
Abraham Solvation Parameter Model: Examination of Possible Intramolecular Hydrogen-Bonding Using Calculated Solute Descriptors



Abstract
:1. Introduction
2. Solute Descriptor Computations
3. Solubility Predictions in Additional Organic Mono-Solvents
4. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnett, E.M.; Joris, L.; Mitchell, E.; Murty, T.S.S.R.; Gorrie, T.M.; Schleyer, P.v.R. Hydrogen-bonded complex formation. III. Thermodynamics of complexing by infrared spectroscopy and calorimetry. J. Am. Chem. Soc. 1970, 92, 2365–2377. [Google Scholar] [CrossRef]
- Spencer, J.N.; Wolbach, W.S.; Hovick, J.W.; Ansel, L.; Modarress, K.J. Hydrogen bonding by alcohols and amines. J. Solut. Chem. 1985, 14, 805–814. [Google Scholar] [CrossRef]
- Spencer, J.N.; Campanella, C.L.; Harris, E.M.; Wolbach, W.S. Solvent effects on hydrogen-bond formation. J. Phys. Chem. 1985, 89, 1888–1891. [Google Scholar] [CrossRef]
- Solomonov, B.N.; Novikov, V.B.; Varfolomeev, M.A.; Mileshko, N.M. A new method for the extraction of specific interaction enthalpy from the enthalpy of solvation. J. Phys. Org. Chem. 2005, 18, 49–61. [Google Scholar] [CrossRef]
- Varfolomeev, M.A.; Abaidullina, D.I.; Solomonov, B.N.; Verevkin, S.P.; Emel’Yanenko, V.N. Pairwise substitution effects, inter- and intramolecular hydrogen bonds in methoxyphenols and dimethoxybenzenes. Thermochemistry, calorimetry, and first-principles calculations. J. Phys. Chem. B 2010, 114, 16503–16516. [Google Scholar]
- Zaitseva, K.V.; Varfolomeev, M.A.; Solomonov, B.N. Thermodynamics of hydrogen bonding of weak bases in alcohol solutions: Calorimetry of solution, IR-spectroscopy and vapor pressure analysis. J. Mol. Struct. 2012, 1018, 14–20. [Google Scholar] [CrossRef]
- Solomonov, B.N.; Nagrimanov, R.N.; Mukhametzyanov, T.A. Additive scheme for calculation of solvation enthalpies of heterocyclic aromatic compounds. Sublimation/vaporization enthalpy at 298.15 K. Thermochim. Acta 2016, 633, 37–47. [Google Scholar] [CrossRef]
- Nagrimanov, R.N.; Samatov, A.A.; Solomonov, B.N. Additive scheme of solvation enthalpy for linear, cyclic and branched-chain aliphatic compounds at 298.15 K. J. Mol. Liq. 2019, 292, 111365/1–111365/7. [Google Scholar] [CrossRef]
- Solomonov, B.N.; Varfolomeev, M.A.; Nagrimanov, R.N.; Novikov, V.B.; Buzyurov, A.V.; Fedorova, Y.V.; Mukhametzyanov, T.A. New method for determination of vaporization and sublimation enthalpy of aromatic compounds at 298.15 K using solution calorimetry technique and group-additivity scheme. Thermochim. Acta 2015, 622, 88–96. [Google Scholar] [CrossRef]
- Golmohammadi, H.; Dashtbozorgi, Z.; Gholam Samani, M.; Acree, W.E., Jr. QSPR prediction of gas-to-methanol solvation enthalpy of organic compounds using replacement method and support vector machine. Phys. Chem. Liq. 2015, 53, 46–66. [Google Scholar] [CrossRef]
- Toubaei, A.; Golmohammadi, H.; Dashtbozorgi, Z.; Acree, W.E., Jr. QSPR studies for predicting gas to acetone and gas to acetonitrile solvation enthalpies using support vector machine. J. Mol. Liq. 2012, 175, 24–32. [Google Scholar] [CrossRef]
- Fariba, S.; Dakhel, A.A.; Shariati, S. Predictive artificial neural network model for solvation enthalpy of organic compounds in N,N-dimethylformamide. Russ. J. Phys. Chem. A 2019, 93, 2661–2668. [Google Scholar] [CrossRef]
- Magsumov, T.I.; Sedov, I.A.; Acree, W.E., Jr. Development of Abraham model correlations for enthalpies of solvation of solutes dissolved in N-methylformamide, 2-pyrrolidone and N-methylpyrrolidone. J. Mol. Liq. 2021, 323, 114609/1–114609/10. [Google Scholar] [CrossRef]
- Huang, J.; Eddula, S.; Tirumala, P.; Casillas, T.; Acree, W.E., Jr.; Abraham, M.H. Updated Abraham model correlations to describe enthalpies of solvation of solutes dissolved in heptane, cyclohexane and N,N-dimethylformamide. Phys. Chem. Liq. 2021, 59, 442–453. [Google Scholar] [CrossRef]
- Lu, J.Z.; Acree, W.E., Jr.; Abraham, M.H. Abraham model correlations for enthalpies of solvation of organic solutes dissolved in methyl acetate and octane. Phys. Chem. Liq. 2020, 58, 18–30. [Google Scholar] [CrossRef]
- Stolov, M.A.; Zaitseva, K.V.; Varfolomeev, M.A.; Acree, W.E. Enthalpies of solution and enthalpies of solvation of organic solutes in ethylene glycol at 298.15 K: Prediction and analysis of intermolecular interaction contributions. Thermochim. Acta 2017, 648, 91–99. [Google Scholar] [CrossRef]
- Hart, E.; Grover, D.; Zettl, H.; Koshevarova, V.; Acree, W.E., Jr.; Abraham, M.H. Development of Abraham model expressions for predicting the enthalpies of solvation of solutes dissolved in acetic acid. Phys. Chem. Liq. 2016, 54, 141–154. [Google Scholar] [CrossRef]
- Wilson, A.; Tian, A.; Dabadge, N.; Acree, W.E., Jr.; Varfolomeev, M.A.; Rakipov, I.T.; Arkhipova, S.M.; Abraham, M.H. Enthalpy of solvation correlations for organic solutes and gases dissolved in dichloromethane and 1,4-dioxane. Struct. Chem. 2013, 24, 1841–1853. [Google Scholar] [CrossRef]
- Acree, W.E., Jr.; Smart, K.; Abraham, M.H. Abraham model solute descriptors reveal strong intramolecular hydrogen bonding in 1,4-dihydroxyanthraquinone and 1,8-dihydroxyanthraquinone. Phys. Chem. Liq. 2018, 56, 416–420. [Google Scholar] [CrossRef]
- Abraham, M.H.; Du, C.M.; Platts, J.A. Lipophilicity of the nitrophenols, J. Org. Chem. 2000, 65, 7114–7118. [Google Scholar] [CrossRef]
- Abraham, M.H.; Acree, W.E., Jr.; Earp, C.E.; Vladimirova, A.; Whaley, W.L. Studies on the hydrogen bond acidity, and other descriptors and properties for hydroxyflavones and hydroxyisoflavones. J. Mol. Liq. 2015, 208, 363–372. [Google Scholar] [CrossRef]
- Ulrich, N.; Endo, S.; Brown, T.N.; Watanabe, N.; Bronner, G.; Abraham, M.H.; Goss, K.-U. UFZ-LSER Database v 3.2.1 [Internet]. Leipzig (Germany): Helmholtz Centre for Environmental Research-UFZ. 2017. Available online: http://www.ufz.de/lserd (accessed on 17 June 2022).
- Wu, Y.; Xie, Y.; Shi, H. Determination and model correlation of rhein in 12 pure solvents from 283.15 K to 323.15 K. J. Chem. Eng. Data 2022, in press. [CrossRef]
- Cheng, Y.; Wang, D.; Zhang, Z.; Wang, Z. Solubility and solution thermodynamics of rhein in eight pure solvents from (288.15 to 313.15) K. RSC Adv. 2015, 5, 80548–80552. [Google Scholar] [CrossRef]
- Platts, J.A.; Butina, D.; Abraham, M.H.; Hersey, A. Estimation of molecular linear free energy relation descriptors using a group contribution approach. J. Chem. Inf. Comp. Sci. 1999, 39, 835–845. [Google Scholar] [CrossRef]
- Platts, J.A.; Abraham, M.H.; Butina, D.; Hersey, A. Estimation of molecular linear free energy relationship descriptors by a group contribution approach. 2. Prediction of partition coefficients. J. Chem. Inf. Comp. Sci. 2000, 40, 71–80. [Google Scholar] [CrossRef]
- Chung, Y.; Vermeire, F.H.; Wu, H.; Walker, P.J.; Abraham, M.H.; Green, W.H. Group contribution and machine learning approaches to predict Abraham solute parameters, solvation free energy, and solvation enthalpy. J. Chem. Inf. Model. 2022, 62, 433–446. [Google Scholar] [CrossRef]
- Ulrich, N.; Ebert, A. Can deep learning algorithms enhance the prediction of solute descriptors for linear solvation energy relationship approaches? Fluid Phase Equilib. 2022, 555, 113349/1–113349/7. [Google Scholar] [CrossRef]
- Yamada, H.; Yajima, K.; Wada, H.; Nakagawa, G. Effects of solvation on partition and dimerization of benzoic acid in mixed solvent systems. Talanta 1995, 42, 789–795. [Google Scholar] [CrossRef]
- Strickland, S.; Ocon, L.; Zhang, A.; Wang, S.; Eddula, S.; Liu, G.; Tirumala, P.; Huang, J.; Dai, J.; Jiang, C.; et al. Abraham model correlations for describing dissolution of organic solutes and inorganic gases in dimethyl carbonate. Phys. Chem. Liq. 2021, 59, 181–195. [Google Scholar] [CrossRef]
- PharmaAlgorithms. ADME Boxes, Version 3.0; PharmaAlgorithms Inc.: Toronto, ON, Canada.
