

ERBB1/EGFR and JAK3 Tyrosine Kinases as Potential Therapeutic Targets in High-Risk Multiple Myeloma



Abstract
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Normalization for Multiple Myeloma (MM) Samples
2.2. Statistical Methods for Differential Gene Expression
2.3. Hierarchical Clustering Analysis
2.4. Overall Survival and Progression-Free Survival Analysis
2.5. Processing of the Multiple Myeloma Research Foundation (MMRF)-CoMMpass RNAseq Dataset
2.6. Overall Survival Analysis and Cox Proportional Hazards Model of the MMRF CoMMpass Study
3. Results
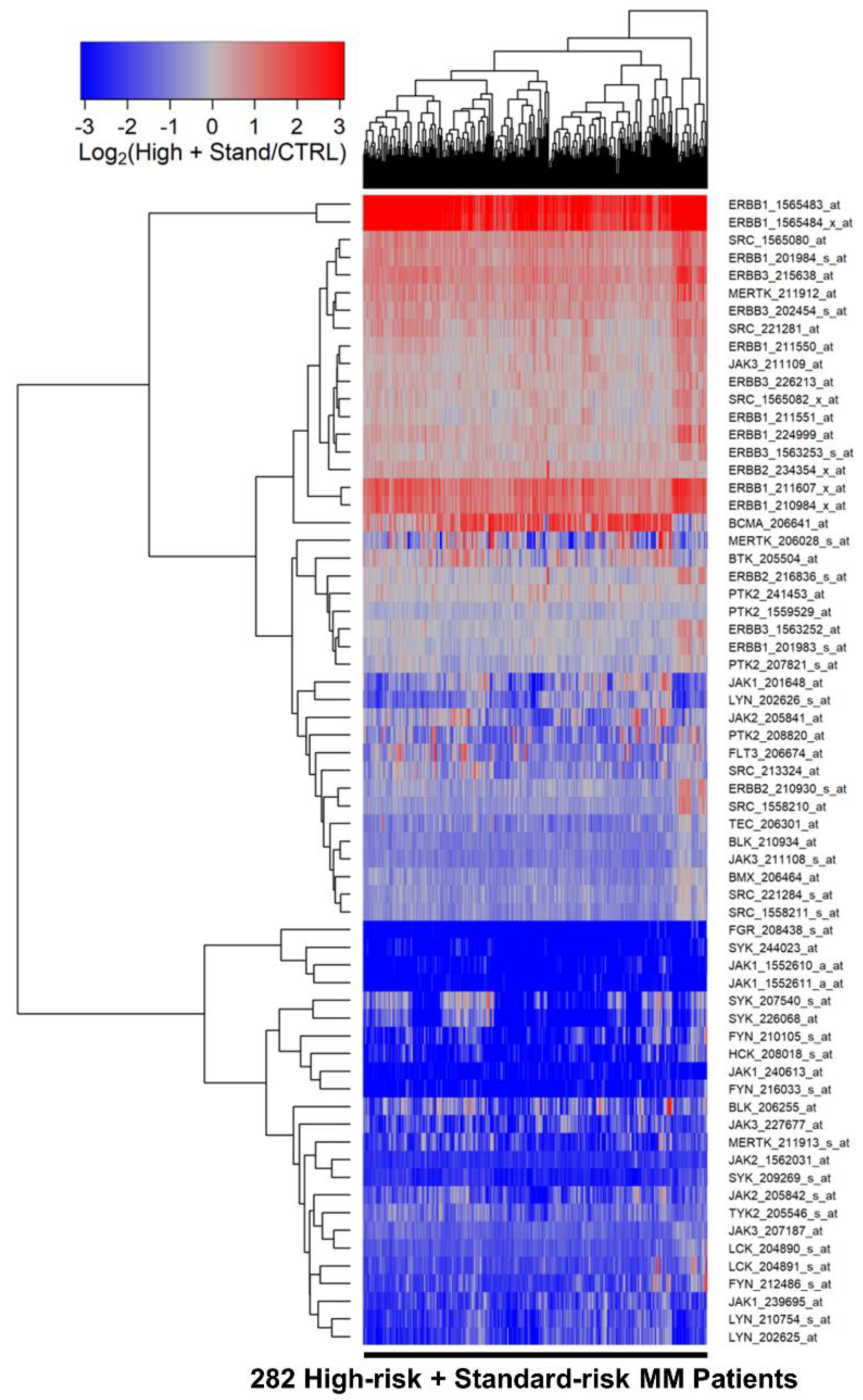
3.1. Amplified Expression of Selective Tyrosine Kinase Genes in Malignant Plasma Cells from MM Patients
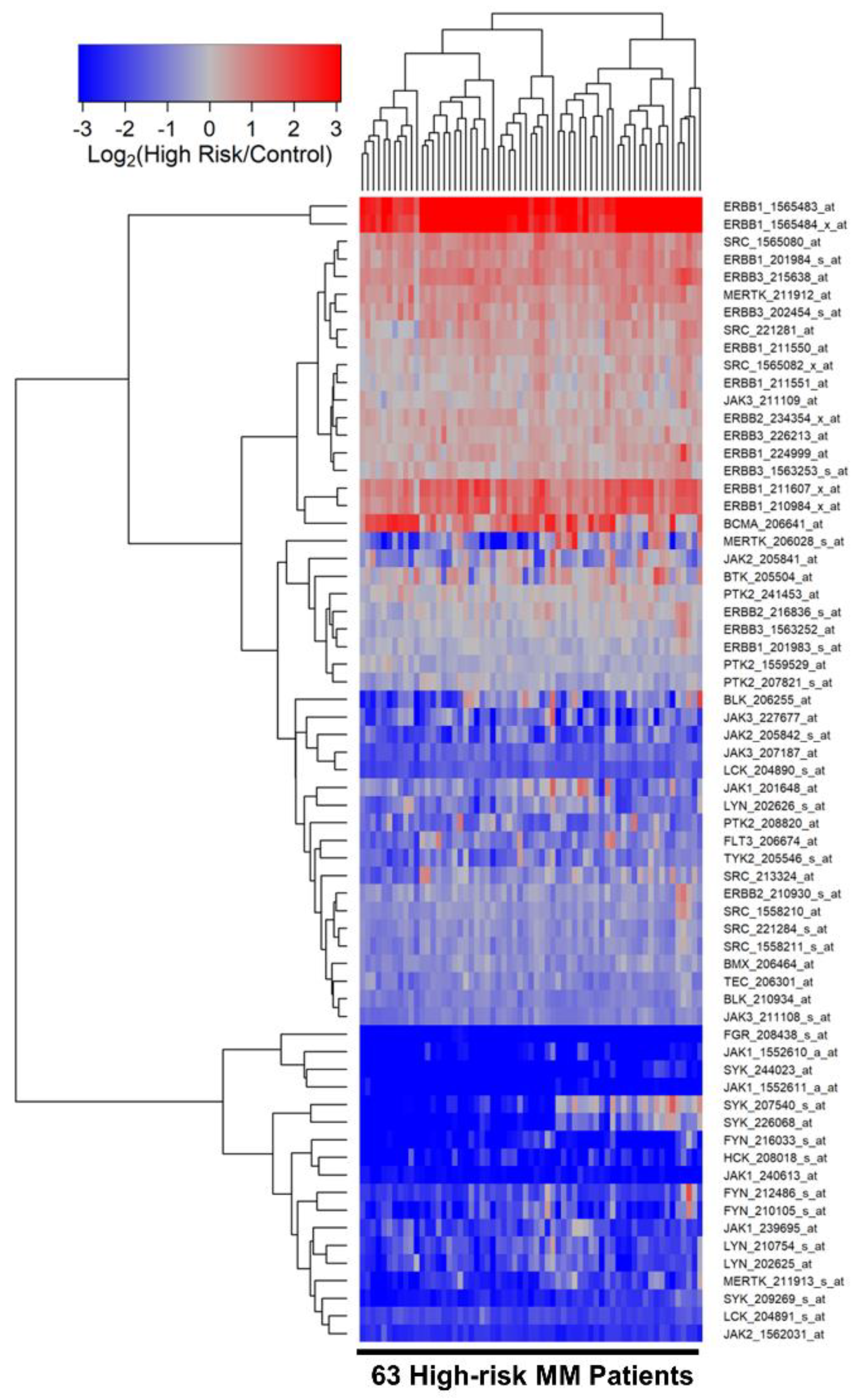
3.2. Differentially Amplified Expression of ERBB1/EGFR and JAK3 Genes in Malignant Plasma Cells from High-Risk MM Patients
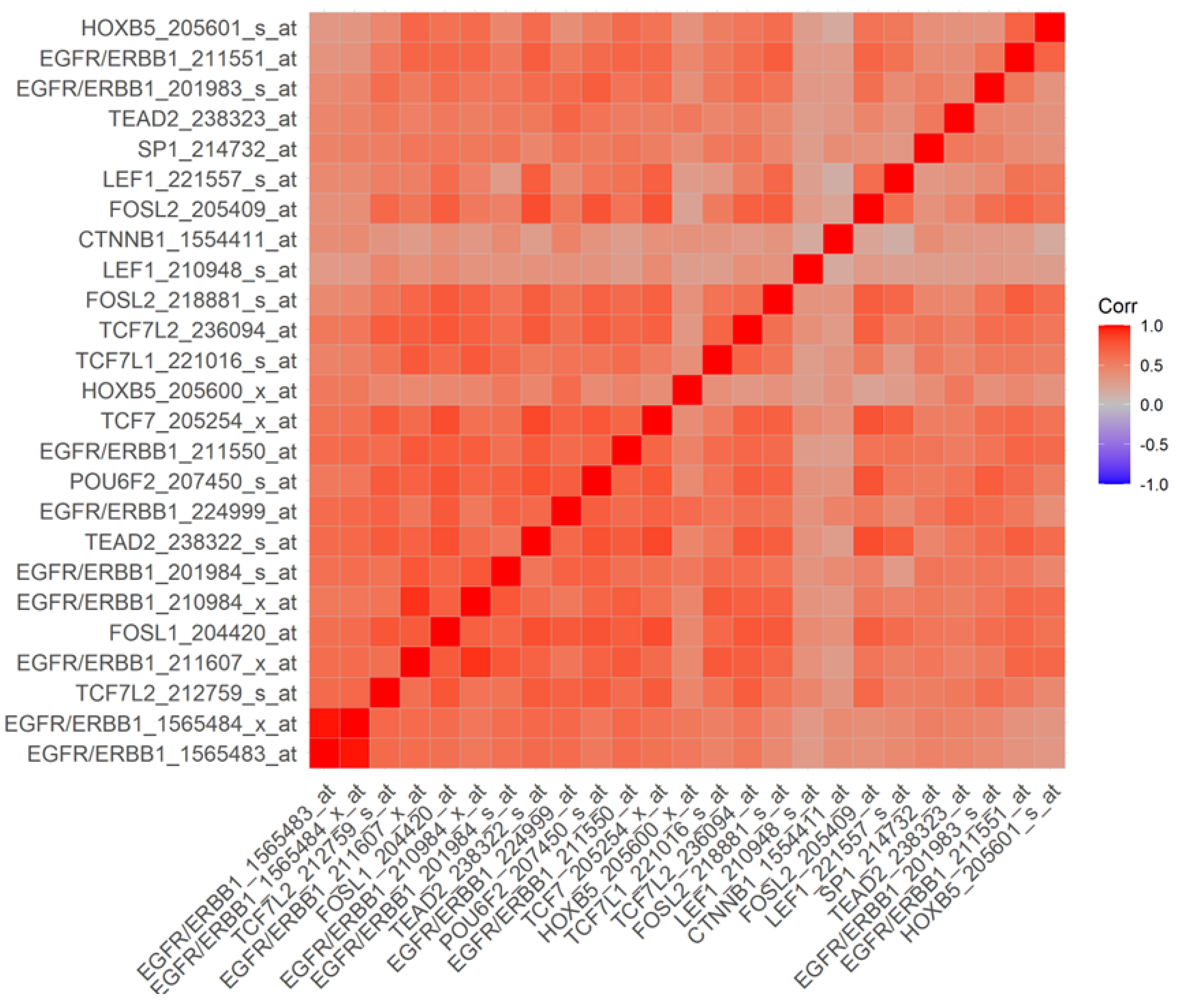
3.3. Overexpression of EGFR/ERBB1 in MM Cells Is Associated with Augmented Expression of Transcription Factors Binding to Multiple ERBB1 Gene Promoter Sites
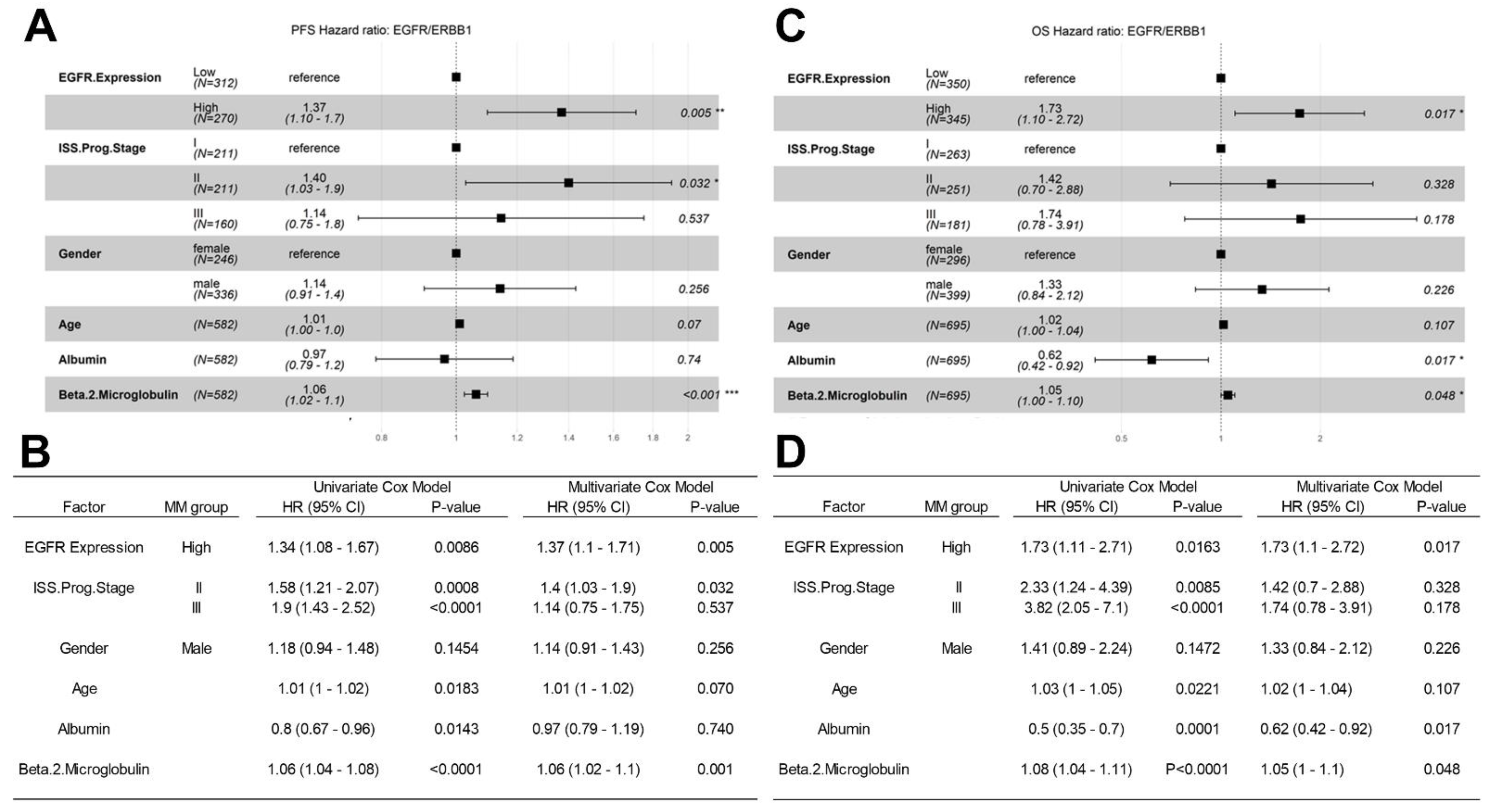
3.4. Amplified Expression of ERBB1/EGFR Is Associated with Poor PFS and OS in Newly Diagnosed MM
3.5. Analyses Using the MMRF-CoMMpass RNAseq Dataset for Validation of the Microarray-Based Findings from the HOVON65/GMMG-HD4 Trial Regarding the Prognostic Effect of EGFR/ERBB1 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Uckun, F.M. Cancer drug resistance in multiple myeloma. Cancer Drug Resist. 2022, 5, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Qazi, S.; Demirer, T.; Champlin, R.E. Contemporary patient-tailored treatment strategies against high risk and re-lapsed or refractory multiple myeloma. EBioMedicine 2019, 39, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, U.H.; Cornell, R.F.; Lakshman, A.; Gahvari, Z.J.; McGehee, E.; Jagosky, M.H.; Gupta, R.; Varnado, W.; Fiala, M.A.; Chhabra, S.; et al. Outcomes of patients with multiple myeloma refractory to CD38-targeted monoclonal antibody therapy. Leukemia 2019, 33, 2266–2275. [Google Scholar] [CrossRef] [PubMed]
- Moreau, P.; Kumar, S.K.; San Miguel, J.; Davies, F.; Zamagni, E.; Bahlis, N.; Ludwig, H.; Mikhael, J.; Terpos, E.; Schjesvold, F.; et al. Treatment of relapsed and refractory multiple myeloma: Recommendations from the International Myeloma Working Group. Lancet Oncol. 2021, 22, e105–e118. [Google Scholar] [CrossRef]
- Uckun, F.M. Dual Targeting of Multiple Myeloma Stem Cells and Myeloid-Derived Suppressor Cells for Treatment of Chemotherapy-Resistant Multiple Myeloma. Front. Oncol. 2021, 11, 760382. [Google Scholar] [CrossRef]
- Uckun, F. Overcoming the Immunosuppressive Tumor Microenvironment in Multiple Myeloma. Cancers 2021, 13, 2018. [Google Scholar] [CrossRef]
- Minnie, S.A.; Hill, G. Immunotherapy of multiple myeloma. J. Clin. Investig. 2020, 130, 1565–1575. [Google Scholar] [CrossRef]
- Braunstein, M.; Weltz, J.; Davies, F. A new decade: Novel immunotherapies on the horizon for relapsed/refractory multiple myeloma. Expert Rev. Hematol. 2021, 14, 377–389. [Google Scholar] [CrossRef]
- Gay, F.; Engelhardt, M.; Terpos, E.; Wäsch, R.; Giaccone, L.; Auner, H.W. From transplant to novel cellular therapies in multiple myeloma: European Myeloma Network guidelines and future perspectives. Haematologica 2018, 103, 197–211. [Google Scholar] [CrossRef]
- Abramson, H.N. The multiple myeloma drug pipeline 2018: A review of small molecules and their therapeutic targets. Clin. Lymphoma Myeloma Leuk. 2018, 18, 611–627. [Google Scholar] [CrossRef]
