Delle Site, L. Ionic liquids studied across different scales: A computational perspective. Faraday Discuss. 2012, 154, 111–132. [Google Scholar] [CrossRef] [PubMed]
- Schröder, C.; Lyons, A.; Rick, S.W. Polarizable MD simulations of ionic liquids: How does additional charge transfer change the dynamics. Phys. Chem. Chem. Phys. 2020, 22, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Heinz, H.; Vaia, R.A.; Koerner, H.; Farmer, B.L. Photoisomerization of azobenzene grafted to layered silicates: Simulation and experimental challenges. Chem. Mater. 2008, 20, 6444–6456. [Google Scholar] [CrossRef]
- Tian, Z.; Wen, J.; Ma, J. Dynamic simulations of stimuli-responsive swithcing of azobenzene derivatives in self-assembled monolayers: Reactive rotation potential and switching functions. Mol. Simul. 2015, 41, 28–42. [Google Scholar] [CrossRef]
- Del Pópolo, M.G.; Lynden-Bell, R.M.; Kohanoff, J. Ab Initio Molecular Dynamics Simulation of a Room Temperature Ionic Liquid. J. Phys. Chem. B 2005, 109, 5895–5902. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous Electron Gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Le Donne, A.; Adenusi, H.; Procelli, F.; Bodo, E. Structural Features of Cholinium Based Protic Ionic Liquids through Molecular Dynamics. J. Phys. Chem. B 2019, 123, 5568–5576. [Google Scholar] [CrossRef]
- Kirchner, B.; Seitsonen, A.P. Ionic Liquids from Car-Parrinello Simulations. 2. Structural Diffusion Leading to Large Anions in Chloraluminate Ionic Liquids. Inorg. Chem. 2007, 46, 2751–2754. [Google Scholar] [CrossRef] [PubMed]
- Salanne, M.; Siqueira, L.J.A.; Seitsonen, A.P.; Madden, P.A.; Kirchner, B. From molten salts to room temperature ionic liquids: Simulation studies on chloroaluminate systems. Faraday Discuss. 2012, 154, 171–188. [Google Scholar] [CrossRef] [PubMed]
- Campetella, M.; Montagna, M.; Gontrani, L.; Bodo, E. Unexpected proton mobility in the bulk phase of cholinium-based ionic liquids: New insights from theoretical calculations. Phys. Chem. Chem. Phys. 2017, 19, 11869. [Google Scholar] [CrossRef]
- Adenusi, H.; Le Donne, A.; Porcelli, F.; Bodo, E. Ab Initio Molecular Dynamics Study of Phospho-Amino Acid-Based Ionic Liquids: Formation of Zwitterionic Anions in the Presence of Acidic Side Chains. J. Phys. Chem. B 2020, 124, 1955–1964. [Google Scholar] [CrossRef]
- Brehm, M.; Weber, H.; Pensado, A.S.; Stark, A.; Kirchner, B. Proton transfer and polarity changes in ionic liquid–water mixtures: A perspective on hydrogen bonds from ab initio molecular dynamics at the example of 1-ethyl-3-methylimidazolium acetate–water mixtures: Part 1. Phys. Chem. Chem. Phys. 2012, 14, 5030–5044. [Google Scholar] [CrossRef]
- Hollóczki, O.; Gerhard, D.; Massone, K.; Szarvas, L.; Németh, B.; Veszprémi, T.; Nyulászi, L. Carbenes in ionic liquids. New J. Chem. 2010, 34, 3004–3009. [Google Scholar] [CrossRef]
- Thomas, M.; Brehm, M.; Hollóczki, O.; Kirchner, B. How Can a Carbene be Active in an Ionic Liquid? Chem. Eur. J. 2014, 20, 1622–1629. [Google Scholar] [CrossRef]
