1. Introduction
The usage of AgNPs has increased dramatically in recent years owing to their wide range of utility in many fields including water purification [
1], air filters [
2], food packaging [
3], electromagnetic shielding [
4], biomedical applications [
5,
6,
7], and smart textiles [
8], to mention a few. On the other hand, the heightened focus on smart textile technology can be attributed to its remarkable advantages, which include significant elasticity, robust mechanical resilience, breathability, a porous structure, lightness, and the capability for tailored surface alteration to match specific requirements [
9,
10]. Nonetheless, for most of the uses above, a functional material coating is essential to bestow specific characteristics upon the fabric. These traits may include electrical conductivity, essential for developing specific smart textile materials, as well as antimicrobial properties in PPE applications achieved through a silver layer coating.
There are two popular techniques for creating functional textiles using metal NPs. The first, known as ex situ coating, coats fabrics with already-formed metal NPs, while the second technique coats and grows metal NPs on the textile substrate in an in situ manner using a metal salt precursor and a reducing agent [
11,
12]. The latter approach relies on the reaction between a reducing agent and a metal salt, regardless of whether the reducing agent is organic or inorganic [
13]. In addition to reducing agents, capping and dispersive agents play a crucial role in controlling the size, shape, and aggregation rate [
14,
15,
16]. Conversely, the temperature, pH, and precursor concentration of the solution significantly influence the nanoparticle shape [
17,
18,
19,
20].
In general, NPs created in situ on fibers have strong adhesion, which can be strengthened even further through surface modification [
21]. Targeting biological applications, researchers have attempted to create an approach for growing NPs both on the surface of the fibers and into the textile structures with greater adhesion to the fibers [
22,
23]. Lakshmanan et al. coated AgNPs on the surface of jute fabric with a silver nitrate solution at a pH of 6.5. In this case, the temperature was raised to 90 °C at a rate of 2 °C/min before being cooled to 40 °C. Finally, the samples were washed with distilled water. To promote the AgNPs’ adhesion to the surface, the fabrics were pre-treated as follows: the samples were soaked in a non-ionic detergent for an hour before being rinsed with distilled water, then dipped in acetic acid, and washed with distilled water again. The fabric samples worked effectively against Bacillus subtilis and Escherichia coli [
24]. Shateri-Khalilabad et al. used KOH as a pre-treatment material, silver nitrate as a precursor, and ascorbic acid as a reducing agent to perform in situ silver coating on cotton fabric. The average size of the silver nanocrystals was 53 nm [
25]. Two types of cotton coated with in situ AgNPs were prepared using silver ammonia as the precursor, with a mixture of dopamine and glucose as the reducing agent. The results indicated that increasing the silver precursor solution concentration from 0.10 to 0.25 mol/L led to a decrease in surface resistivity from 0.32 to 0.12 Ω [
26]. In an ex situ study, four different concentrations of colloidal AgNP solutions were coated on the cotton substrate and dried at room temperature [
27]. The AgNP solution was prepared using silver nitrate (AgNO
3), sodium hydroxide (NaOH), ammonium hydroxide (NH
4OH), and ascorbic acid (C
6H
8O
6). The average size of the NPs was 12.4 ± 5.1 nm [
27].
In several studies, sodium borohydride (NaBH
4) has been utilized as a powerful reducing agent. In this regard, AgNPs of an average size of 20 nm were grown on cotton yarns. The growth of such particles was achieved by using 250 mL of AgNO
3 solution (1 g·L
−1, soaking for 24 h) as the precursor, and 250 mL of NaBH
4 solution (250 mg·L
−1) as the reducer [
28]. The size-controlled AgNPs were synthesized on a cotton yarn surface by varying the silver precursor (AgNO
3) concentration. When the AgNO
3 concentration was increased from 0.1 mM to 10 mM, the particle size increased accordingly [
29].