- Abraham, M.H.; McGowan, J.C. The use of characteristic volumes to measure cavity terms in reversed phase liquid chromatography. Chromatographia 1987, 23, 243–246. [Google Scholar] [CrossRef]
- Abraham, M.H.; Abraham, R.J.; Acree, W.E.; Aliev, A.E.; Leo, A.J.; Whaley, W.L. An NMR method for the quantitative assessment of intramolecular hydrogen bonding; Application to physicochemical, environmental, and biochemical properties. J. Org. Chem. 2014, 79, 11075–11083. [Google Scholar] [CrossRef] [PubMed]
- Whaley, W.L.; Okoso-amaa, E.M.; Womack, C.L.; Vladimirova, A.; Rogers, L.B.; Risher, M.J.; Abraham, M.H. Summation solute hydrogen bonding acidity values for hydroxyl substituted flavones determined by NMR spectroscopy. Nat. Prod. Comm. 2013, 8, 85–98. [Google Scholar] [CrossRef] [Green Version]
- Padhye, S.B.; Kulkarni, B.A. PMR evidence for hydrogen bonding in naturally occurring isomeric juglones. J. Mag. Res. 1974, 16, 150–152. [Google Scholar] [CrossRef]
- Bradley, J.-C.; Abraham, M.H.; Acree, W.E., Jr.; Lang, A.S.I.D.; Beck, S.N.; Bulger, D.A.; Clark, E.A.; Condron, L.N.; Costa, S.T.; Curtin, E.M.; et al. Determination of Abraham model solute descriptors for the monomeric and dimeric forms of trans-cinnamic acid using measured solubilities from the Open Notebook Science Challenge. Chem. Cent. J. 2015, 9, 1–6. [Google Scholar] [CrossRef] [Green Version]
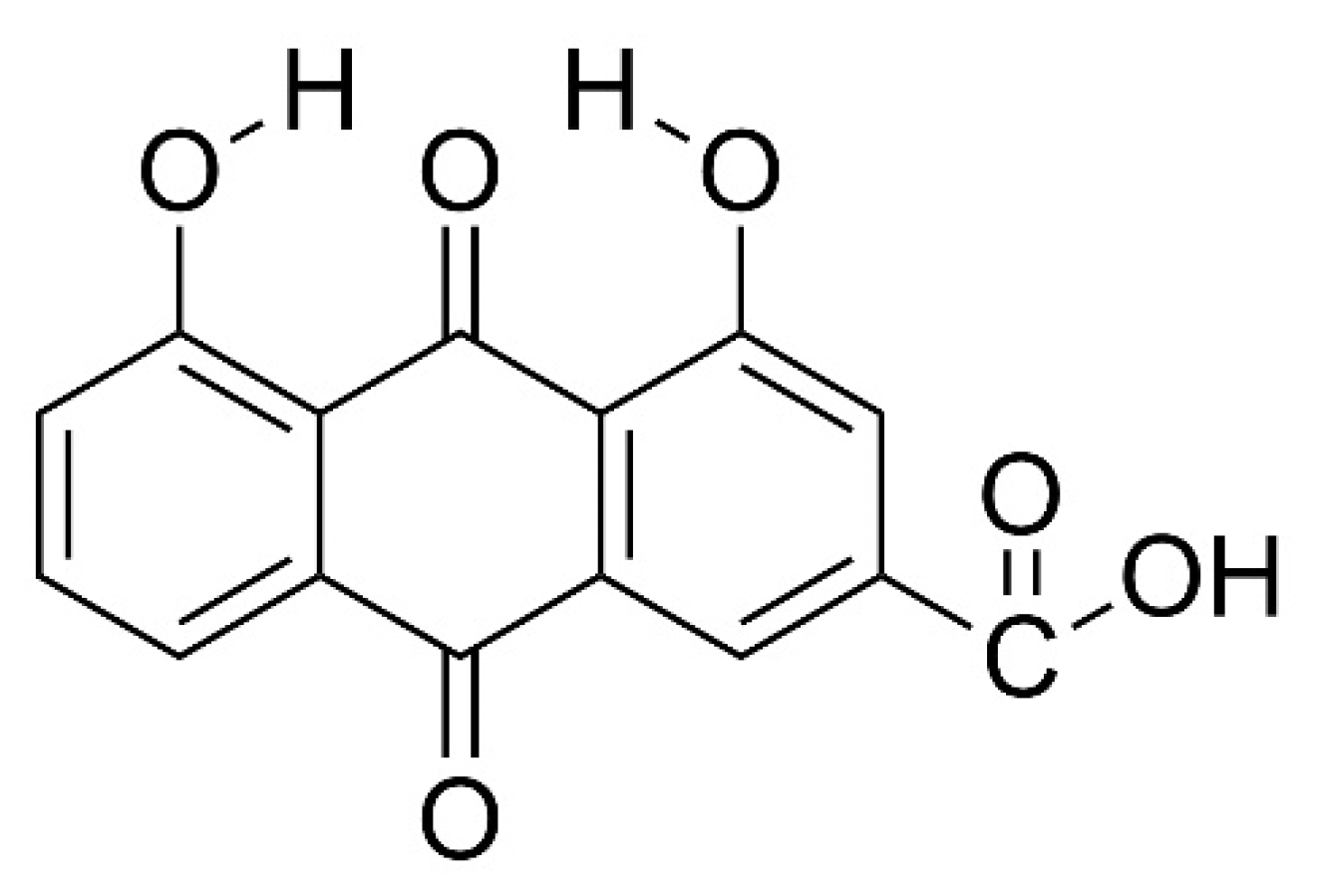
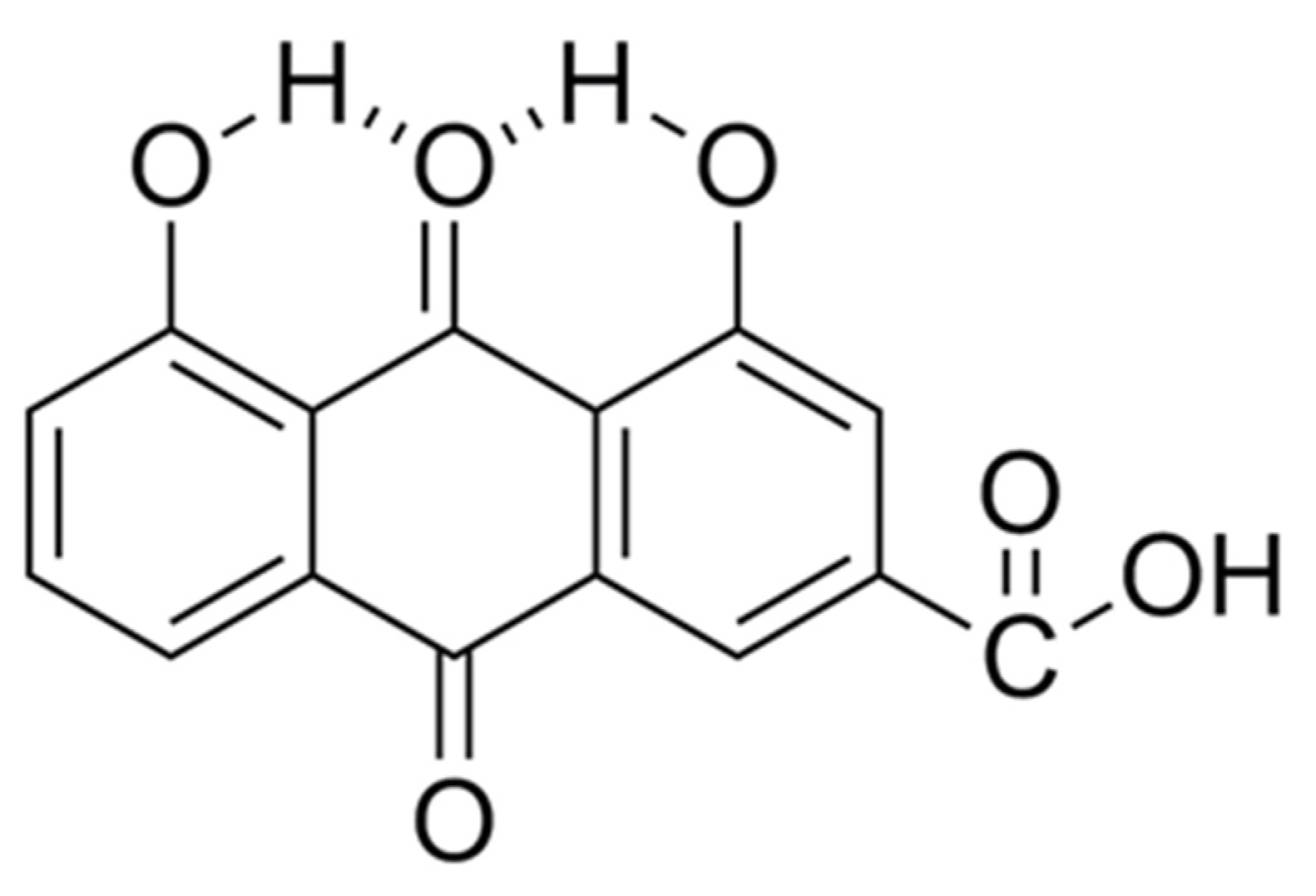
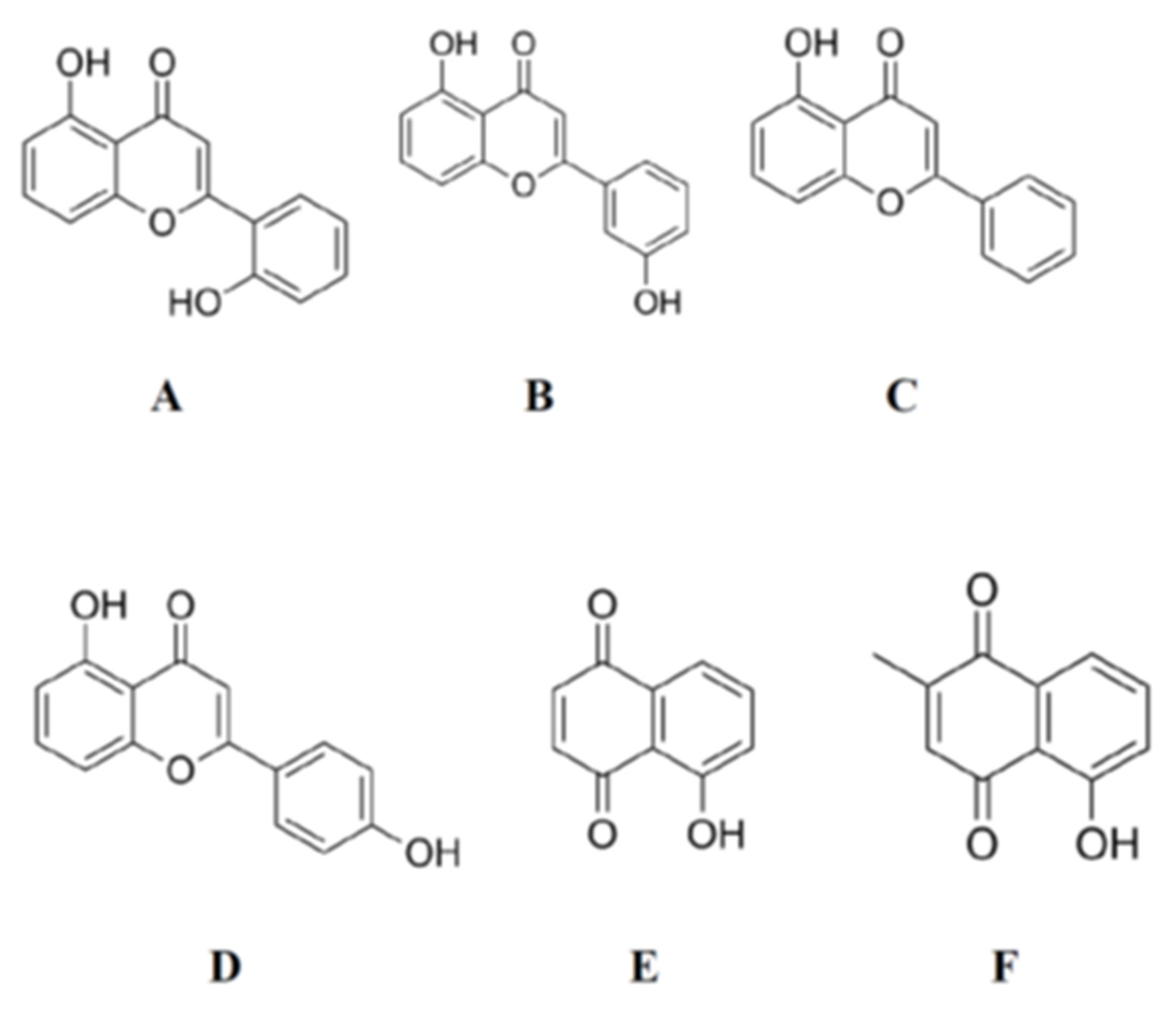

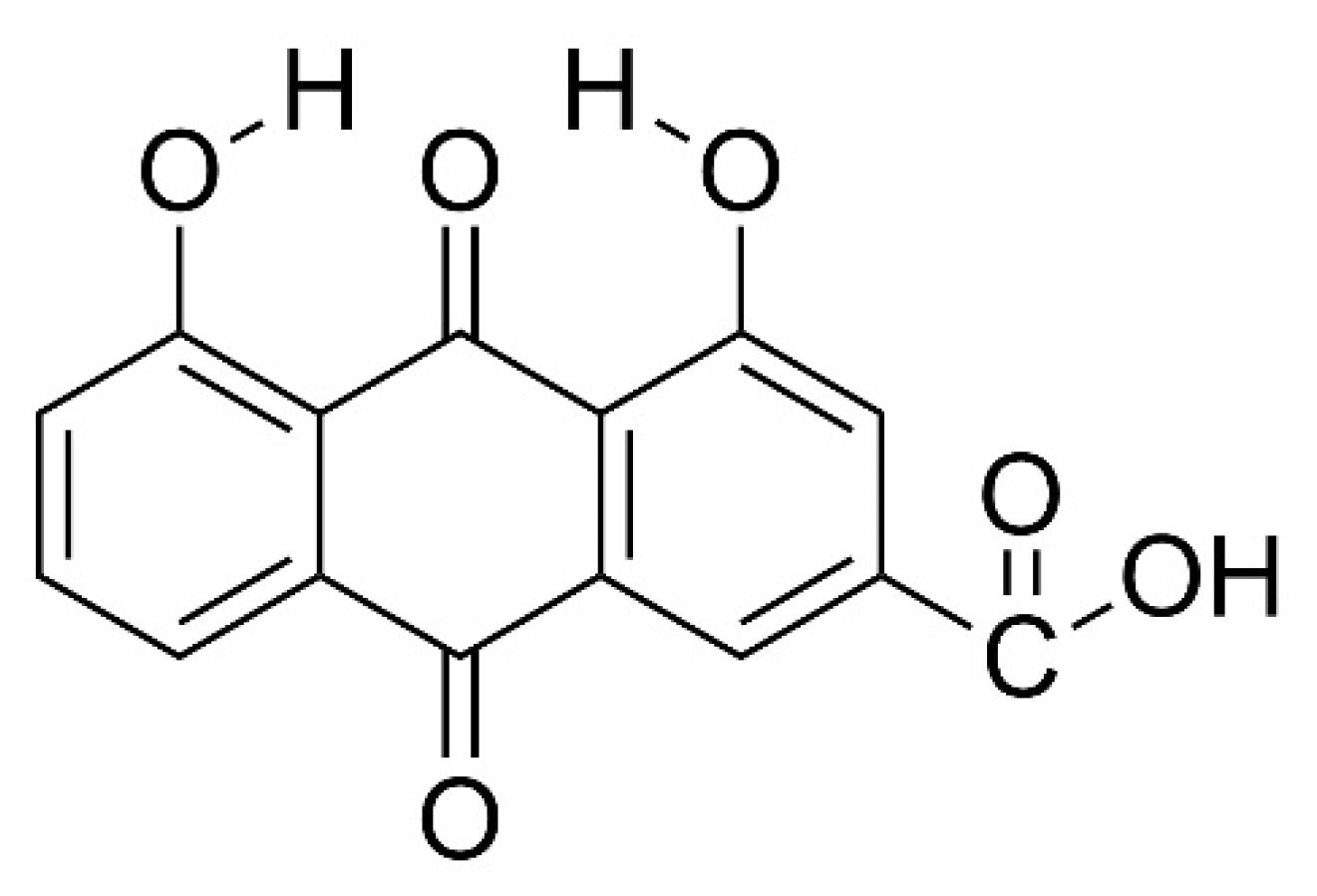{kind=link}
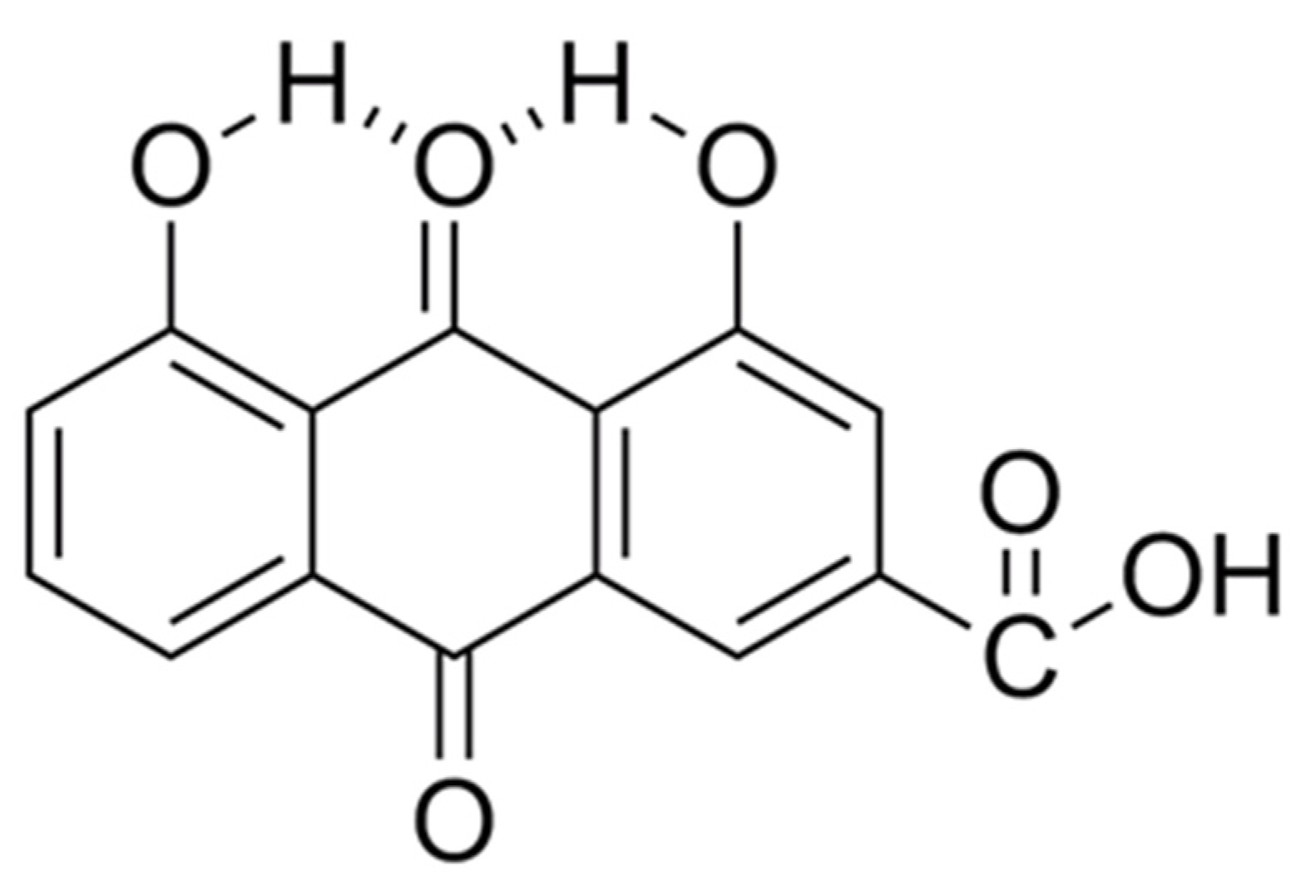{kind=link}
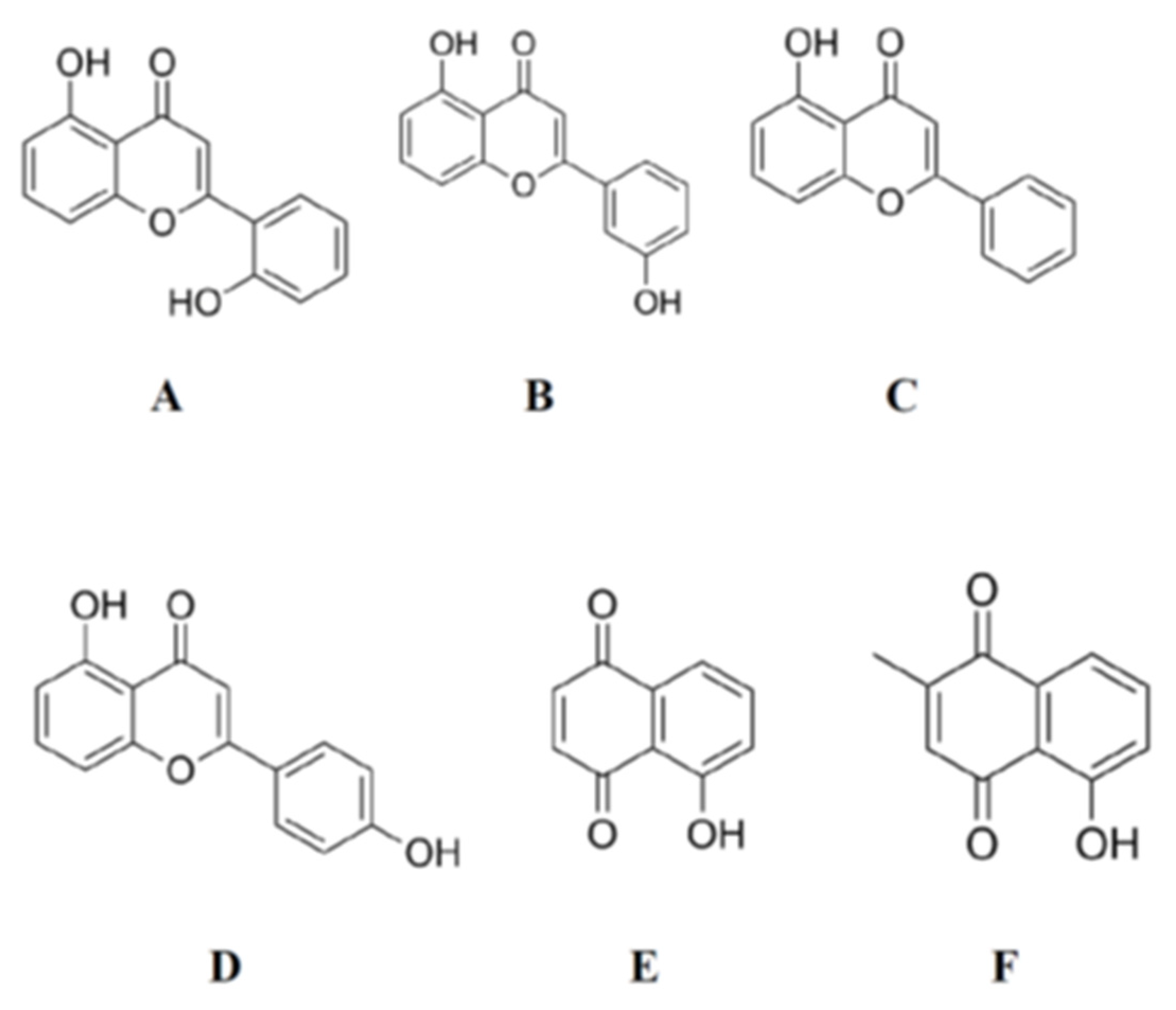{kind=link}
| Solvent | Log CS,organic | Log P | Ref. |
|---|---|---|---|
| 1-Octanol (wet) | 4.580 | ACD a | |
| Methanol | −3.495 | [23] | |
| Ethanol | −3.588 | [23] | |
| Propan-1-ol | −3.618 | [23] | |
| Propan-2-ol | −3.547 | [23] | |
| Butan-1-ol | −3.568 | [23] | |
| 2-Methylpropan-1-ol | −3.824 | [23] | |
| Ethylene glycol | −3.738 | [24] | |
| Acetone | −2.800 | [23] | |
| Butanone | −2.995 | [23] | |
| Acetonitrile | −3.770 | [23] | |
| Ethyl acetate | −3.680 | [24] |
| Log (P CS,org/CS,water) | c3 | e3 | s3 | a3 | b3 | v3 |
|---|---|---|---|---|---|---|
| Octanol (wet) | 0.088 | 0.562 | −1.054 | 0.034 | −3.460 | 3.814 |
| Dichloromethane | 0.319 | 0.102 | −0.187 | −3.058 | −4.090 | 4.324 |
| Trichloromethane | 0.191 | 0.105 | −0.403 | −3.112 | −3.514 | 4.395 |
| Tetrachloromethane | 0.199 | 0.523 | −1.159 | −3.560 | −4.594 | 4.618 |
| 1,2-Dichloroethane | 0.183 | 0.294 | −0.134 | −2.801 | −4.291 | 4.180 |
| 1-Chlorobutane | 0.222 | 0.273 | −0.569 | −2.918 | −4.883 | 4.456 |
| Hexane | 0.333 | 0.560 | −1.710 | −3.578 | −4.939 | 4.463 |
| Heptane | 0.297 | 0.634 | −1.755 | −3.571 | −4.946 | 4.488 |
| Octane | 0.241 | 0.690 | −1.769 | −3.545 | −5.011 | 4.511 |
| Nonane | 0.240 | 0.619 | −1.713 | −3.532 | −4.921 | 4.482 |
| Decane | 0.172 | 0.726 | −1.750 | −3.446 | −4.496 | 4.489 |
| Undecane | 0.058 | 0.603 | −1.661 | −3.421 | −5.120 | 4.619 |
| Dodecane | 0.114 | 0.668 | −1.644 | −3.545 | −5.006 | 4.459 |
| Hexadecane | 0.087 | 0.667 | −1.617 | −3.587 | −4.869 | 4.433 |
| Cyclohexane | 0.159 | 0.784 | −1.678 | −3.740 | −4.929 | 4.577 |
| Methylcyclohexane | 0.246 | 0.782 | −1.982 | −3.517 | −4.293 | 4.528 |
| Isooctane | 0.318 | 0.555 | −1.737 | −3.677 | −4.864 | 4.417 |