- Hollóczki, O.; Firaha, D.S.; Friedrich, J.; Brehm, M.; Cybik, R.; Wild, M.; Stark, A.; Kirchner, B. Carbene Formation in Ionic Liquids: Spontaneous, Induced, or Prohibited? J. Phys. Chem. B 2013, 117, 5898–5907. [Google Scholar] [CrossRef]
- Gabl, S.; Schröder, C.; Steinhauser, O. Computational studies of ionic liquids: Size does matter and time too. J. Chem. Phys. 2012, 137, 094501. [Google Scholar] [CrossRef]
- Gray-Weale, A. Correlations in the Structure and Dynamics of Ionic Liquids. Aust. J. Chem. 2009, 62, 288–297. [Google Scholar] [CrossRef]
- Bowron, D.T.; D’Agostino, C.; Gladden, L.F.; Hardacre, C.; Holbrey, J.D.; Lagunas, M.C.; McGregor, J.; Mantle, M.D.; Mullan, C.L.; Youngs, T.G.A. Structure and Dynamics of 1-Ethyl-3-methylimidazolium Acetate via Molecular Dynamicsand Neutron Diffraction. J. Phys. Chem. B 2010, 114, 7760–7768. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.L.; Corchado, J.C.; Martín, M.E.; Galván, I.F.; Barata-Morgado, R.; Aguilar, M.A. A New QM/MM Method Oriented to the Study of Ionic Liquids. J. Comput. Chem. 2015, 36, 1893–1901. [Google Scholar] [CrossRef] [PubMed]
- Seduraman, A.; Klähn, M.; Wu, P. Characterization of nano-domains in ionic liquids with molecular simulations. Calphad 2009, 33, 605–613. [Google Scholar] [CrossRef]
- Chiappe, C.; Pomelli, C.S. Computational Studies on Organic Reactivity in Ionic Liquids. Phys. Chem. Chem. Phys. 2013, 15, 412–423. [Google Scholar] [CrossRef]
- Acevedo, O. Simulating chemical reactions in ionic liquids using QM/MM methodology. J. Phys. Chem. A 2014, 118, 11653–11666. [Google Scholar] [CrossRef]
- Vázquez-Montelongo, E.A.; Vázquez-Cervantes, J.E.; Cisneros, G.A. Polarizable ab initio QM/MM Study of the Reaction Mechanism of N-tert-Butyloxycarbonylation of Aniline in [EMIm][BF4]. Molecules 2018, 23, 2830. [Google Scholar] [CrossRef]
- Bakowies, D.; Thiel, W. Hybrid Models for Combined Quantum Mechanical and Molecular Mechanical Approaches. J. Phys. Chem. 1996, 100, 10580–10594. [Google Scholar] [CrossRef]
- Zhang, Y.; Lin, H. Flexible-boundary QM/MM calculations: II. Partial charge transfer across the QM/MM boundary that passes through a covalent bond. Theor. Chem. Acc. 2010, 126, 315–322. [Google Scholar] [CrossRef]
- Shen, Z.; Glover, W.J. Flexible boundary layer using exchange for embedding theories. I. Theory and implementation. J. Chem. Phys. 2021, 155, 224112. [Google Scholar] [CrossRef]
- Vreven, T.; Morokuma, K. Chapter 3: Hybrid methods: ONIOM (QM:MM) and QM/MM. Annu. Rep. Comput. Chem. 2006, 2, 35–51. [Google Scholar]
