
Fraction of Genome Altered, Age, Microsatellite Instability Score, Tumor Mutational Burden, Cancer Type, Metastasis Status, and Choice of Cancer Therapy Predict Overall Survival in Multiple Machine Learning Models


{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Methods
2.1. Patient Data
2.2. Statistics
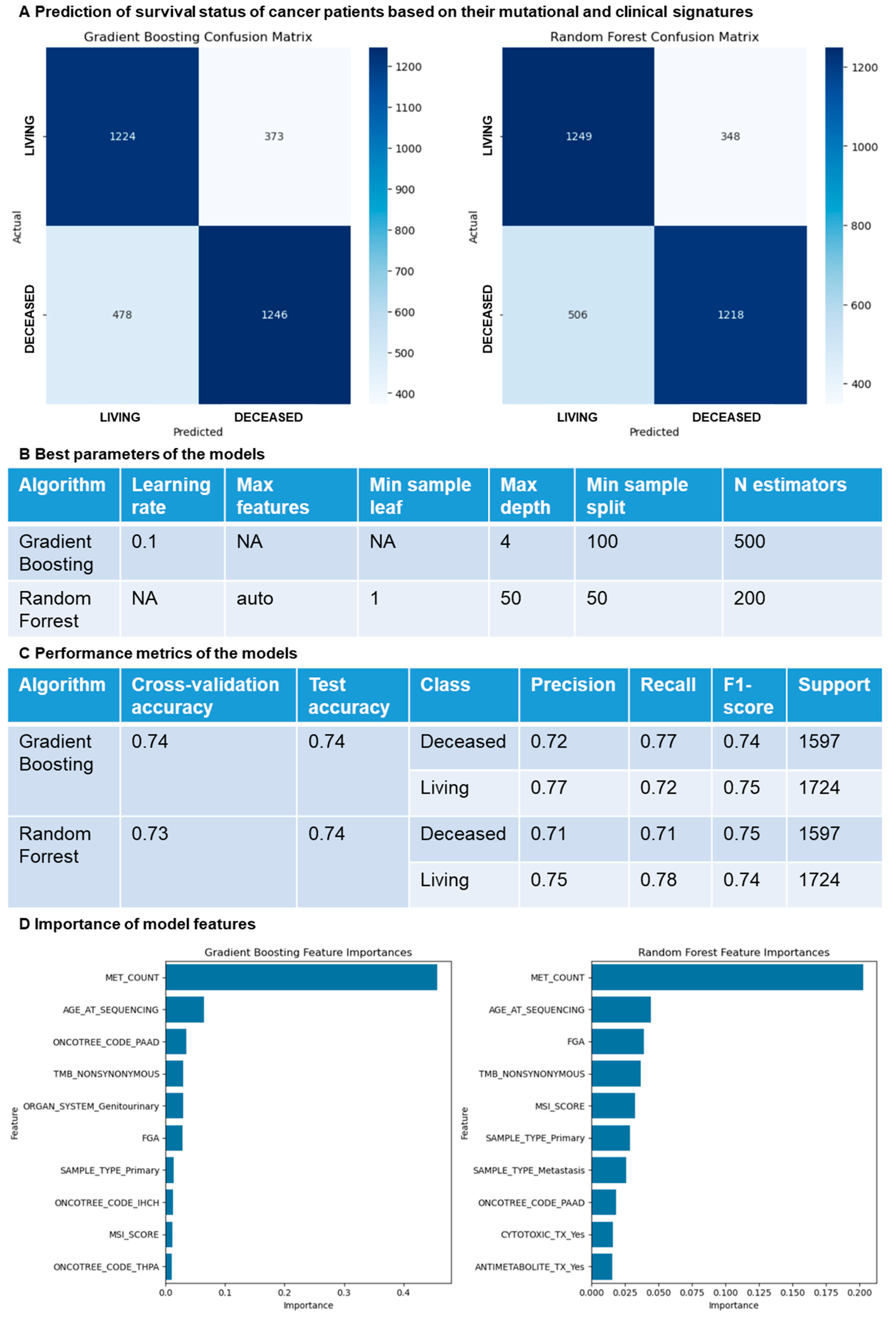
2.3. Machine Learning Models
3. Results
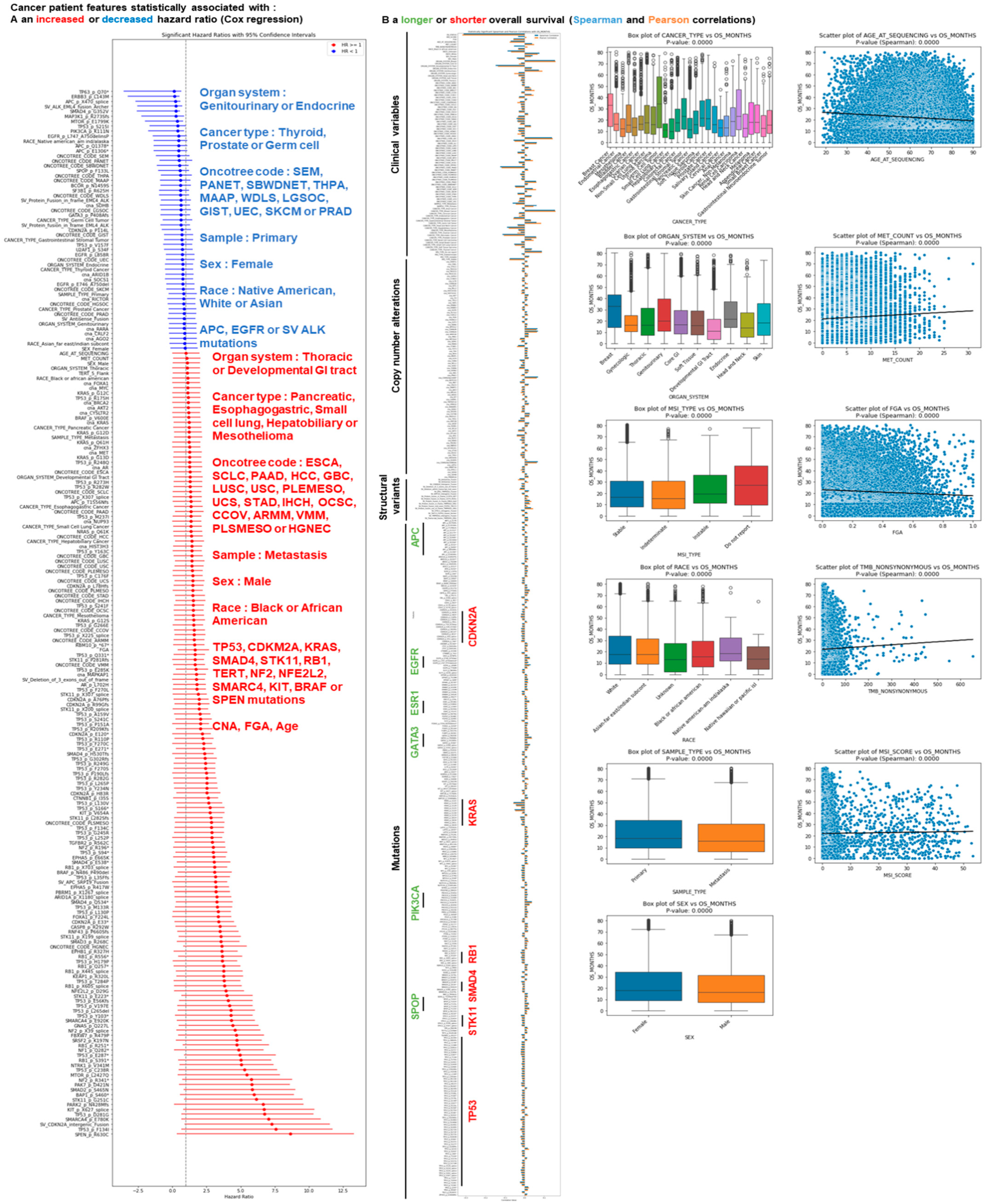
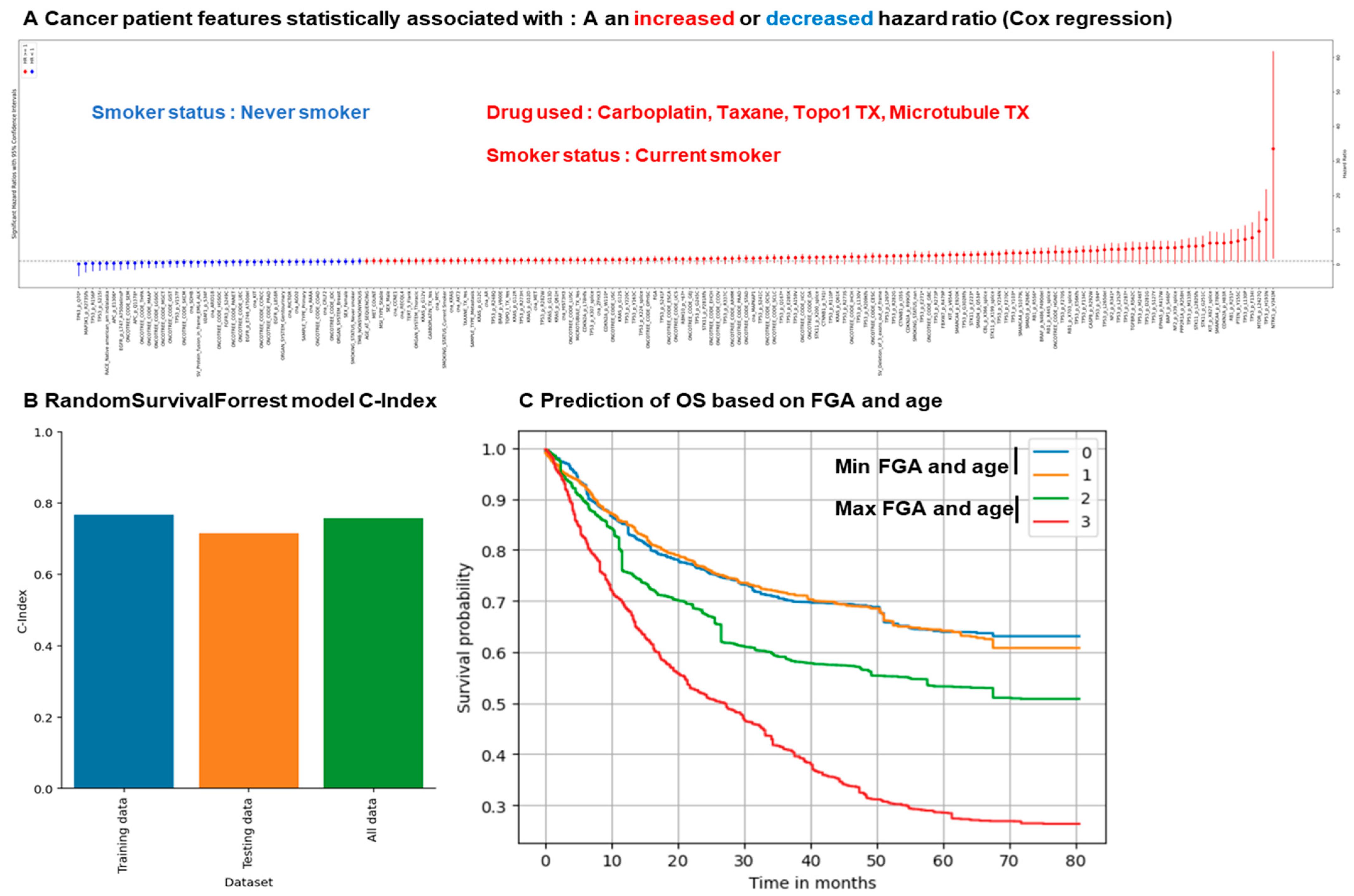
3.1. Identification of Cancer Patient Mutations Associated with a Difference in Survival
3.2. Identification of Cancer Patient Clinical Features Associated with a Difference in Survival
3.3. Identification of Cancer Types Associated with a Difference in Survival
3.4. Random Survival Forest Approach Predicts Overall Survival Based on Clinical and Mutational Data
4. Discussion
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mestrallet, G. Predicting Immunotherapy Outcomes in Glioblastoma Patients through Machine Learning. Cancers 2024, 16, 408. [Google Scholar] [CrossRef]
- Mestrallet, G. Prediction of Glioma Resistance to Immune Checkpoint Inhibitors Based on Mutation Profile. Neuroglia 2024, 5, 145–154. [Google Scholar] [CrossRef]
- Mestrallet, G. Predicting Resistance to Immunotherapy in Melanoma, Glioblastoma, Renal, Stomach and Bladder Cancers by Machine Learning on Immune Profiles. Onco 2024, 4, 192–206. [Google Scholar] [CrossRef]
- Sung, J.-Y.; Cheong, J.-H. Machine Learning Predictor of Immune Checkpoint Blockade Response in Gastric Cancer. Cancers 2022, 14, 3191. [Google Scholar] [CrossRef] [PubMed]