| Hexadec-1-ene | 0.116 | 0.706 | −1.616 | −3.181 | −4.796 | 4.322 |
| Deca-1,9-diene | 0.185 | 0.468 | −1.888 | −2.911 | −3.972 | 4.382 |
| Air-to-water | −0.994 | 0.577 | 2.549 | 3.813 | 4.841 | −0.869 |
| Benzene | 0.142 | 0.464 | −0.588 | −3.099 | −4.625 | 4.491 |
| Toluene | 0.125 | 0.431 | −0.644 | −3.002 | −4.748 | 4.524 |
| 1,2-Dimethylbenzene | 0.083 | 0.518 | −0.813 | −2.884 | −4.821 | 4.559 |
| 1,3-Dimethylbenzene | 0.122 | 0.377 | −0.603 | −2.981 | −4.961 | 4.535 |
| 1,4-Dimethylbenzene | 0.166 | 0.477 | −0.812 | −2.939 | −4.874 | 4.532 |
| Ethylbenzene | 0.093 | 0.467 | −0.723 | −3.001 | −4.844 | 4.514 |
| Fluorobenzene | 0.139 | 0.152 | −0.374 | −3.030 | −4.601 | 4.540 |
| Chlorobenzene | 0.065 | 0.381 | −0.521 | −3.183 | −4.700 | 4.614 |
| Bromobenzene | −0.017 | 0.436 | −0.424 | −3.174 | −4.558 | 4.445 |
| Iodobenzene | −0.192 | 0.298 | −0.308 | −3.213 | −4.653 | 4.588 |
| Nitrobenzene | −0.152 | 0.525 | 0.081 | −2.332 | −4.494 | 4.187 |
| Benzonitrile | 0.097 | 0.285 | 0.059 | −1.605 | −4.562 | 4.028 |
| Diethyl ether (wet) | 0.248 | 0.561 | −1.016 | −0.226 | −4.553 | 4.075 |
| Diisopropyl ether (wet) | 0.472 | 0.413 | −0.745 | −0.632 | −5.251 | 4.059 |
| Dibutyl ether (wet) | 0.252 | 0.677 | −1.506 | −0.807 | −5.249 | 4.815 |
| Ethyl acetate (wet) | 0.441 | 0.591 | −0.669 | −0.325 | −4.261 | 3.666 |
| Butyl acetate (wet) | −0.475 | 0.428 | −0.094 | −0.241 | −4.151 | 4.046 |
| PGDP a | 0.256 | 0.501 | −0.828 | −1.022 | −4.640 | 4.033 |
| Olive oil | −0.035 | 0.574 | −0.798 | −1.422 | −4.984 | 4.210 |
| Carbon disulfide | 0.047 | 0.686 | −0.943 | −3.603 | −5.818 | 4.921 |
| Isopropyl myristate | −0.605 | 0.930 | −1.153 | −1.682 | −4.093 | 4.240 |
| Methyl ethyl ketone (wet) | 0.350 | 0.315 | −0.196 | 0.040 | −2.502 | 1.947 |
| Methyl isobutyl ketone (wet) | 0.383 | 0.801 | −0.831 | −0.121 | −4.441 | 3.876 |
| o-Nitrophenyl octyl ether (wet) | 0.182 | 0.631 | −0.447 | −2.254 | −3.973 | 3.559 |
| Methanol | 0.276 | 0.334 | −0.714 | 0.243 | −3.320 | 3.549 |
| Ethanol | 0.222 | 0.471 | −1.035 | 0.326 | −3.596 | 3.857 |
| Propan-1-ol | 0.139 | 0.405 | −1.029 | 0.247 | −3.767 | 3.986 |
| Butan-1-ol | 0.165 | 0.401 | −1.011 | 0.056 | −3.958 | 4.044 |
| Pentan-1-ol | 0.150 | 0.536 | −1.229 | 0.141 | −3.864 | 4.077 |
| Hexan-1-ol | 0.115 | 0.492 | −1.164 | 0.054 | −3.978 | 4.131 |
| Heptan-1-ol | 0.035 | 0.398 | −1.063 | 0.002 | −4.342 | 4.317 |
| Octan-1-ol | −0.034 | 0.489 | −1.044 | −0.024 | −4.235 | 4.218 |
| Decan-1-ol | −0.058 | 0.616 | −1.319 | 0.026 | −4.153 | 4.279 |
| Propan-2-ol | 0.099 | 0.344 | −1.049 | 0.406 | −3.827 | 4.033 |
| 2-Methylpropan-1-ol | 0.188 | 0.354 | −1.127 | 0.016 | −3.568 | 3.986 |
| Butan-2-ol | 0.127 | 0.253 | −0.976 | 0.158 | −3.882 | 4.114 |
| 2-Methylpropan-2-ol | 0.211 | 0.171 | −0.947 | 0.331 | −4.085 | 4.109 |
| 2-Methylbutan-1-ol | 0.143 | 0.388 | −1.173 | −0.024 | −3.817 | 4.129 |
| 3-Methylbutan-1-ol | 0.073 | 0.360 | −1.273 | 0.090 | −3.770 | 4.273 |
| 2-Methylbutan-2-ol | 0.225 | 0.361 | −1.180 | 0.473 | −3.944 | 4.159 |
| Pentan-2-ol | 0.115 | 0.455 | −1.331 | 0.206 | −3.745 | 4.201 |
| 4-Methylpentan-2-ol | 0.096 | 0.301 | −1.100 | 0.039 | −4.081 | 4.242 |
| 2-Ethylhexan-1-ol | −0.033 | 0.566 | −1.233 | −0.068 | −3.912 | 4.153 |
| Cyclopentanol | 0.332 | 0.522 | −1.034 | −0.106 | −3.756 | 3.892 |
| Benzyl alcohol | −0.071 | 0.337 | −0.323 | −0.778 | −3.548 | 3.938 |