- Mohty, M.; Terpos, E.; Mateos, M.V.; Cavo, M.; Lejniece, S.; Beksac, M. Multiple myeloma treatment in real-world clinical prac-tice: Results of a prospective, multinational, noninterventional study. Clin. Lymphoma Myeloma Leuk. 2018, 18, e401–e409. [Google Scholar] [CrossRef] [PubMed]
- Cavo, M.; Terpos, E.; Bargay, J.; Einsele, H.; Cavet, J.; Greil, R. The multiple myeloma treatment landscape: International guide-line recommendations and clinical practice in Europe. Expert Rev. Hematol. 2018, 11, 219–237. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.S.; Karasic, T.B.; Demichele, A.; Vaughn, D.J.; O’Hara, M.; Perini, R. Palbociclib (PD0332991)-a selective and potent cy-clin-dependent kinase inhibitor: A review of pharmacodynamics and clinical development. JAMA Oncol. 2016, 2, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Heuck, C.J.; Jethava, Y.; Khan, R.; van Rhee, F.; Zangari, M.; Chavan, S. Inhibiting MEK in MAPK pathway-activated myeloma. Leukemia 2016, 30, 976–980. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagarón, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef]
- Scheijen, B.; Griffin, J.D. Tyrosine kinase oncogenes in normal hematopoiesis and hematological disease. Oncogene 2002, 21, 3314–3333. [Google Scholar] [CrossRef]
- Bhanumathy, K.K.; Balagopal, A.; Vizeacoumar, F.S.; Vizeacoumar, F.J.; Freywald, A.; Giambra, V. Protein Tyrosine Kinases: Their Roles and Their Targeting in Leukemia. Cancers 2021, 13, 184. [Google Scholar] [CrossRef]
- Kuiper, R.; Broyl, A.; De Knegt, Y.; Van Vliet, M.H.; Van Beers, E.H.; Van Der Holt, B.; El Jarari, L.; Mulligan, G.; Gregory, W.; Morgan, G.; et al. A gene expression signature for high-risk multiple myeloma. Leukemia 2012, 26, 2406–2413. [Google Scholar] [CrossRef]
- van Beers, E.H.; Huigh, D.; Bosman, L.; de Best, L.; Kuiper, R.; Spaan, M.; van Duin, M.; Sonneveld, P.; Dumee, B.; van Vliet, M.H. Analytical Validation of SKY92 for the Identification of High-Risk Multiple Myeloma. J. Mol. Diagn. 2020, 23, 120–129. [Google Scholar] [CrossRef]
- van Beers, E.H.; van Vliet, M.H.; Kuiper, R.; de Best, L.; Anderson, K.C.; Chari, A.; Jagannath, S.; Jakubowiak, A.; Kumar, S.K.; Levy, J.B.; et al. Prognostic Validation of SKY92 and Its Combination with ISS in an Independent Cohort of Patients with Multiple Myeloma. Clin. Lymphoma Myeloma Leuk. 2017, 17, 555–562. [Google Scholar] [CrossRef]
- Kuiper, R.; Zweegman, S.; van Duin, M.; van Vliet, M.H.; van Beers, E.H.; Dumee, B.; Vermeulen, M.; Koenders, J.; van der Holt, B.; Visser-Wisselaar, H.; et al. Prognostic and predictive performance of R-ISS with SKY92 in older patients with multiple myeloma: The HOVON-87/NMSG-18 trial. Blood Adv. 2020, 4, 6298–6309. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Qazi, S. Identification and targeting of CD22ΔE12 as a molecular RNAi target to overcome drug resistance in high-risk B-lineage leukemias and lymphomas. Cancer Drug Resist. 2018, 1, 30–47. [Google Scholar] [CrossRef] [PubMed]
- Uckun, F.M.; Myers, D.E.; Qazi, S.; Ozer, Z.; Rose, R.; D’Cruz, O.J.; Ma, H. Recombinant human CD19L-sTRAIL effectively targets B cell precursor acute lymphoblastic leukemia. J. Clin. Investig. 2015, 125, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Yekutieli, D. The control of the false discovery rate in multiple testing under dependency. Ann. Stat. 2001, 29, 1165–1188. [Google Scholar] [CrossRef]
- Uckun, F.M.; Mitchell, L.G.; Qazi, S.; Liu, Y.; Zheng, N.; Myers, D.E.; Song, Z.; Ma, H.; Cheng, J. Development of Polypeptide-based Nanoparticles for Non-viral Delivery of CD22 RNA Trans-splicing Molecule as a New Precision Medicine Candidate Against B-lineage ALL. EBioMedicine 2015, 2, 649–659. [Google Scholar] [CrossRef][Green Version]