- Tonneau, M.; Phan, K.; Mane Tonneau, M.; Phan, K.; Manem, V.S.K.; Low-Kam, C.; Dutil, F.; Kazandjian, S.; Vanderweyen, D.; Panasci, J.; et al. Generalization optimizing machine learning to improve CT scan radiomics and assess immune checkpoint inhibitors’ response in non-small cell lung cancer: A multicenter cohort study. Front. Oncol. 2023, 13, 1196414. [Google Scholar] [CrossRef] [PubMed]
- Wiesweg, M.; Mairinger, F.; Reis, H.; Goetz, M.; Walter, R.F.H.; Hager, T.; Metzenmacher, M.; Eberhardt, W.E.E.; McCutcheon, A.; Köster, J.; et al. Machine learning-based predictors for immune checkpoint inhibitor therapy of non-small-cell lung cancer. Ann. Oncol. 2019, 30, 655–657. [Google Scholar] [CrossRef] [PubMed]
- Jee, J.; Fong, C.; Pichotta, K.; Tran, T.N.; Luthra, A.; Waters, M.; Fu, C.; Altoe, M.; Liu, S.Y.; Maron, S.B.; et al. Automated real-world data integration improves cancer outcome prediction. Nature 2024, 636, 728–736. [Google Scholar] [CrossRef]
- Mestrallet, G. Leveraging Tumor Mutation Profiles to Forecast Immune Checkpoint Blockade Resistance in Melanoma, Lung, Head and Neck, Bladder and Renal Cancers. Onco 2024, 4, 439–457. [Google Scholar] [CrossRef]
- Mestrallet, G.; Brown, M.; Bozkus, C.C.; Bhardwaj, N. Immune escape and resistance to immunotherapy in mismatch repair deficient tumors. Front. Immunol. 2023, 14, 1210164. [Google Scholar] [CrossRef] [PubMed]
- Gillies, R.J.; Schabath, M.B. Radiomics Improves Cancer Screening and Early Detection. Cancer Epidemiol. Biomark. Prev. Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2020, 29, 2556–2567. [Google Scholar] [CrossRef]