In our earlier study [
5], we demonstrated that AgNPs averaging 24 ± 6 nm in size were successfully coated onto cotton fabric, achieving a 99.89% effectiveness in eliminating HCoV-229E, a virus that serves as a surrogate for SARS-CoV-2. Since AgNPs of varying sizes exhibit different levels of effectiveness against various viruses and bacteria [
30], we were inspired to conduct a thorough and systematic investigation on how size variations of AgNPs, influenced by several parameters, could optimize the packing density of AgNPs on cotton yarn surfaces. In order to develop a simple and cost-effective synthesis method, we employed a self-assembled in situ reactive coating strategy to synthesize AgNPs with specific size control directly on the fibers’ surface of a cotton yarn. The principal benefit of our research is the markedly short time required for synthesizing AgNPs on cotton yarn. A dense and even coating of AgNPs was successfully formed on the substrate within 5 min. Furthermore, in this method, at the molecular level, the reducer agent and silver precursor are dissolved in the solvent, which then can penetrate deep inside the cotton yarn structure. As such, the AgNPs are thus synthesized deep from inside the cotton yarn rather than from its surface only. The following setup offers more stability in that the nanoparticles will be entrapped within the cotton fibers and, therefore, less vulnerable to detachment upon application or washing.
The work presented here reports a new method of synthesizing AgNPs by using a reactive in situ self-assembly technique. To the best of our knowledge, none of the previously conducted studies focused on the growth mechanisms of AgNPs through this technique coupled with an investigation into the impact of seven different parameters on its development. This investigation uniquely presents comprehensive new insights into the precision of controlling AgNP synthesis. We offer a detailed analysis of how various factors—such as the concentrations of reducer and silver precursor, oven temperature, reaction time, vacuum conditions, and ice bath treatment—affect the synthesis of AgNPs on cotton yarn substrates. Each of these factors plays a crucial role in determining the size of the AgNPs formed directly on the cotton, making our investigation into AgNPs size control essential for optimizing the parameters necessary for customized AgNPs coatings.
2. Materials and Methods
2.1. Materials
Silver perchlorate (AgClO4) (anhydrous, 97%), sodium borohydride (NaBH4), and isopropyl alcohol (IPA 99.5%) were obtained from Sigma-Aldrich, and ethanol (EtOH 99%) from Fisher. Commercially available ring-spun cotton yarn with a diameter of 0.8 mm was bought from Etsy.
2.2. Self-Assembled Reactive In Situ Growth of AgNPs on the Cotton Substrate
The cotton yarns were initially cleaned using a standard laundry detergent solution, prepared by mixing 1 part detergent with 30 parts water and heated to 100 °C. The yarns were stirred in this solution for 3 min to remove oil, dust, and other contaminants. Afterward, they were thoroughly rinsed with distilled water and allowed to air-dry for 24 h. Individual cotton yarn samples, each measuring 4 cm in length, were selected for every growth condition.
To synthesize AgNPs, AgClO4 was dissolved in IPA at various concentrations, while NaBH4 dissolved in EtOH served as the reducing agent. The size, shape, and surface coverage of the AgNPs on the cotton yarn were investigated by varying the NaBH4 concentration from 1.5 to 7.5 g·L−1 and the AgClO4 concentration from 3.33 to 10 g·L−1. Additionally, the effects of vacuum and non-vacuum curing conditions were examined, along with four different curing temperatures (room temperature, 40 °C, 70 °C, and 110 °C) and three different curing durations (1 min, 3 min, and 6 min).
The experimental procedure began by immersing the yarn samples in the reducing solution at room temperature for 30 s. The samples were then carefully removed from this solution and immediately immersed in the precursor solution for 15 s. Afterward, the samples were taken out and placed horizontally in an oven. A schematic illustration of the self-assembled in situ reactive synthesis of AgNPs on cotton yarn is shown in
Figure 1. To remove any loosely bound AgNPs from the yarn surface, the samples were rinsed with distilled water. The samples were mounted on a silicon substrate for subsequent analysis using field emission scanning electron microscopy (FESEM). Furthermore, a 5 nm gold layer was sputter-coated onto the sample surface to reduce surface charging effects and facilitate effective morphological analysis.
2.3. Characterization
A System Zeiss Gemini SEM 500 is used to examine the morphology, shape, and distribution of AgNPs on the surface of cotton yarn. The acceleration voltage is adjusted to 10 kV. X-ray diffraction (XRD) was employed to analyze the crystal structure of AgNPs. The analysis was conducted with a Rigaku Ultima IV Diffractometer, utilizing copper Kα radiation (λ = 1.5406 Å). The 2θ range was set from 30° to 68°, with a scan speed of 5° per minute and a step size of 0.04°. The elemental analysis of AgNPs on the cotton yarn is provided using an energy-dispersive X-ray spectrometer (EDS) located in the SEM chamber. To obtain the analytical results, the acceleration voltage is adjusted to 20 kV. Fourier-transform infrared spectroscopy (FTIR) is used to analyze the chemical interactions and bonding of AgNPs on the cotton fiber surface. For this purpose, a Thermo Scientific Nicolet 6700 FTIR/ATR is used.