- Uckun, F.M.; Goodman, P.; Ma, H.; Dibirdik, I.; Qazi, S. CD22 EXON 12 deletion as a pathogenic mechanism of human B-precursor leukemia. Proc. Natl. Acad. Sci. USA 2010, 107, 16852–16857. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Tran, T.H.; Langlois, S.; Meloche, C.; Caron, M.; Saint-Onge, P.; Rouette, A.; Bataille, A.R.; Jimenez-Cortes, C.; Sontag, T.; Bittencourt, H.; et al. Whole-transcriptome analysis in acute lymphoblastic leukemia: A report from the DFCI ALL Consortium Protocol 16-001. Blood Adv. 2022, 6, 1329–1341. [Google Scholar] [CrossRef]
- Eshibona, N.; Giwa, A.; Rossouw, S.C.; Gamieldien, J.; Christoffels, A.; Bendou, H. Upregulation of FHL1, SPNS3, and MPZL2 predicts poor prognosis in pediatric acute myeloid leukemia patients with FLT3-ITD mutation. Leuk. Lymphoma 2022, 1–10. [Google Scholar] [CrossRef]
- Hetzel, S.; Mattei, A.L.; Kretzmer, H.; Qu, C.; Chen, X.; Fan, Y.; Wu, G.; Roberts, K.G.; Luger, S.; Litzow, M.; et al. Acute lymphoblastic leukemia displays a distinct highly methylated genome. Nat. Cancer 2022, 1–15. [Google Scholar] [CrossRef]
- Huber, W.; von Heydebreck, A.; Sueltmann, H.; Poustka, A.; Vingron, M. Variance Stabilization Applied to Microarray Data Calibration and to the Quantification of Differential Expression. Bioinformatics 2002, 18 (Suppl. S1), S96–S104. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K.; Marioni, J.; Pai, A.A.; Degner, J.F.; Engelhardt, B.E.; Nkadori, E.; Veyrieras, J.-B.; Stephens, M.; Gilad, Y.; Pritchard, J.K. Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 2010, 464, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Foltz, S.M.; Gao, Q.; Yoon, C.J.; Sun, H.; Yao, L.; Li, Y.; Jayasinghe, R.G.; Cao, S.; King, J.; Kohnen, D.R.; et al. Evolution and structure of clinically relevant gene fusions in multiple myeloma. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Kageyama, R.; Merlino, G.T.; Pastan, I. A transcription factor active on the epidermal growth factor receptor gene. Proc. Natl. Acad. Sci. USA 1988, 85, 5016–5020. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, R.; Merlino, G.T.; Pastan, I. Epidermal growth factor (EGF) receptor gene transcription. Requirement for Sp1 and an EGF receptor-specific factor. J. Biol. Chem. 1988, 263, 6329–6336. [Google Scholar] [CrossRef]
- Song, S.; Honjo, S.; Jin, J.; Chang, S.-S.; Scott, A.W.; Chen, Q.; Kalhor, N.; Correa, A.M.; Hofstetter, W.L.; Albarracin, C.T.; et al. The Hippo Coactivator YAP1 Mediates EGFR Overexpression and Confers Chemoresistance in Esophageal Cancer. Clin. Cancer Res. 2015, 21, 2580–2590. [Google Scholar] [CrossRef]
- Haley, J.; Whittle, N.; Bennet, P.; Kinchington, D.; Ullrich, A.; Waterfield, M. The human EGF receptor gene: Structure of the 110 kb locus and identification of sequences regulating its transcription. Oncogene Res. 1987, 1, 375–396. [Google Scholar]
- Johnson, A.C.; Ishii, S.; Jinno, Y.; Pastan, I.; Merlino, G.T. Epidermal growth factor receptor gene promoter. Deletion analysis and identification of nuclear protein binding sites. J. Biol. Chem. 1988, 263, 5693–5699. [Google Scholar] [CrossRef]
- Kitadai, Y.; Yasui, W.; Yokozaki, H.; Kuniyasu, H.; Haruma, K.; Kajiyama, G.; Tahara, E. The level of a transcription factor Sp1 is correlated with the expression of EGF receptor in human gastric carcinomas. Biochem. Biophys. Res. Commun. 1992, 189, 1342–1348. [Google Scholar] [CrossRef]