- Zhu, Y.; Bi, D.; Saunders, M.; Ji, Y. Prediction of chronic kidney disease progression using recurrent neural network and electronic health records. Sci. Rep. 2023, 13, 22091. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Zanini, J.; Pietzner, M.; Koprulu, M.; Wheeler, E.; Kerrison, N.D.; Wareham, N.J.; Langenberg, C. Proteomic prediction of diverse incident diseases: A machine learning-guided biomarker discovery study using data from a prospective cohort study. Lancet Digit. Health 2024, 6, e470-9. [Google Scholar] [CrossRef]
- Feretzakis, G.; Sakagianni, A.; Loupelis, E.; Kalles, D.; Skarmoutsou, N.; Martsoukou, M.; Christopoulos, C.; Lada, M.; Petropoulou, S.; Velentza, A.; et al. Machine Learning for Antibiotic Resistance Prediction: A Prototype Using Off-the-Shelf Techniques and Entry-Level Data to Guide Empiric Antimicrobial Therapy. Healthc. Inform. Res. 2021, 27, 214–221. [Google Scholar] [CrossRef]
- Nguyen, B.; Fong, C.; Luthra, A.; Smith, S.A.; DiNatale, R.G.; Nandakumar, S.; Walch, H.; Chatila, W.K.; Madupuri, R.; Kundra, R.; et al. Genomic characterization of metastatic patterns from prospective clinical sequencing of 25,000 patients. Cell 2022, 185, 563–575.e11. [Google Scholar] [CrossRef]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020, 52, 1219–1226. [Google Scholar] [CrossRef]
- Zhu, G.; Pei, L.; Xia, H.; Tang, Q.; Bi, F. Role of oncogenic KRAS in the prognosis, diagnosis and treatment of colorectal cancer. Mol. Cancer 2021, 20, 143. [Google Scholar] [CrossRef]