- Acevedo, O.; Jorgensen, W.L. Advances in Quantum and Molecular Mechanical (QM/MM) Simulations for Organic and Enzymatic Reactions. Acc. Chem. Res. 2010, 43, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, O.; Jorgensen, W.L. Quantum and Molecular Mechanical (QM/MM) Monte Carlo Techniques for Modeling Condensed-Phase Reactions. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 422–435. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; McCann, B.W.; Acevedo, O. Ionic Liquid Effects on Nucleophilic Aromatic Substitution Reactions from QM/MM Simulations. J. Phys. Chem. B 2015, 119, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Sambasivarao, S.V.; Acevedo, O. An Ionic Liquid Dependent Mechanism for Base Catalyzed B-Elimination Reactions from QM/MM Simulations. J. Am. Chem. Soc. 2013, 135, 1065–1072. [Google Scholar] [CrossRef]
- Acevedo, O.; Jorgensen, W.L.; Evanseck, J.D. Elucidation of Rate Variations for a Diels-Alder Reaction in Ionic Liquids from QM/MM Simulations. J. Chem. Theory Comput. 2007, 3, 132–138. [Google Scholar] [CrossRef]
- Chen, Y.; Roux, B. Constant-pH Hybrid Nonequilibrium Molecular Dynamics-Monte Carlo Simulation Method. J. Chem. Theory Comput. 2015, 11, 3919–3931. [Google Scholar] [CrossRef]
- Warshel, A.; Weiss, R.M. An Empirical Valence Bond Approach for Comparing Reactions in Solutions and in Enzymes. J. Am. Chem. Soc. 1980, 102, 6218–6226. [Google Scholar] [CrossRef]
- Jensen, F.; Norrby, P.O. Transition states from empirical force fields. Theor. Chem. Acc. 2003, 109, 1–7. [Google Scholar]
- Chang, Y.-T.; Minichino, C.; Miller, W.H. Classical trajectory studies of the molecular dissociation dynamics of formaldehyde: H2CO → H2 + CO. J. Chem. Phys. 1992, 96, 4341–4355. [Google Scholar] [CrossRef]
- Kim, Y.; Corchado, J.C.; Villa, J.; Xing, J.; Truhlar, D.G. Multiconfiguration molecular mechanics algorithm for potential energy surfaces of chemical reactions. J. Chem. Phys. 2000, 112, 2718–2735. [Google Scholar] [CrossRef]
- Sonnenberg, J.L.; Wong, K.F.; Voth, G.A.; Schlegel, H.B. Distributed Gaussian Valence Bond Surface Derived from Ab Initio Calculations. J. Chem. Theory Comput. 2009, 5, 949–961. [Google Scholar] [CrossRef] [PubMed]
- Åqvist, J.; Warshel, A. Simulation of Enzyme Reactions Using Valence Bond Force Fields and Other Hybrid Quantum/Classical Approaches. Chem. Rev. 1993, 93, 2523–2544. [Google Scholar] [CrossRef]
- Hinsen, K.; Roux, B. Potential of mean force and reaction rates for proton transfer in acetylacetone. J. Chem. Phys. 1997, 106, 3567–3577. [Google Scholar] [CrossRef]
- Nagaoka, M.; Okuyama-Yoshida, N.; Yamabe, T. Origin of the Transition State on the Free Energy Surface: Intramolecular Proton Transfer Reaction of Glycine in Aqueous Solution. J. Phys. Chem. A 1998, 102, 8202–8208. [Google Scholar] [CrossRef]
- Okuyama-Yoshida, N.; Nagaoka, M.; Yamabe, T. Potential Energy Function for Intramolecular Proton Transfer Reaction of Glycine in Aqueous Solution. J. Phys. Chem. A 1998, 102, 285–292. [Google Scholar] [CrossRef]
- Luzkov, V.B. Empirical valence bond study of radical reactions: Hydrogen atom transfer in peroxidation of phenol. Chem. Phys. Lett. 2001, 345, 345–352. [Google Scholar] [CrossRef]
- Warshel, A. Dynamics of Reactions in Polar Solvents. Semiclassical Trajectory Studies of Electron-Transfer and Proton-Transfer Reactions. J. Phys. Chem. 1982, 86, 2218–2224. [Google Scholar] [CrossRef]