- Carpentier, C.; Laigle-Donadey, F.; Marie, Y.; Auger, N.; Benouaich-Amiel, A.; Lejeune, J.; Kaloshi, G.; Delattre, J.-Y.; Thillet, J.; Sanson, M. Polymorphism in Sp1 recognition site of the EGF receptor gene promoter and risk of glioblastoma. Neurology 2006, 67, 872–874. [Google Scholar] [CrossRef]
- Beishline, K.; Azizkhan-Clifford, J. Sp1 and the ‘hallmarks of cancer’. FEBS J. 2015, 282, 224–258. [Google Scholar] [CrossRef] [PubMed]
- Guturi, K.K.N.; Mandal, T.; Chatterjee, A.; Sarkar, M.; Bhattacharya, S.; Chatterjee, U.; Ghosh, M.K. Mechanism of β-Catenin-mediated Transcriptional Regulation of Epidermal Growth Factor Receptor Expression in Glycogen Synthase Kinase 3 β-inactivated Prostate Cancer Cells. J. Biol. Chem. 2012, 287, 18287–18296. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, Y.; Ding, Y.; Zhang, S.; Pan, L. Characteristics of TGFBR1–EGFR–CTNNB1–CDH1 Signaling Axis in Wnt-Regulated Invasion and Migration in Lung Cancer. Cell Transplant. 2020, 29, 0963689720969167. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-Y.; Kim, J.M.; Jeong, D.S.; Kim, M.H. Transcriptional activation of EGFR by HOXB5 and its role in breast cancer cell invasion. Biochem. Biophys. Res. Commun. 2018, 503, 2924–2930. [Google Scholar] [CrossRef]
- Lee, J.-Y.; Hur, H.; Yun, H.J.; Kim, Y.; Yang, S.; Kim, S.I.; Kim, M.H. HOXB5 Promotes the Proliferation and Invasion of Breast Cancer Cells. Int. J. Biol. Sci. 2015, 11, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Hudson, L.G.; Thompson, K.L.; Xu, J.; Gill, G.N. Identification and characterization of a regulated promoter element in the epidermal growth factor receptor gene. Proc. Natl. Acad. Sci. USA 1990, 87, 7536–7540. [Google Scholar] [CrossRef]
- Johnson, A.C.; Murphy, B.A.; Matelis, C.M.; Rubinstein, Y.; Piebenga, E.C.; Akers, L.M.; Neta, G.; Vinson, C.; Birrer, M. Activator pro-tein-1 mediates induced but not basal epidermal growth factor receptor gene expression. Mol. Med. 2000, 6, 17–27. [Google Scholar] [CrossRef]
- Kharman-Biz, A.; Gao, H.; Ghiasvand, R.; Zhao, C.; Zendehdel, K.; Dahlman-Wright, K. Expression of activator protein-1 (AP-1) family members in breast cancer. BMC Cancer 2013, 13, 441. [Google Scholar] [CrossRef]
- Fan, F.; Podar, K. The Role of AP-1 Transcription Factors in Plasma Cell Biology and Multiple Myeloma Pathophysiology. Cancers 2021, 13, 2326. [Google Scholar] [CrossRef]
- Goldschmidt, H.; Lokhorst, H.M.; Mai, E.K.; Van Der Holt, B.; Blau, I.W.; Zweegman, S.; Weisel, K.C.; Vellenga, E.; Pfreundschuh, M.; Kersten, M.J.; et al. Bortezomib before and after high-dose therapy in myeloma: Long-term results from the phase III HOVON-65/GMMG-HD4 trial. Leukemia 2018, 32, 383–390. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, H.; Yoo, S.; Lee, E.; Laganà, A.; Parekh, S.; Schadt, E.E.; Wang, L.; Zhu, J. A Network Analysis of Multiple Myeloma Related Gene Signatures. Cancers 2019, 11, 1452. [Google Scholar] [CrossRef] [PubMed]
- Settino, M.; Arbitrio, M.; Scionti, F.; Caracciolo, D.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P.; Cannataro, M. MMRF-CoMMpass Data Integration and Analysis for Identifying Prognostic Markers. Int. Conf. Comput. Sci. 2020, 12139, 564–571. [Google Scholar] [CrossRef]
- Laganà, A.; Perumal, D.; Melnekoff, D.; Readhead, B.; A Kidd, B.; Leshchenko, V.; Kuo, P.-Y.; Keats, J.; Derome, M.; Yesil, J.; et al. Integrative network analysis identifies novel drivers of pathogenesis and progression in newly diagnosed multiple myeloma. Leukemia 2017, 32, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.; Laskin, J. EGFR-directed therapies to treat non-small-cell lung cancer. Expert Opin. Investig. Drugs 2009, 18, 1133–1145. [Google Scholar] [CrossRef]