- Robles, A.I.; Harris, C.C. Clinical Outcomes and Correlates of TP53 Mutations and Cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001016. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Lee, C.-H.; Shoushtari, A.N.; Hellmann, M.D.; Shen, R.; Janjigian, Y.Y.; Barron, D.A.; Zehir, A.; Jordan, E.J.; Omuro, A.; et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat. Genet. 2019, 51, 202–206. [Google Scholar] [CrossRef]
- Cheng, S.; Han, Z.; Dai, D.; Li, F.; Zhang, X.; Lu, M.; Lu, Z.; Wang, X.; Zhou, J.; Li, J.; et al. Multi-omics of the gut microbial ecosystem in patients with microsatellite-instability-high gastrointestinal cancer resistant to immunotherapy. Cell Rep. Med. 2024, 5, 101355. [Google Scholar] [CrossRef] [PubMed]
- Mestrallet, G. Software development for severe burn diagnosis and autologous skin substitute production. Comput. Methods Programs Biomed. Update 2022, 2, 100069. [Google Scholar] [CrossRef]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Cutler, D.R.; Edwards, T.C., Jr.; Beard, K.H.; Cutler, A.; Hess, K.T.; Gibson, J.; Lawler, J.J. Random Forests for Classification in Ecology. Ecology 2007, 88, 2783–2792. [Google Scholar] [CrossRef]
- Prasad, A.M.; Iverson, L.R.; Liaw, A. Newer Classification and Regression Tree Techniques: Bagging and Random Forests for Ecological Prediction. Ecosystems 2006, 9, 181–199. [Google Scholar] [CrossRef]
- Pal, M.; Mather, P.M. An assessment of the effectiveness of decision tree methods for land cover classification. Remote Sens. Environ. 2003, 86, 554–565. [Google Scholar] [CrossRef]