| Trifluoroethanol | 0.395 | −0.094 | −0.594 | −1.280 | −1.274 | 3.088 |
| Ethylene glycol | −0.270 | 0.578 | −0.511 | 0.715 | −2.619 | 2.729 |
| 1,2-Propanediol | −0.149 | 0.754 | −0.966 | 0.684 | −3.134 | 3.247 |
| 2-Methoxyethanol | 0.175 | 0.326 | −0.140 | 0.000 | −4.086 | 3.630 |
| 2-Ethoxyethanol | 0.133 | 0.392 | −0.419 | 0.125 | −4.200 | 3.888 |
| 2-Propoxyethanol | 0.053 | 0.419 | −0.569 | 0.000 | −4.327 | 4.095 |
| 2-Butoxyethanol | −0.055 | 0.377 | −0.607 | −0.080 | −4.371 | 4.234 |
| 2-Isopropoxyethanol | 0.107 | 0.391 | −0.525 | 0.071 | −4.439 | 4.051 |
| 3-Methoxybutan-1-ol | −0.094 | 0.400 | −0.565 | 0.072 | −4.240 | 4.149 |
| 1-tert-Butoxypropan-2-ol | 0.172 | 0.436 | −0.612 | −0.044 | −4.367 | 3.922 |
| Diethylene glycol | −0.096 | 0.580 | −0.145 | 0.138 | −3.718 | 3.072 |
| Triethylene glycol | −0.071 | 0.501 | 0.074 | 0.157 | −3.957 | 3.106 |
| Diethyl ether | 0.330 | 0.401 | −0.814 | −0.457 | −4.959 | 4.320 |
| Diisopropyl ether | 0.181 | 0.285 | −0.954 | −0.956 | −5.077 | 4.542 |
| Dibutyl ether | 0.203 | 0.369 | −0.954 | −1.488 | −5.426 | 4.508 |
| Methyl tert-butyl ether | 0.376 | 0.264 | −0.788 | −1.078 | −5.030 | 4.410 |
| Tetrahydrofuran | 0.207 | 0.372 | −0.392 | −0.236 | −4.934 | 4.447 |
| Dioxane | 0.098 | 0.350 | −0.083 | −0.556 | −4.826 | 4.172 |
| Methyl acetate | 0.351 | 0.223 | −0.150 | −1.035 | −4.527 | 3.972 |
| Ethyl acetate | 0.328 | 0.369 | −0.446 | −0.700 | −4.904 | 4.150 |
| Propyl acetate | 0.288 | 0.363 | −0.474 | −0.784 | −4.938 | 4.216 |
| Isopropyl acetate | 0.307 | 0.314 | −0.481 | −0.952 | −4.779 | 4.159 |
| Butyl acetate | 0.248 | 0.356 | −0.501 | −0.867 | −4.973 | 4.281 |
| Pentyl acetate | 0.182 | 0.261 | −0.474 | −1.017 | −4.952 | 4.388 |
| Methyl butyrate | 0.238 | 0.368 | −0.538 | −1.031 | −4.623 | 4.253 |
| Dimethyl carbonate | 0.114 | 0.109 | −0.083 | −1.405 | −4.578 | 4.163 |
| Diethyl carbonate | 0.133 | 0.135 | −0.309 | −1.532 | −4.816 | 4.398 |
| Propylene carbonate | 0.004 | 0.168 | 0.504 | −1.283 | −4.407 | 3.421 |
| Propanone | 0.313 | 0.312 | −0.121 | −0.608 | −4.753 | 3.942 |
| Butanone | 0.276 | 0.296 | −0.174 | −0.714 | −4.868 | 4.138 |
| Cyclopentanone | −0.016 | 0.386 | −0.201 | −0.766 | −4.454 | 4.321 |
| Cyclohexanone | 0.077 | 0.249 | 0.028 | −0.891 | −4.917 | 4.283 |
| N,N-Dimethylformamide | −0.305 | −0.058 | 0.343 | 0.358 | −4.865 | 4.486 |
| N,N-Dimethylacetamide | −0.271 | 0.084 | 0.209 | 0.915 | −5.003 | 4.557 |
| N-Methylpyrolidinone | 0.147 | 0.532 | 0.225 | 0.840 | −4.794 | 4.374 |
| Formamide | −0.171 | 0.070 | 0.308 | 0.589 | −3.152 | 2.432 |
| Acetonitrile | 0.413 | 0.077 | 0.326 | −1.566 | −4.391 | 3.364 |
| Propanenitrile | 0.357 | 0.188 | 0.061 | −1.515 | −4.539 | 3.760 |
| Butanenitrile | 0.316 | 0.224 | −0.065 | −1.369 | −4.608 | 3.944 |
| Nitromethane | 0.023 | −0.091 | 0.793 | −1.463 | −4.364 | 3.460 |
| Dimethyl sulfoxide | −0.194 | 0.327 | 0.791 | 1.260 | −4.540 | 3.361 |
| Pyridine | −0.046 | 0.298 | 0.000 | 0.558 | −4.504 | 4.292 |
| 2-Pyrrolidone | 0.000 | 0.378 | 0.465 | 1.220 | −4.621 | 3.423 |
| Aniline | −0.157 | 0.322 | 0.000 | −1.287 | −3.519 | 3.842 |
| Methoxybenzene | 0.071 | 0.416 | −0.317 | −2.538 | −4.479 | 4.278 |
| Acetophenone | 0.177 | 0.365 | 0.000 | −1.537 | −4.658 | 3.958 |
| Acetic acid | 0.175 | 0.174 | −0.454 | −1.073 | −2.789 | 3.725 |