- Ciardiello, F.; Tortora, G. EGFR Antagonists in Cancer Treatment. N. Engl. J. Med. 2008, 358, 1160–1174. [Google Scholar] [CrossRef]
- Marquez-Medina, D.; Popat, S. Afatinib: A second-generation EGF receptor and ErbB tyrosine kinase inhibitor for the treatment of advanced non-small-cell lung cancer. Futur. Oncol. 2015, 11, 2525–2540. [Google Scholar] [CrossRef]
- Santos, E.D.S.; Nogueira, K.A.B.; Fernandes, L.C.C.; Martins, J.R.P.; Reis, A.V.F.; Neto, J.D.B.V.; Júnior, I.J.D.S.; Pessoa, C.; Petrilli, R.; Eloy, J.O. EGFR targeting for cancer therapy: Pharmacology and immunoconjugates with drugs and nanoparticles. Int. J. Pharm. 2020, 592, 120082. [Google Scholar] [CrossRef]
- Singh, D.; Attri, B.K.; Gill, R.K.; Bariwal, J. Review on EGFR Inhibitors: Critical Updates. Mini-Rev. Med. Chem. 2016, 16, 1134–1166. [Google Scholar] [CrossRef]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef]
- Cai, W.-Q.; Zeng, L.-S.; Wang, L.-F.; Wang, Y.-Y.; Cheng, J.-T.; Zhang, Y.; Han, Z.-W.; Zhou, Y.; Huang, S.-L.; Wang, X.-W.; et al. The Latest Battles Between EGFR Monoclonal Antibodies and Resistant Tumor Cells. Front. Oncol. 2020, 10, 1249. [Google Scholar] [CrossRef]
- Kumagai, S.; Koyama, S.; Nishikawa, H. Antitumour immunity regulated by aberrant ERBB family signalling. Nat. Cancer 2021, 21, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Friedlaender, A.; Subbiah, V.; Russo, A.; Banna, G.L.; Malapelle, U.; Rolfo, C.; Addeo, A. EGFR and HER2 exon 20 insertions in solid tumours: From biology to treatment. Nat. Rev. Clin. Oncol. 2021, 19, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, T.; Yamaoka, T.; Ando, K.; Kusumoto, S.; Kishino, Y.; Manabe, R.; Sagara, H. Molecular and Clinical Features of EGFR-TKI-Associated Lung Injury. Int. J. Mol. Sci. 2021, 22, 792. [Google Scholar] [CrossRef]
- Zhao, Y.; Cheng, B.; Chen, Z.; Li, J.; Liang, H.; Chen, Y.; Zhu, F.; Li, C.; Xu, K.; Xiong, S.; et al. Toxicity profile of epidermal growth factor receptor tyrosine kinase inhibitors for patients with lung cancer: A systematic review and network meta-analysis. Crit. Rev. Oncol. 2021, 160, 103305. [Google Scholar] [CrossRef] [PubMed]
- Mahtouk, K.; Hose, D.; Rème, T.; De Vos, J.; Jourdan, M.; Moreaux, J.; Fiol, G.; Raab, M.; Jourdan, E.; Grau, V.; et al. Expression of EGF-family receptors and amphiregulin in multiple myeloma. Amphiregulin is a growth factor for myeloma cells. Oncogene 2005, 24, 3512–3524. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Zhang, D.; Wang, F.; Wang, Q.; Wu, Y.; Gou, M.; Hu, Y.; Zhang, W.; Huang, J.; Gong, Y.; et al. ALCAM-EGFR interaction regulates myelomagenesis. Blood Adv. 2021, 5, 5269–5282. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, R.; Ding, J.; Ji, D.; Song, B.; Yuan, L.; Chang, H.; Chen, G. Multiple myeloma acquires resistance to EGFR inhibitor via induction of pentose phosphate pathway. Sci. Rep. 2015, 5, 9925. [Google Scholar] [CrossRef]
- Raimondo, S.; Saieva, L.; Vicario, E.; Pucci, M.; Toscani, D.; Manno, M.; Raccosta, S.; Giuliani, N.; Alessandro, R.; Raccosta, S. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J. Hematol. Oncol. 2019, 12, 2. [Google Scholar] [CrossRef]
- Von Tresckow, B.; Peine, D.; Böll, B.; A Eichenauer, D.; Von Strandmann, E.P.; Knop, S.; Goebeler, M.; Hallek, M.; Engert, A.; Hübel, K. Anti-EGFR Antibody Cetuximab in Refractory or Relapsed Multiple Myeloma: Preliminary Results and Evaluation of Response Prediction in a Phase II Clinical Trial. Blood 2008, 112, 3686. [Google Scholar] [CrossRef]