- Rodriguez-Galiano, V.F.; Ghimire, B.; Rogan, J.; Chica-Olmo, M.; Rigol-Sanchez, J.P. An assessment of the effectiveness of a random forest classifier for land-cover classification. ISPRS J. Photogramm. Remote Sens. 2012, 67, 93–104. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mestrallet, G. Fraction of Genome Altered, Age, Microsatellite Instability Score, Tumor Mutational Burden, Cancer Type, Metastasis Status, and Choice of Cancer Therapy Predict Overall Survival in Multiple Machine Learning Models. Onco 2025, 5, 8. https://doi.org/10.3390/onco5010008
Mestrallet G. Fraction of Genome Altered, Age, Microsatellite Instability Score, Tumor Mutational Burden, Cancer Type, Metastasis Status, and Choice of Cancer Therapy Predict Overall Survival in Multiple Machine Learning Models. Onco. 2025; 5(1):8. https://doi.org/10.3390/onco5010008
Chicago/Turabian StyleMestrallet, Guillaume. 2025. "Fraction of Genome Altered, Age, Microsatellite Instability Score, Tumor Mutational Burden, Cancer Type, Metastasis Status, and Choice of Cancer Therapy Predict Overall Survival in Multiple Machine Learning Models" Onco 5, no. 1: 8. https://doi.org/10.3390/onco5010008
APA StyleMestrallet, G. (2025). Fraction of Genome Altered, Age, Microsatellite Instability Score, Tumor Mutational Burden, Cancer Type, Metastasis Status, and Choice of Cancer Therapy Predict Overall Survival in Multiple Machine Learning Models. Onco, 5(1), 8. https://doi.org/10.3390/onco5010008