3. Results and Discussion
Table 1 outlines the sample codes, and the specific experimental conditions used during sample preparation. For detailed information on the modified variables, corresponding average sizes, and standard deviations of the AgNPs, please refer to the extended
Table S1 in the Supplementary Information, which also provides a comprehensive description of the methodologies and quantitative metrics related to the samples.
3.1. Ag Precursor (AgClO4) Concentration and Vacuum Effect on AgNPs Average Size
The synthesis of AgNPs on cotton yarn was carried out under non-vacuum conditions to study the effect of AgClO
4 concentration on the size of the resulting nanoparticles. Three distinct samples, designated T1, T2, and T3, were prepared for this purpose. Each sample was immersed in the AgClO
4 solution and then placed horizontally in a preheated oven at 40 °C for 3 min without applying a vacuum. High-magnification FESEM images, shown in
Figure 2a–c, reveal the size and distribution of the AgNPs. In this experiment, the concentration of the Ag precursor was the only variable altered, with all the other conditions, such as the reducer concentration (maintained at 3.75 g·L
−1), temperature, and curing duration were kept constant. The particle size distribution obtained from the FESEM images demonstrates that increasing the AgClO
4 concentration from 3.33 g·L
−1 (
Figure 2a) to 10 g·L
−1 (
Figure 2c) resulted in a 1.5-fold increase in the average size of the AgNPs.
The same synthesis technique was repeated under vacuum conditions, with the oven pressure precisely set to 254 Torr (10 inches of mercury). High-magnification FESEM images of three additional samples, labeled T4 to T6, are shown in
Figure 3a,b. The images reveal that increasing the silver precursor concentration from 3.33 g·L
−1 (
Figure 3a) to 10 g·L
−1 (
Figure 3c) resulted in an approximate doubling of the average size of the AgNPs. A comparative analysis between the vacuum and non-vacuum samples demonstrates that the average diameter of AgNPs synthesized under vacuum conditions is approximately 1.5 times larger than those produced without a vacuum. Additionally, FESEM imaging shows a denser aggregation of AgNPs on the cotton yarn substrate, which correlates with the increased concentration of the silver precursor. This trend is consistently observed in both vacuum-assisted and non-vacuum synthesis methods.
In the non-vacuum synthesis (samples T1 to T3), increasing the silver precursor concentration leads to a higher availability of silver ions, which accelerates the nucleation process and promotes the growth of larger nanoparticles. The introduction of a vacuum during synthesis (samples T4 to T6) further amplifies this effect by accelerating solvent evaporation and removing dissolved gases, thereby creating a highly concentrated and reactive environment. This results in an accelerated reduction reaction, increased nucleation rates, and enhanced nanoparticle formation due to reduced solvation of silver ions and a higher frequency of collisions among reactive species. Although the vacuum applied is not high, the pressure of 254 Torr in the presence of heat effectively reduces potential oxidizers, such as oxygen and residual water, which favors particle growth over nucleation, leading to the formation of larger AgNPs. Moreover, the density of nanoparticles on the cotton yarn surface increases with the concentration of the silver precursor, regardless of vacuum use. This suggests that the higher local concentration of silver ions promotes nanoparticle aggregation, resulting in more compact clusters due to reduced particle mobility and leading to concentrated growth in specific regions.
3.2. Reducer Concentration (NaBH4) Effect on AgNPs Average Size
The impact of varying the concentration of the reducing agent on the size of AgNPs was investigated across four distinct concentrations. The experimental conditions were kept constant, with the oven temperature set at 40 °C for 3 min, and no vacuum setting was used. FESEM analysis, as shown in
Figure 4a–d, reveals a direct correlation between the concentration of the reducing agent and the average size of the synthesized AgNPs. Specifically, the average nanoparticle size increased approximately 2.6-fold when the reducing agent concentration was raised from 1.5 g·L
−1 to 3.75 g·L
−1 (samples T7 to T8). Further increases in the reducer concentration to 5 g·L
−1 resulted in a 50% size increase (samples T8 to T9), and raising it to 7.5 g·L
−1 led to an additional 25% increase in average size (samples T9 to T10).