- Harvey, R.C.; Tasian, S.K. Clinical diagnostics and treatment strategies for Philadelphia chromosome–like acute lymphoblastic leukemia. Blood Adv. 2020, 4, 218–228. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.; Jabbour, E. Optimizing the treatment of acute lymphoblastic leukemia in younger and older adults: New drugs and evolving paradigms. Leukemia 2021, 35, 3044–3058. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Kantarjian, H.; Jabbour, E. SOHO State of the Art Updates & Next Questions: Intensive and Non–Intensive Approaches for Adults with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Clin. Lymphoma Myeloma Leuk. 2021, 22, 61–66. [Google Scholar] [CrossRef] [PubMed]
- Slayton, W.B.; Schultz, K.R.; Kairalla, J.A.; Devidas, M.; Mi, X.; Pulsipher, M.A.; Chang, B.H.; Mullighan, C.; Iacobucci, I.; Silverman, L.B.; et al. Dasatinib plus intensive chemotherapy in children, adolescents, and young adults with Philadelphia chromosome-positive acute lymphoblastic leukemia: Results of Children’s Oncology Group Trial AALL0622. J. Clin. Oncol. 2018, 36, 2306–2314. [Google Scholar] [CrossRef] [PubMed]
- Schultz, K.R.; Carroll, A.; Heerema, N.A.; Bowman, W.P.; Aledo, A.; Slayton, W.B.; Sather, H.; Devidas, M.; Zheng, H.W.; Davies, S.M.; et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia 2014, 28, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
- Warraich, Z.; Tenneti, P.; Thai, T.; Hubben, A.; Amin, H.; McBride, A.; Warraich, S.; Hannan, A.; Warraich, F.; Majhail, N.; et al. Relapse Prevention with Tyrosine Kinase Inhibitors after Allogeneic Transplantation for Philadelphia Chromosome–Positive Acute Lymphoblast Leukemia: A Systematic Review. Biol. Blood Marrow Transplant. 2019, 26, e55–e64. [Google Scholar] [CrossRef] [PubMed]
- Foà, R.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Puzzolo, M.-C.; Canichella, M.; Viero, P.; Ferrara, F.; Lunghi, M.; et al. Dasatinib–Blinatumomab for Ph-Positive Acute Lymphoblastic Leukemia in Adults. N. Engl. J. Med. 2020, 383, 1613–1623. [Google Scholar] [CrossRef]
- Foà, R.; Vitale, A.; Vignetti, M.; Meloni, G.; Guarini, A.; De Propris, M.S.; Elia, L.; Paoloni, F.; Fazi, P.; Cimino, G.; et al. Dasatinib as first-line treatment for adult patients with Philadelphia chromosome–positive acute lymphoblastic leukemia. Blood 2011, 118, 6521–6528. [Google Scholar] [CrossRef]
- Samra, B.; Jabbour, E.; Ravandi, F.; Kantarjian, H.; Short, N.J. Evolving therapy of adult acute lymphoblastic leukemia: State-of-the-art treatment and future directions. J. Hematol. Oncol. 2020, 13, 1–17. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H.; Ravandi, F.; Thomas, D.; Huang, X.; Faderl, S.; Pemmaraju, N.; Daver, N.; Garcia-Manero, G.; Sasaki, K.; et al. Combination of hyper-CVAD with ponatinib as first-line therapy for patients with Philadelphia chromosome-positive acute lymphoblastic leukaemia: A single-centre, phase 2 study. Lancet Oncol. 2015, 16, 1547–1555. [Google Scholar] [CrossRef]
- Short, N.J.; Kantarjian, H.M.; Ravandi, F.; Huang, X.; Daver, N.G.; Dinardo, M.C.D.; Konopleva, M.Y.; Pemmaraju, N.; Wierda, W.G.; Garcia-Manero, G.; et al. Long-Term Safety and Efficacy of Hyper-CVAD Plus Ponatinib as Frontline Therapy for Adults with Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Blood 2019, 134, 283. [Google Scholar] [CrossRef]