- Vendrell, O.; Moreno, M.; Lluch, J.M.; Hammes-Schiffer, S. Molecular Dynamics of Excited State Intramolecular Proton Transfer: 2-(2’-Hydroxylphenyl)-4-methyloxazole in Gas Phase, Solution, and Protein Environments. J. Phys. Chem. B 2004, 108, 6616–6623. [Google Scholar] [CrossRef]
- Voth, G.A. Computer Simulation of Proton Solvation and Transport in Aqueous and Biomolecular Systems. Acc. Chem. Res. 2006, 39, 143–150. [Google Scholar] [CrossRef]
- Romero, M.; Kushnir, J.S.; Mochi, B.; Velez, C.; Acevedo, O. Monte Carlo QM/MM simulation studies of the Cannizzaro reaction in ionic liquids for improved biofuel production. J. Chem. Phys. 2024, 161, 084117. [Google Scholar] [CrossRef]
- Schmitt, U.W.; Voth, G.A. Multistate Empirical Valence Bond Model for Proton Transport in Water. J. Phys. Chem. B 1998, 102, 5547–5551. [Google Scholar] [CrossRef]
- Schmitt, U.W.; Voth, G.A. The computer simulation of proton transport in water. J. Chem. Phys. 1999, 111, 9361–9381. [Google Scholar] [CrossRef]
- Day, T.J.F.; Soudackov, A.V.; Čuma, M.; Schmitt, U.W.; Voth, G.A. A second generation multistate empirical valence bond model for proton transport in aqueous systems. J. Chem. Phys. 2002, 117, 5839–5849. [Google Scholar] [CrossRef]
- Wu, Y.; Chen, H.; Wang, F.; Paesani, F.; Voth, G.A. An Improved Multistate Empirical Valence Bond Model for Aqueous Proton Solvation and Transport. J. Phys. Chem. B 2008, 112, 467–482. [Google Scholar] [CrossRef]
- Chen, H.; Liu, P.; Voth, G.A. Efficient Multistate Reactive Molecular Dynamics Approach Based on Short-Range Effective Potentials. J. Chem. Theory Comput. 2010, 6, 3039–3047. [Google Scholar] [CrossRef]
- Čuma, M.; Schmitt, U.W.; Voth, G.A. A multi-state empirical valence bond model for acid-base chemistry in aqueous solution. Chem. Phys. 2000, 258, 187–199. [Google Scholar] [CrossRef]
- Patil, A.B.; Bhanage, B.M. Modern ab initio valence bond theory calculations reveal charge shift bonding in protic ionic liquids. Phys. Chem. Chem. Phys. 2016, 18, 15783–15790. [Google Scholar] [CrossRef]
- Patila, A.B.; Bhanage, B.M. Brønsted acidity of protic ionic liquids: A modern ab initio valence bond theory perspective. Phys. Chem. Chem. Phys. 2016, 18, 26020–26025. [Google Scholar] [CrossRef]
- Brenner, D.W.; Shenderova, O.A.; Harrison, J.A.; Stuart, S.J.; Ni, B.; Sinnott, S.B. A second-generation reactive empirical bond order (REBO) potential energy expression for hydrocarbons. J. Phys. Condens. Matter 2002, 14, 783–802. [Google Scholar] [CrossRef]
- Xiao, Y.; Dong, W.; Busnengo, H.F. Reactive force fields for surface chemical reactions: A case study with hydrogen dissociation on Pd surfaces. J. Chem. Phys. 2010, 132, 014704. [Google Scholar] [CrossRef]
- Tersoff, J. New empirical approach for the structure and energy of covalent systems. Phys. Rev. B 1988, 37, 6991–7000. [Google Scholar] [CrossRef] [PubMed]