| Tributyl phosphate | 0.054 | 0.411 | −0.474 | 0.252 | −4.810 | 4.181 |
| 95 vol% Methanol b | 0.270 | 0.278 | −0.520 | 0.230 | −3.368 | 3.365 |
| 90 vol% Methanol | 0.258 | 0.250 | −0.452 | 0.229 | −3.206 | 3.175 |
| 80 vol% Methanol | 0.172 | 0.197 | −0.319 | 0.241 | −2.912 | 2.842 |
| 70 vol% Methanol | 0.098 | 0.192 | −0.260 | 0.266 | −2.558 | 2.474 |
| 60 vol% Methanol | 0.053 | 0.207 | −0.238 | 0.272 | −2.157 | 2.073 |
| 50 vol% Methanol | 0.023 | 0.223 | −0.222 | 0.264 | −1.747 | 1.662 |
| 40 vol% Methanol | 0.020 | 0.222 | −0.205 | 0.218 | −1.329 | 1.257 |
| 30 vol% Methanol | 0.016 | 0.187 | −0.172 | 0.165 | −0.953 | 0.898 |
| 20 vol% Methanol | 0.022 | 0.142 | −0.138 | 0.088 | −0.574 | 0.557 |
| 10 vol% Methanol | 0.012 | 0.072 | −0.081 | 0.026 | −0.249 | 0.226 |
| 96 vol% Ethanol c | 0.238 | 0.353 | −0.833 | 0.297 | −3.533 | 3.724 |
| 95 vol% Ethanol | 0.239 | 0.328 | −0.795 | 0.294 | −3.514 | 3.697 |
| 90 vol% Ethanol | 0.243 | 0.213 | −0.575 | 0.262 | −3.450 | 3.545 |
| 80 vol% Ethanol | 0.172 | 0.175 | −0.465 | 0.260 | −3.212 | 3.323 |
| 70 vol% Ethanol | −0.063 | 0.085 | −0.368 | 0.311 | −2.936 | 3.102 |
| 60 vol% Ethanol | −0.040 | 0.138 | −0.335 | 0.293 | −2.675 | 2.812 |
| 50 vol% Ethanol | −0.142 | 0.124 | −0.252 | 0.251 | −2.275 | 2.415 |
| 40 vol% Ethanol | −0.221 | 0.131 | −0.159 | 0.171 | −1.809 | 1.918 |
| 30 vol% Ethanol | −0.269 | 0.107 | −0.098 | 0.133 | −1.316 | 1.414 |
| 20 vol% Ethanol | −0.252 | 0.042 | −0.040 | 0.096 | −0.823 | 0.916 |
| 10 vol% Ethanol | −0.173 | −0.023 | −0.001 | 0.065 | −0.372 | 0.454 |
| Log (K or CS,org/CS,gas) | c4 | e4 | s4 | a4 | b4 | l4 |
| Octanol (wet) | −0.198 | 0.002 | 0.709 | 3.519 | 1.429 | 0.858 |
| Dichloromethane | 0.192 | −0.572 | 1.492 | 0.460 | 0.847 | 0.965 |
| Trichloromethane | 0.157 | −0.560 | 1.259 | 0.374 | 1.333 | 0.976 |
| Tetrachloromethane | 0.217 | −0.435 | 0.554 | 0.000 | 0.000 | 1.069 |
| 1,2-Dichloroethane | 0.017 | −0.337 | 1.600 | 0.774 | 0.637 | 0.921 |
| 1-Chlorobutane | 0.130 | −0.581 | 1.114 | 0.724 | 0.000 | 1.016 |
| Hexane | 0.320 | 0.000 | 0.000 | 0.000 | 0.000 | 0.945 |
| Heptane | 0.284 | 0.000 | 0.000 | 0.000 | 0.000 | 0.950 |
| Octane | 0.219 | 0.000 | 0.000 | 0.000 | 0.000 | 0.960 |
| Nonane | 0.193 | 0.000 | 0.000 | 0.000 | 0.000 | 0.964 |
| Decane | 0.159 | 0.000 | 0.000 | 0.000 | 0.000 | 0.972 |
| Undecane | 0.113 | 0.000 | 0.000 | 0.000 | 0.000 | 0.971 |
| Dodecane | 0.017 | 0.000 | 0.000 | 0.000 | 0.000 | 0.989 |
| Hexadecane | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 1.000 |
| Cyclohexane | 0.163 | −0.110 | 0.000 | 0.000 | 0.000 | 1.013 |
| Methylcyclohexane | 0.318 | −0.215 | 0.000 | 0.000 | 0.000 | 1.012 |
| Isooctane | 0.264 | −0.230 | 0.000 | 0.000 | 0.000 | 0.975 |
| Hexadec-1-ene | −0.021 | 0.027 | 0.093 | 0.223 | −0.119 | 0.976 |
| Deca-1,9-diene | −0.006 | −0.098 | −0.249 | 0.505 | 0.713 | 1.021 |
| Air-to-water | −1.271 | 0.822 | 2.743 | 3.904 | 4.814 | −0.213 |
| Benzene | 0.107 | −0.313 | 1.053 | 0.457 | 0.169 | 1.020 |
| Toluene | 0.085 | −0.400 | 1.063 | 0.501 | 0.154 | 1.011 |
| 1,2-Dimethylbenzene | 0.064 | −0.296 | 0.934 | 0.647 | 0.000 | 1.010 |
| 1,3-Dimethylbenzene | 0.071 | −0.423 | 1.068 | 0.552 | 0.000 | 1.014 |
| 1,4-Dimethylbenzene | 0.113 | −0.302 | 0.826 | 0.651 | 0.000 | 1.011 |
| Ethylbenzene | 0.059 | −0.295 | 0.924 | 0.573 | 0.098 | 1.010 |
| Fluorobenzene | 0.181 | −0.621 | 1.432 | 0.647 | 0.000 | 0.986 |