- Rousselot, P.; Coudé, M.M.; Goekbuget, N.; Gambacorti Passerini, C.; Hayette, S.; Cayuela, J.-M.; Huguet, F.; Leguay, T.; Chevallier, P.; Salanoubat, C.; et al. Dasatinib and low-intensity chemotherapy in elderly patients with Philadelphia chromosome–positive ALL. Blood 2016, 128, 774–782. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, S.; Bassan, R.; Vitale, A.; Elia, L.; Piciocchi, A.; Puzzolo, C.; Canichella, M.; Ferrara, F.; Lunghi, M.; Fabbiano, F.; et al. Dasatinib-blinatumomab combination for the front-line treatment of adult Ph + ALL patients. Updated results of the Gimema LAL2116 D-Alba trial. Blood 2019, 134 (Suppl. 1), 740. [Google Scholar] [CrossRef]
- Jabbour, E.; DerSarkissian, M.; Duh, M.S.; McCormick, N.; Cheng, W.Y.; McGarry, L.J.; Souroutzidis, A.; Huang, H.; O’Brien, S.; Ravandi, F.; et al. Efficacy of ponatinib versus earlier gen-eration tyrosine kinase inhibitors for front-line treatment of newly diagnosed Philadelphia-positive acute lymphoblastic leukemia. Clin. Lymphoma Myeloma Leuk. 2018, 18, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Cortes, J.E.; Ravandi, F.; Konopleva, M.; Alvarado, Y.; Kadia, T.; Wierda, W.G.; Borthakur, G.; Naqvi, K.; Pemmaraju, N.; et al. Inotuzumab ozogamicin in combination with bosu-tinib for patients with relapsed or refractory Ph + ALL or CML in lymphoid blast phase. Blood 2017, 130 (Suppl. S1), 143. [Google Scholar]
- Chen, M.; Zhu, Y.; Lin, Y.; Tengwang, T.; Zhang, L. Use of tyrosine kinase inhibitors for paediatric Philadelphia chromosome-positive acute lymphoblastic leukaemia: A systematic review and meta-analysis. BMJ Open 2021, 11, e042814. [Google Scholar] [CrossRef]
- Li, B.; Wan, Q.; Li, Z.; Chng, W.-J. Janus Kinase Signaling: Oncogenic Criminal of Lymphoid Cancers. Cancers 2021, 13, 5147. [Google Scholar] [CrossRef]
- Qazi, S.; Uckun, F.M. Gene expression profiles of infant acute lymphoblastic leukaemia and its prognostically distinct subsets. Br. J. Haematol. 2010, 149, 865–873. [Google Scholar] [CrossRef]
- Uckun, F.M.; Pitt, J.; Qazi, S. JAK3 pathway is constitutively active in B-lineage acute lymphoblastic leukemia. Expert Rev. Anticancer Ther. 2011, 11, 37–48. [Google Scholar] [CrossRef]
- Leonard, W.J.; Lin, J.-X.; O’Shea, J.J. The γc Family of Cytokines: Basic Biology to Therapeutic Ramifications. Immunity 2019, 50, 832–850. [Google Scholar] [CrossRef]
- Mahapatra, D.K.; Asati, V.; Bharti, S.K. Discovery of small-molecule ATR inhibitors for potential cancer treatment: A patent review from 2014 to present. Expert Opin. Ther. Pat. 2017, 27, 887–906. [Google Scholar] [CrossRef]
- Vyas, D.; O’Dell, K.M.; Bandy, J.L.; Boyce, E.G. Tofacitinib. Ann. Pharmacother. 2013, 47, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S. Tofacitinib: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1987–2001. [Google Scholar] [CrossRef] [PubMed]
- Lam, C.; Ferguson, I.D.; Mariano, M.C.; Lin, Y.-H.T.; Murnane, M.; Liu, H.; Smith, G.A.; Wong, S.W.; Taunton, J.; Liu, J.O.; et al. Repurposing tofacitinib as an anti-myeloma therapeutic to reverse growth-promoting effects of the bone marrow microenvironment. Haematologica 2018, 103, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, M.B.; Fook-Alves, V.L.; Eugenio, A.I.; Fernando, R.C.; Sanson, L.F.G.; de Carvalho, M.F.; Braga, W.M.; Davies, F.E.; Colleoni, G.W. Anti-myeloma effects of ruxolitinib combined with bortezomib and lenalidomide: A rationale for JAK/STAT pathway inhibition in myeloma patients. Cancer Lett. 2017, 403, 206–215. [Google Scholar] [CrossRef]
- Stubbs, M.C.; Burn, T.C.; Sparks, R.B.; Maduskuie, T.; Diamond, S.; Rupar, M.; Wen, X.; Volgina, A.; Zolotarjova, N.; Waeltz, P.; et al. The Novel Bromodomain and Extraterminal Domain Inhibitor INCB054329 Induces Vulnerabilities in Myeloma Cells That Inform Rational Combination Strategies. Clin. Cancer Res. 2019, 25, 300–311. [Google Scholar] [CrossRef]