- Stuart, S.J.; Tutein, A.B.; Harrison, J.A. A reactive potential for hydrocarbons with intermolecular interactions. J. Chem. Phys. 2000, 112, 6472–6486. [Google Scholar] [CrossRef]
- Ilyin, D.V.; Goddard, W.A.; Oppenheim, J.J.; Cheng, T. First-principles–based reaction kinetics from reactive molecular dynamics simulations: Application to hydrogen peroxide decomposition. Proc. Natl. Acad. Sci. USA 2019, 116, 18202–18208. [Google Scholar] [CrossRef]
- Majd Sayed Ahmad, E.M.K.; Giusti, A. A Reactive Molecular Dynamics Investigation of Nanoparticle Interactions in Hydrocarbon Combustion. Combust. Sci. Technol. 2023, 195, 3281–3295. [Google Scholar] [CrossRef]
- Aktulga, H.M.; Pandit, S.A.; van Duin, A.C.T.; Grama, A.Y. Reactive Molecular Dynamics: Numerical Methods and Algorithmic Techniques. SIAM J. Sci. Comput. 2012, 34, C1–C23. [Google Scholar] [CrossRef]
- Brenner, D.W. Empirical potential for hydrocarbons for use in simulating the chemical vapor deposition of diamond films. Phys. Rev. B 1990, 42, 9458–9471. [Google Scholar] [CrossRef]
- Biedermann, M.; Diddens, D.; Heuer, A. rs@md: Introducing Reactive Steps at the Molecular Dynamics Simulation Level. J. Chem. Theory Comput. 2021, 17, 1074–1085. [Google Scholar] [CrossRef]
- Rouha, M.; Cummings, P.T. Thickness-dependent structural arrangement in nano-confined imidazolium-based ionic liquid films. Phys. Chem. Chem. Phys. 2015, 17, 4152–4159. [Google Scholar] [CrossRef]
- Prakash, K.; Sathian, S.P. Temperature-dependent differential capacitance of an ionic liquid-graphene-based supercapacitor. Phys. Chem. Chem. Phys. 2024, 26, 4657–4667. [Google Scholar] [CrossRef]
- Senftle, T.P.; Hong, S.; Islam, M.M.; Kylasa, S.B.; Zheng, Y.; Shin, Y.K.; Junkermeier, C.; Engel-Herbert, R.; Janik, M.J.; Aktulga, H.M.; et al. The ReaxFF reactive force-field: Development, applications and future directions. npj Comput. Mater. 2016, 2, 15011. [Google Scholar] [CrossRef]
- Grimme, S. A General Quantum Mechanically Derived Force Field (QMDFF) for Molecules and Condensed Phase Simulations. J. Chem. Theory Comput. 2014, 10, 4497–4514. [Google Scholar] [CrossRef] [PubMed]
- van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Goddard, W.A., III. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Yang, M.Y.; Zybin, S.V.; Das, T.; Merinov, B.V.; Goddard, W.A., III; Mok, E.K.; Hah, H.J.; Han, H.E.; Choi, Y.C.; Kim, S.H. Characterization of the Solid Electrolyte Interphase at the LiMetal–Ionic Liquid Interface. Adv. Energy Mater. 2023, 13, 2202949. [Google Scholar] [CrossRef]
- Wu, G.; Tang, H.; Ma, X.; Luo, Z.; Chen, W.; Yu, B. Investigation of lubrication mechanism of phosphate ionic liquid by ReaxFF molecular dynamics simulations. J. Mol. Liq. 2024, 401, 124715. [Google Scholar] [CrossRef]
- Zhang, B.; van Duin, A.C.T.; Johnson, J.K. Development of a ReaxFF Reactive Force Field for Tetrabutylphosphonium Glycinate/CO2 Mixtures. J. Phys. Chem. B 2014, 118, 12008–12016. [Google Scholar] [CrossRef]