| Chlorobenzene | 0.064 | −0.399 | 1.151 | 0.313 | 0.171 | 1.032 |
| Bromobenzene | −0.064 | −0.326 | 1.261 | 0.323 | 0.292 | 1.002 |
| Iodobenzene | −0.171 | −0.192 | 1.197 | 0.245 | 0.245 | 1.002 |
| Nitrobenzene | −0.296 | 0.092 | 1.707 | 1.147 | 0.443 | 0.912 |
| Benzonitrile | −0.075 | −0.341 | 1.798 | 2.030 | 0.291 | 0.880 |
| Diethyl ether (wet) | 0.206 | −0.169 | 0.873 | 3.402 | 0.000 | 0.882 |
| Dipropyl ether (wet) | 0.065 | −0.202 | 0.776 | 3.074 | 0.000 | 0.948 |
| Diisopropyl ether (wet) | 0.114 | −0.032 | 0.685 | 3.108 | 0.000 | 0.941 |
| Dibutyl ether (wet) | 0.369 | −0.216 | 0.026 | 2.626 | −0.499 | 1.124 |
| Ethyl acetate (wet) | 0.130 | 0.031 | 1.202 | 3.199 | 0.463 | 0.828 |
| Butyl acetate (wet) | −0.664 | 0.061 | 1.671 | 3.373 | 0.824 | 0.832 |
| PGDP a (wet) | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Olive oil | −0.156 | −0.254 | 0.859 | 1.656 | 0.000 | 0.873 |
| Carbon disulfide | 0.101 | 0.251 | 0.177 | 0.027 | 0.095 | 1.068 |
| Methyl ethyl ketone (wet) | −0.360 | 0.413 | 2.104 | 3.782 | 2.214 | 0.324 |
| Methyl isobutyl ketone (wet) | 0.244 | 0.183 | 0.987 | 3.418 | 0.323 | 0.854 |
| o-Nitrophenyl octyl ether (wet) | −0.104 | 0.290 | 1.333 | 1.306 | 0.967 | 0.759 |
| Methanol | −0.039 | −0.338 | 1.317 | 3.826 | 1.396 | 0.773 |
| Ethanol | 0.017 | −0.232 | 0.867 | 3.894 | 1.192 | 0.846 |
| Propan-1-ol | −0.042 | −0.246 | 0.749 | 3.888 | 1.076 | 0.874 |
| Butan-1-ol | −0.004 | −0.285 | 0.768 | 3.705 | 0.879 | 0.890 |
| Pentan-1-ol | −0.002 | −0.161 | 0.535 | 3.778 | 0.960 | 0.900 |
| Hexan-1-ol | −0.014 | −0.205 | 0.583 | 3.621 | 0.891 | 0.913 |
| Heptan-1-ol | −0.056 | −0.216 | 0.554 | 3.596 | 0.803 | 0.933 |
| Octan-1-ol | −0.147 | −0.214 | 0.561 | 3.507 | 0.749 | 0.943 |
| Decan-1-ol | −0.139 | −0.090 | 0.356 | 3.547 | 0.727 | 0.958 |
| Propan-2-ol | −0.048 | −0.324 | 0.713 | 4.036 | 1.055 | 0.884 |
| 2-Methylpropan-1-ol | −0.003 | −0.357 | 0.699 | 3.595 | 1.247 | 0.881 |
| Butan-2-ol | −0.034 | −0.387 | 0.719 | 3.736 | 1.088 | 0.905 |
| 2-Methylpropan-2-ol | 0.053 | −0.443 | 0.699 | 4.026 | 0.882 | 0.907 |
| 2-Methylbutan-1-ol | −0.055 | −0.348 | 0.601 | 3.565 | 0.996 | 0.925 |
| 3-Methylbutan-1-ol | −0.052 | −0.430 | 0.628 | 3.661 | 0.932 | 0.937 |
| 2-Methylbutan-2-ol | 0.097 | −0.375 | 0.653 | 3.975 | 0.875 | 0.914 |
| Pentan-2-ol | −0.031 | −0.325 | 0.496 | 3.792 | 1.024 | 0.934 |
| 4-Methylpentan-2-ol | −0.013 | −0.606 | 0.687 | 3.622 | 0.436 | 0.985 |
| 2-Ethylhexan-1-ol | −0.127 | −0.339 | 0.551 | 3.397 | 0.722 | 0.963 |
| Cyclopentanol | −0.151 | −0.314 | 0.693 | 3.549 | 0.914 | 0.956 |
| Benzyl alcohol | −0.305 | −0.344 | 1.533 | 2.773 | 1.372 | 0.860 |
| Trifluoroethanol | −0.092 | −0.547 | 1.339 | 2.213 | 3.807 | 0.645 |
| Ethylene glycol | −0.887 | 0.132 | 1.657 | 4.457 | 2.355 | 0.565 |
| 1,2-Propanediol | −0.607 | 0.239 | 1.008 | 4.278 | 1.755 | 0.706 |
| 2-Methoxyethanol | −0.141 | −0.265 | 1.810 | 3.641 | 0.590 | 0.790 |
| 2-Ethoxyethanol | −0.064 | 0.257 | 1.452 | 3.672 | 0.662 | 0.843 |
| 2-Propoxyethanol | −0.091 | −0.288 | 1.265 | 3.566 | 0.390 | 0.902 |
| 2-Butoxyethanol | −0.109 | −0.304 | 1.126 | 3.407 | 0.660 | 0.914 |
| 2-Isopropoxyethanol | −0.045 | −0.264 | 1.296 | 3.646 | 0.352 | 0.880 |
| 3-Methoxybutan-1-ol | −0.252 | −0.364 | 1.182 | 3.622 | 0.594 | 0.934 |
| 1-tert-Butoxypropan-2-ol | −0.043 | −0.343 | 1.197 | 3.525 | 0.216 | 0.904 |