- Ogiya, D.; Liu, J.; Ohguchi, H.; Kurata, K.; Samur, M.K.; Tai, Y.-T.; Adamia, S.; Ando, K.; Hideshima, T.; Anderson, K.C. The JAK-STAT pathway regulates CD38 on myeloma cells in the bone marrow microenvironment: Therapeutic implications. Blood 2020, 136, 2334–2345. [Google Scholar] [CrossRef]
- Berenson, J.R.; To, J.; Spektor, T.M.; Martinez, D.; Turner, C.; Sanchez, A.; Ghermezi, M.; Eades, B.M.; Swift, R.A.; Schwartz, G.; et al. A Phase I Study of Ruxolitinib, Lenalidomide, and Steroids for Patients with Relapsed/Refractory Multiple Myeloma. Clin. Cancer Res. 2020, 26, 2346–2353. [Google Scholar] [CrossRef]
- Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death (accessed on 7 January 2022).
- Qiu, Q.; Feng, Q.; Tan, X.; Guo, M. JAK3-selective inhibitor peficitinib for the treatment of rheumatoid arthritis. Expert Rev. Clin. Pharmacol. 2019, 12, 547–554. [Google Scholar] [CrossRef]
- Pei, H.; He, L.; Shao, M.; Yang, Z.; Ran, Y.; Li, D.; Zhou, Y.; Tang, M.; Wang, T.; Gong, Y.; et al. Discovery of a highly selective JAK3 inhibitor for the treatment of rheumatoid arthritis. Sci. Rep. 2018, 8, 5273. [Google Scholar] [CrossRef]
- Bahekar, R.; Panchal, N.; Soman, S.; Desai, J.; Patel, D.; Argade, A.; Gite, A.; Gite, S.; Patel, B.; Kumar, J.; et al. Discovery of diaminopyrimidine-carboxamide derivatives as JAK3 inhibitors. Bioorganic Chem. 2020, 99, 103851. [Google Scholar] [CrossRef]
- Yin, Y.; Chen, C.-J.; Yu, R.-N.; Shu, L.; Wang, Z.-J.; Zhang, T.-T.; Zhang, D.-Y. Novel 1H-pyrazolo [3,4-d]pyrimidin-6-amino derivatives as potent selective Janus kinase 3 (JAK3) inhibitors. Evaluation of their improved effect for the treatment of rheumatoid arthritis. Bioorganic Chem. 2020, 98, 103720. [Google Scholar] [CrossRef] [PubMed]
- Sahin, K.; Yabas, M.; Orhan, C.; Tuzcu, M.; Sahin, T.K.; Ozercan, I.H.; Qazi, S.; Uckun, F.M. Prevention of DMBA-induced mammary gland tumors in mice by a dual-function inhibitor of JAK3 and EGF receptor tyrosine kinases. Expert Opin. Ther. Targets 2020, 24, 379–387. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Kinase | Drug | Brand |
|---|---|---|
| ABL1, SRC | Bosutinib | Bosulif, SKI-606 |
| ABL1, SRC, CKIT | Dasatinib | BMS-354825, Sprycell |
| ABL1, SRC | Ponatinib | Iclusig |
| EGFR | Gefitinib | ZD1839, Iressa |
| EGFR | Dacomitinib | PF-00299804, Visimpro |
| EGFR | Erlotinib | OSI-744, Tarceva |
| EGFR T790M | Osimertinib | AZD-9292, Tagrisso |
| EGFR with exon 20 insertions | Mobocertinib | TAK-788, AP-32788, EXKIVITY |
| ErbB1/2/4 | Afatinib | Gilotrif, Tovok |
| ErbB1/2/HER2 | Lapatinib | Tykerb |
| JAK1 | Upadacitinib | ABT-494, Rinvoq |
| JAK1/2 | Baricitinib | Olumiant, LY 3009104 |
| JAK1/2/3, Tyk | Ruxolitinib | Jakafi |
| JAK2 | Fedratinib | TG101348, Inrebic |
| JAK3 | Tofacitinib | Xeljanz |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uckun, F.M.; Qazi, S. ERBB1/EGFR and JAK3 Tyrosine Kinases as Potential Therapeutic Targets in High-Risk Multiple Myeloma. Onco 2022, 2, 282-304. https://doi.org/10.3390/onco2040016
Uckun FM, Qazi S. ERBB1/EGFR and JAK3 Tyrosine Kinases as Potential Therapeutic Targets in High-Risk Multiple Myeloma. Onco. 2022; 2(4):282-304. https://doi.org/10.3390/onco2040016
Chicago/Turabian StyleUckun, Fatih M., and Sanjive Qazi. 2022. "ERBB1/EGFR and JAK3 Tyrosine Kinases as Potential Therapeutic Targets in High-Risk Multiple Myeloma" Onco 2, no. 4: 282-304. https://doi.org/10.3390/onco2040016
APA StyleUckun, F. M., & Qazi, S. (2022). ERBB1/EGFR and JAK3 Tyrosine Kinases as Potential Therapeutic Targets in High-Risk Multiple Myeloma. Onco, 2(4), 282-304. https://doi.org/10.3390/onco2040016