- Thompson, A.P.; Aktulga, H.M.; Berger, R.; Bolintineanu, D.S.; Brown, W.M.; Crozier, P.S.; in ’t Veld, P.J.; Kohlmeyer, A.; Moore, S.G.; Nguyen, T.D.; et al. LAMMPS—A flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales. Comput. Phys. Commun. 2022, 271, 108–171. [Google Scholar] [CrossRef]
- Winetrout, J.J.; Kanhaiya, K.; Kemppainen, J.; in ‘t Veld, P.J.; Sachdeva, G.; Pandey, R.; Damirchi, B.; van Duin, A.; Odegard, G.M.; Heinz, H. Implementing reactivity in molecular dynamics simulations with harmonic force fields. Nat. Commun. 2024, 15, 7945. [Google Scholar] [CrossRef]
- Joerg, F.; Wieder, M.; Schröder, C. Protex—A Python utility for proton exchange in molecular dynamics simulations. Front. Chem. 2023, 11, 1140896. [Google Scholar] [CrossRef]
- Gődény, M.; Joerg, F.; Kovar, M.P.P.; Schröder, C. Updates to Protex for Simulating Proton Transfers in an Ionic Liquid. J. Phys. Chem. B 2024, 128, 3416–3426. [Google Scholar] [CrossRef]
- Schmidt, R.G.; Brickmann, J. Molecular Dynamics Simulation of the Proton Transport in Water. Ber. Bunsenges. Phys. Chem. 1997, 101, 1816–1827. [Google Scholar] [CrossRef]
- Boresch, S.; Karplus, M. The Role of Bonded Terms in Free Energy Simulations: 1. Theoretical Analysis. J. Phys. Chem. A 1999, 103, 103–118. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Gissinger, J.R.; Jensen, B.D.; Wise, K.E. Modeling chemical reactions in classical molecular dynamics simulations. Polymer 2017, 128, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Gissinger, J.R.; Jensen, B.D.; Wise, K.E. REACTER: A Heuristic Method for Reactive Molecular Dynamics. Macromolecules 2020, 53, 9953–9961. [Google Scholar] [CrossRef]
- Gissinger, J.R.; Jensen, B.D.; Wise, K.E. Molecular modeling of reactive systems with REACTER. Comp. Phys. Commun. 2024, 304, 109287. [Google Scholar] [CrossRef]
- Kemppainen, J.; Gissinger, J.R.; Gowtham, S.; Odegard, G.M. LUNAR: Automated Input Generation and Analysis for Reactive LAMMPS Simulations. J. Chem. Inf. Model. 2024, 64, 5108–5126. [Google Scholar] [CrossRef]
- Danielsson, J.; Meuwly, M. Atomistic Simulation of Adiabatic Reactive Processes Based on Multi-State Potential Energy Surfaces. J. Chem. Theory Comput. 2008, 4, 1083–1093. [Google Scholar] [CrossRef]
- Cazade, P.A.; Huang, J.; Yosa, J.; Szymczak, J.J.; Meuwly, M. Atomistic simulations of reactive processes in the gas- and condensed phase. Int. Rev. Phys. Chem. 2012, 31, 235–264. [Google Scholar] [CrossRef]
- Nagy, T.; Reyes, J.Y.; Meuwly, M. Multisurface Adiabatic Reactive Molecular Dynamics. J. Chem. Theory Comput. 2014, 10, 1366–1375. [Google Scholar] [CrossRef]
- Steward, J.J.P. Optimization of Parameters for Semiempirical Methods: I. Method. J. Comp. Chem. 1989, 10, 209–220. [Google Scholar] [CrossRef]
- Zheng, M.; Li, X.; Liu, J.; Wang, Z.; Gong, X.; Guo, L.; Song, W. Pyrolysis of Liulin Coal Simulated by GPU-Based ReaxFF MD with Cheminformatics Analysis. Energy Fuels 2014, 28, 522–534. [Google Scholar] [CrossRef]
- Hartke, B.; Grimme, S. Reactive force fields made simple. Phys. Chem. Chem. Phys. 2015, 17, 16715–16718. [Google Scholar] [CrossRef] [PubMed]