| Diethylene glycol | −0.496 | 0.167 | 1.961 | 3.831 | 1.057 | 0.617 |
| Triethylene glycol | −0.469 | 0.235 | 2.079 | 3.824 | 0.775 | 0.626 |
| Diethyl ether | 0.288 | −0.347 | 0.775 | 2.985 | 0.000 | 0.973 |
| Diisopropyl ether | 0.139 | −0.473 | 0.610 | 2.568 | 0.000 | 1.016 |
| Dibutyl ether | 0.165 | −0.421 | 0.760 | 2.102 | −0.664 | 1.002 |
| Methyl tert-butyl ether | 0.278 | −0.489 | 0.801 | 2.495 | 0.000 | 0.993 |
| Tetrahydrofuran | 0.189 | −0.347 | 1.238 | 3.289 | 0.000 | 0.982 |
| Dioxane | −0.034 | −0.354 | 1.674 | 3.021 | 0.000 | 0.919 |
| Methyl acetate | 0.134 | −0.477 | 1.749 | 2.678 | 0.000 | 0.876 |
| Ethyl acetate | 0.182 | −0.352 | 1.316 | 2.891 | 0.000 | 0.916 |
| Propyl acetate | 0.165 | −0.383 | 1.264 | 2.757 | 0.000 | 0.935 |
| Isopropyl acetate | 0.233 | −0.495 | 1.324 | 2.550 | 0.000 | 0.928 |
| Butyl acetate | 0.147 | −0.414 | 1.212 | 2.623 | 0.000 | 0.954 |
| Pentyl acetate | 0.154 | −0.424 | 1.172 | 2.506 | 0.000 | 0.962 |
| Methyl butyrate | 0.201 | −0.502 | 1.290 | 2.469 | 0.000 | 0.958 |
| Dimethyl carbonate | 0.000 | −0.616 | 1.905 | 2.123 | 0.000 | 0.892 d |
| Diethyl carbonate | 0.092 | −0.598 | 1.527 | 1.942 | 0.000 | 0.948 |
| Propylene carbonate | −0.356 | −0.413 | 2.587 | 2.207 | 0.455 | 0.719 |
| Propanone | 0.127 | −0.387 | 1.733 | 3.060 | 0.000 | 0.866 |
| Butanone | 0.124 | −0.429 | 1.601 | 2.843 | 0.000 | 0.916 |
| Cyclopentanone | −0.072 | −0.414 | 1.678 | 2.843 | 0.000 | 0.954 |
| Cyclohexanone | −0.052 | −0.445 | 1.716 | 2.758 | 0.000 | 0.948 |
| N,N-Dimethylformamide | −0.391 | −0.869 | 2.107 | 3.774 | 0.000 | 1.011 |
| N,N-Dimethylacetamide | −0.308 | −0.736 | 1.802 | 4.361 | 0.000 | 1.028 |
| N-Methylpyrrolidinone | −0.128 | −0.029 | 2.217 | 4.429 | 0.000 | 0.777 |
| Formamide | −0.800 | 0.310 | 2.292 | 4.130 | 1.933 | 0.442 |
| Acetonitrile | −0.007 | −0.595 | 2.461 | 2.085 | 0.418 | 0.738 |
| Propanenitrile | 0.101 | −0.433 | 1.981 | 2.509 | 0.399 | 0.801 |
| Butanenitrile | 0.154 | −0.262 | 1.694 | 2.214 | 0.349 | 0.834 |
| Nitromethane | −0.340 | −0.297 | 2.689 | 2.193 | 0.514 | 0.728 |
| Dimethyl sulfoxide | −0.556 | −0.223 | 2.903 | 5.037 | 0.000 | 0.719 |
| Pyridine | −0.129 | −0.571 | 1.963 | 4.486 | 0.000 | 0.948 |
| 2-Pyrrolidone | −0.490 | 0.087 | 2.275 | 4.997 | 0.262 | 0.771 |
| Aniline | −0.394 | −0.362 | 1.895 | 2.421 | 1.334 | 0.842 |
| Methoxybenzene | −0.054 | −0.376 | 1.558 | 1.280 | 0.000 | 0.957 |
| Acetophenone | −0.062 | −0.178 | 1.677 | 2.167 | 0.340 | 0.863 |
| Acetic acid | −0.070 | −0.366 | 1.300 | 2.736 | 2.117 | 0.796 |
| Tributyl phosphate | −0.036 | −0.285 | 1.385 | 3.735 | 0.000 | 0.899 |
| 95 vol% Methanol b | 0.013 | −0.300 | 1.517 | 3.811 | 1.463 | 0.706 |
| 90 vol% Methanol | −0.050 | −0.200 | 1.615 | 3.827 | 1.637 | 0.647 |
| 80 vol% Methanol | −0.238 | −0.086 | 1.790 | 3.895 | 1.931 | 0.554 |
| 70 vol% Methanol | −0.378 | 0.040 | 1.941 | 3.943 | 2.365 | 0.440 |
| 60 vol% Methanol | −0.530 | 0.149 | 2.081 | 3.995 | 2.778 | 0.330 |
| 50 vol% Methanol | −0.694 | 0.236 | 2.226 | 4.051 | 3.157 | 0.224 |
| 40 vol% Methanol | −0.811 | 0.311 | 2.370 | 4.053 | 3.575 | 0.117 |
| 30 vol% Methanol | −0.927 | 0.338 | 2.431 | 4.047 | 3.869 | 0.053 |
| 20 vol% Methanol | −1.035 | 0.389 | 2.655 | 4.004 | 4.291 | −0.064 |
| 10 vol% Methanol | −1.107 | 0.500 | 2.722 | 3.974 | 4.658 | −0.151 |