- Tubert-Brohman, I.; Acevedo, O.; Jorgensen, W.L. Elucidation of Hydrolysis Mechanisms for Fatty Acid Amide Hydrolase and Its Lys142ala Variant Via QM/MM Simulations. J. Am. Chem. Soc. 2006, 128, 16904–16913. [Google Scholar] [CrossRef] [PubMed]
- Vilseck, J.Z.; Acevedo, O. Chapter 3—Computing Free-Energy Profiles Using Multidimensional Potentials of Mean Force and Polynomial Quadrature Methods. Ann. Rep. Comput. Chem. 2010, 6, 37–49. [Google Scholar]
- Lill, M.A.; Helms, V. Compact parameter set for fast estimation of proton transfer rates. J. Chem. Phys. 2001, 114, 1125–1132. [Google Scholar] [CrossRef]
- Hänggi, P.; Talkner, P.; Borkovec, M. Reaction-rate theory: Fifty years after Kramers. Rev. Mod. Phys. 1990, 62, 251–341. [Google Scholar] [CrossRef]
- Connors, K.A. Chemical Kinetics; Wiley: Weinheim, Germany, 1990. [Google Scholar]
- Antoniou, D.; Schwartz, S.D. Large kinetic isotope effects in enzymatic proton transfer and therole of substrate oscillations. Proc. Natl. Acad. Sci. USA 1997, 94, 12360–12365. [Google Scholar] [CrossRef]
- Knox, J.H. Molecular Thermodynamics; Wiley: Weinheim, Germany, 1971. [Google Scholar]
- Lill, M.A.; Helms, V. Molecular dynamics simulation of proton transport with quantum mechanically derived proton hopping rates (Q-HOP MD). J. Chem. Phys. 2001, 115, 7993–8005. [Google Scholar] [CrossRef]
- Miron, R.A.; Fichthorn, K.A. Accelerated molecular dynamics with the bond-boost method. J. Chem. Phys. 2003, 119, 6210–6216. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Asmussen, S. Applied Probability and Queues; Springer: New York, NY, USA, 2003; Volume 51. [Google Scholar]
- Gillespie, D.T. A General Method for Numerically Simulating the Stochastic Time Evolution of Coupled Chemical Reactions. J. Comput. Phys. 1976, 22, 403–434. [Google Scholar] [CrossRef]
- Gorecki, J.; Gryko, J. Molecular dynamics simulation of a chemical reaction. Comput. Phys. Commun. 1989, 54, 245–249. [Google Scholar] [CrossRef]
- Kirchner, B.; Hollóczki, O.; Canongia Lopes, J.N.; Pádua, A.A.H. Multiresolution calculation of ionic liquids. WIREs Comput. Mol. Sci. 2015, 5, 202–214. [Google Scholar] [CrossRef]
- Byrne, A.; Bringa, E.M.; Del Pópolo, M.G.; Kohanoff, J.J.; Galassi, V.; English, N.J. Mechanisms of Iodide–Triiodide Exchange Reactions in Ionic Liquids: A Reactive Molecular-Dynamics Exploration. Int. J. Mol. Sci. 2019, 20, 1123. [Google Scholar] [CrossRef]
- Cheng, T.; Jaramillo-Botero, A.; Goddard, W.A., III; Sun, H. Adaptive Accelerated ReaxFF Reactive Dynamics with Validationfrom Simulating Hydrogen Combustion. J. Am. Chem. Soc. 2014, 136, 9434–9442. [Google Scholar] [CrossRef]
- Zheng, M.; Li, X.; Guo, L. Algorithms of GPU-enabled reactive force field (ReaxFF) molecular dynamics. J. Mol. Graph. Model. 2013, 41, 1–11. [Google Scholar] [CrossRef]
- Li, X.; Mo, Z.; Liu, J.; Guo, L. Recent advances in the molecular simulation of chemical reactions. Mol. Sim. 2015, 41, 13–27. [Google Scholar] [CrossRef]
- Dral, P.O. Quantum Chemistry in the Age of Machine Learning. J. Phys. Chem. Lett. 2020, 11, 2336–2347. [Google Scholar] [CrossRef]
- Guo, F.; Wen, Y.S.; Feng, S.Q.; Li, X.D.; Li, H.S.; Cui, S.X.; Zhang, Z.R.; Hu, H.Q.; Zhang, G.Q.; Cheng, X.L. Intelligent-ReaxFF: Evaluating the reactive force field parameters with machine learning. Comp. Mat. Sci. 2020, 172, 109393. [Google Scholar] [CrossRef]
- Yilmaz, D.E.; Woodward, W.H.; van Duin, A.C.T. Machine Learning-Assisted Hybrid ReaxFF Simulations. J. Chem. Theory Comput. 2021, 17, 6705–6712. [Google Scholar] [CrossRef]
- Xue, L.Y.; Guo, F.; Wen, Y.S.; Feng, S.Q.; Huang, X.N.; Guo, L.; Li, H.S.; Cui, S.X.; Zhang, G.Q.; Wang, Q.L. ReaxFF-MPNN machine learning potential: A combination of reactive force field and message passing neural networks. Phys. Chem. Chem. Phys. 2021, 23, 19457–19464. [Google Scholar] [CrossRef] [PubMed]
- Sengul, M.Y.; Song, Y.; Nayir, N.; Gao, Y.; Hung, Y.; Dasgupta, T.; van Duin, A.C.T. INDEEDopt: A deep learning-based ReaxFF parameterization framework. npj Comput. Mater. 2021, 7, 68. [Google Scholar] [CrossRef]
- Koutsoukos, S.; Philipps, F.; Malaret, F.; Welton, T. A review on machine learning algorithms for the ionic liquid chemical space. Chem. Sci. 2021, 12, 6820. [Google Scholar] [CrossRef]
- Montes-Campos, H.; Carrete, J.; Bichelmaier, S.; Varela, L.M.; Madsen, G.K.H. A Differentiable Neural-Network Force Field for Ionic Liquids. J. Chem. Inf. Model. 2021, 62, 88–101. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, D.; Xia, Y.; Huang, Y.P.; Lin, X.; Han, X.; Ni, N.; Wang, Z.; Yu, F.; Yang, L.; et al. Artificial Intelligence Enhanced Molecular Simulations. J. Chem. Theory Comput. 2023, 19, 4338–4350. [Google Scholar] [CrossRef]
- Goodwin, Z.A.H.; Wenny, M.B.; Yang, J.H.; Cepellotti, A.; Ding, J.; Bystrom, K.; Duschatko, B.R.; Johansson, A.; Sun, L.; Batzner, S.; et al. Transferability and Accuracy of Ionic Liquid Simulations with Equivariant Machine Learning Interatomic Potentials. J. Phys. Chem. Lett. 2024, 15, 7539–7547. [Google Scholar] [CrossRef]


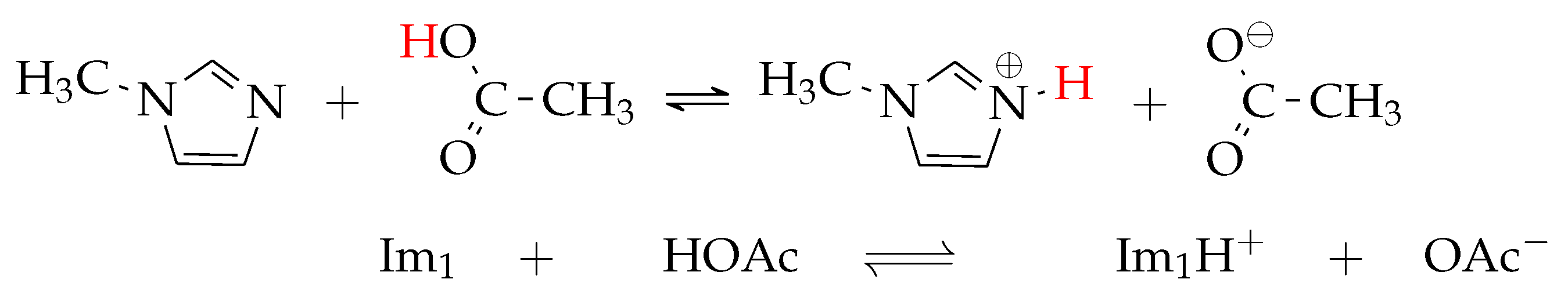
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gődény, M.; Schröder, C. Reactive Molecular Dynamics in Ionic Liquids: A Review of Simulation Techniques and Applications. Liquids 2025, 5, 8. https://doi.org/10.3390/liquids5010008
Gődény M, Schröder C. Reactive Molecular Dynamics in Ionic Liquids: A Review of Simulation Techniques and Applications. Liquids. 2025; 5(1):8. https://doi.org/10.3390/liquids5010008
Chicago/Turabian StyleGődény, Márta, and Christian Schröder. 2025. "Reactive Molecular Dynamics in Ionic Liquids: A Review of Simulation Techniques and Applications" Liquids 5, no. 1: 8. https://doi.org/10.3390/liquids5010008
APA StyleGődény, M., & Schröder, C. (2025). Reactive Molecular Dynamics in Ionic Liquids: A Review of Simulation Techniques and Applications. Liquids, 5(1), 8. https://doi.org/10.3390/liquids5010008