| 96 vol% Ethanol c | −0.032 | −0.181 | 0.980 | 3.940 | 1.379 | 0.802 |
| 95 vol% Ethanol | −0.040 | −0.200 | 1.024 | 3.950 | 1.400 | 0.795 |
| 90 vol% Ethanol | −0.084 | −0.280 | 1.180 | 3.959 | 1.474 | 0.757 |
| 80 vol% Ethanol | −0.253 | −0.278 | 1.400 | 4.000 | 1.775 | 0.715 |
| 70 vol% Ethanol | −0.438 | −0.255 | 1.548 | 4.040 | 2.074 | 0.659 |
| 60 vol% Ethanol | −0.631 | −0.186 | 1.646 | 4.054 | 2.355 | 0.584 |
| 50 vol% Ethanol | −0.851 | −0.063 | 1.806 | 4.050 | 2.745 | 0.479 |
| 40 vol% Ethanol | −1.074 | 0.075 | 2.076 | 4.020 | 3.196 | 0.347 |
| 30 vol% Ethanol | −1.258 | 0.194 | 2.300 | 4.000 | 3.713 | 0.206 |
| 20 vol% Ethanol | −1.364 | 0.383 | 2.385 | 3.950 | 4.280 | 0.065 |
| 10 vol% Ethanol | −1.447 | 0.446 | 2.536 | 3.905 | 4.750 | −0.052 |
| Organic Solvent | Log CS,organic; Equation (3) | Log CS,organic; Equation (4) |
|---|---|---|
| Methanol | −3.466 | −3.522 |
| Ethanol | −3.433 | −3.468 |
| Propan-1-ol | −3.578 | −3.582 |
| Butan-1-ol | −3.673 | −3.672 |
| Pentan-1-ol | −3.668 | −3.677 |
| Hexan-1-ol | −3.697 | −3.702 |
| Heptan-1-ol | −3.685 | −3.684 |
| Octan-1-ol | −3.640 | −3.743 |
| Decan-1-ol | −3.770 | −3.712 |
| Propan-2-ol | −3.633 | −3.639 |
| 2-Methylpropan-1-ol | −3.920 | −3.954 |
| Butan-2-ol | −3.727 | −3.734 |
| 2-Methylpropan-2-ol | −3.771 | −3.704 |
| 2-Methylbutan-2-ol | −3.516 | −3.513 |
| 3-Methylbutan-1-ol | −3.871 | −3.848 |
| 4-Methylpentan-2-ol | −3.964 | −3.987 |
| Pentan-2-ol | −3.766 | −3.750 |
| 2-Ethylhexan-1-ol | −3.983 | −3.916 |
| Cyclopentanol | −3.558 | −3.418 |
| 1,2-Propylene glycol | −3.580 | −3.604 |
| Ethylene glycol | −3.748 | −3.779 |
| 2-Methoxyethanol | −2.816 | −2.803 |
| 2-Ethoxyethanol | −2.809 | −2.840 |
| 2-Propoxyethanol | −2.940 | −2.939 |
| 2-Isopropoxyethanol | −2.950 | −2.975 |
| 2-Butoxyethanol | −3.057 | −3.126 |
| Methyl acetate | −3.311 | −3.285 |
| Ethyl acetate | −3.282 | −3.291 |
| Propyl acetate | −3.494 | −3.476 |
| Isopropyl acetate | −3.608 | −3.682 |
| Butyl acetate | −3.437 | −3.481 |
| Pentyl acetate | −3.568 | −3.585 |
| Dimethyl carbonate | −3.622 | −3.644 |
| Diethyl carbonate | −3.839 | −3.856 |
| Acetone | −2.945 | −2.939 |
| Butanone | −2.924 | −2.940 |
| Acetonitrile | −3.998 | −3.867 |
| Propanenitrile | −3.688 | −3.766 |
| Butanenitrile | −3.510 | −3.490 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinha, S.; Yang, C.; Wu, E.; Acree, W.E., Jr. Abraham Solvation Parameter Model: Examination of Possible Intramolecular Hydrogen-Bonding Using Calculated Solute Descriptors. Liquids 2022, 2, 131-146. https://doi.org/10.3390/liquids2030009
Sinha S, Yang C, Wu E, Acree WE Jr. Abraham Solvation Parameter Model: Examination of Possible Intramolecular Hydrogen-Bonding Using Calculated Solute Descriptors. Liquids. 2022; 2(3):131-146. https://doi.org/10.3390/liquids2030009
Chicago/Turabian StyleSinha, Sneha, Chelsea Yang, Emily Wu, and William E. Acree, Jr. 2022. "Abraham Solvation Parameter Model: Examination of Possible Intramolecular Hydrogen-Bonding Using Calculated Solute Descriptors" Liquids 2, no. 3: 131-146. https://doi.org/10.3390/liquids2030009
APA StyleSinha, S., Yang, C., Wu, E., & Acree, W. E., Jr. (2022). Abraham Solvation Parameter Model: Examination of Possible Intramolecular Hydrogen-Bonding Using Calculated Solute Descriptors. Liquids, 2(3), 131-146. https://doi.org/10.3390/liquids2030009