The phenomenon of diffusion-limited growth becomes more pronounced at higher reducing agent concentrations [
31,
32]. In this context, the rate-limiting step occurs when the reactant diffuses to the nanoparticle surface, which tends to favor the growth of existing particles rather than the formation of new nuclei, mainly when the reducer is abundant. As the concentration of the reducing agent increases, the rate of silver ion reduction to metallic silver accelerates, resulting in faster particle growth. Additionally, at higher reducer concentrations, the surface area coverage of the nanoparticles increases, stabilizing them and promoting further growth rather than the formation of new energetically unstable smaller nanoparticles. Moreover, higher concentrations of the reducing agent can trigger secondary reduction processes [
33,
34], leading to the deposition of additional silver atoms onto the nanoparticles, thereby increasing their size. The excess of reducing agents also introduces more electrons into the system, enhancing the reduction of silver ions and facilitating the continued growth of pre-existing nanoparticles. This process can result in the larger nanoparticles absorbing smaller ones, further increasing their size.
3.3. Ice Bath Effect on AgNPs Average Size
To investigate the effect of lower temperatures on the morphology of AgNPs at constant reducer concentration (3.75 g·L
−1) and constant precursor concentration (6.66 g·L
−1), the synthesis was performed on cotton yarn immersed in separate ice-cooled baths containing the reducing and precursor solutions, as illustrated in
Figure 1b. The primary objective was to utilize the ice bath to moderate the rapid reduction reaction typically driven by sodium borohydride, a potent reducing agent. The reduced reaction kinetics at the lower temperature significantly influenced the average size of the AgNPs.
Figure 5 and
Figure 6 show the SEM images and histograms of samples prepared in the ice bath at fixed reducer concentration and fixed precursor concentration, respectively. For example,
Figure 5 shows a 25% increase in particle size when the concentration of the silver precursor was raised from 3.33 g·L
−1 to 10 g·L
−1 (comparing samples T11 to T13). Further analysis indicated that, at a silver precursor concentration of 3.33 g·L
−1, the ice bath treatment resulted in a 20% reduction in the average size of AgNPs (as observed in samples T4 and T11 from
Figure 3a and
Figure 5a). A more pronounced size reduction of 28% was recorded at a silver precursor concentration of 6.66 g·L
−1 (as shown by samples T5 and T12 from
Figure 3b and
Figure 5b). Notably, at the highest concentration of 10 g·L
−1, the ice bath treatment led to a substantial 48% decrease in average nanoparticle size (as evidenced by samples T6 and T13 in
Figure 3c and
Figure 5c), highlighting the critical role of a preheat treatment temperature (coating step) in the synthesis process.
The average size of the AgNPs increased more than four-fold in the ice bath experiment when the reducer concentration was raised from 1.5 g·L
−1 to 7.5 g·L
−1, as shown in
Figure 6 (samples T14 to T17). Further analysis revealed a notable reduction in the average size of AgNPs in the ice bath treatment experiments compared to the no-ice treatment conditions at varying reducer concentrations. For example, at 1.5 g·L
−1, the treatment achieved a 29% size reduction, as observed in samples T7 and T14 (
Figure 4a and
Figure 6a). A greater reduction of 34% was recorded at a concentration of 3.75 g·L
−1, corresponding to samples T8 and T15 (
Figure 4b and
Figure 6b). When the reducer concentration reached 5 g·L
−1, the average nanoparticle size decreased by 38%, as seen in samples T9 and T16 (
Figure 4c and
Figure 6c). Interestingly, at the highest tested concentration of 7.5 g·L
−1, the ice bath treatment resulted in a 35% reduction in size, as demonstrated by samples T10 and T17 (
Figure 4d and
Figure 6d).
These observations can be attributed to the slower initial chemical reaction rates induced by the ice bath’s low temperature, which delays nucleation and results in smaller initial nanoparticle sizes due to restricted molecular mobility. The process of transferring samples from an ice bath to the oven allows for a gradual temperature increase, which helps to control the nanoparticle formation rate [
35]. This gradual warming reduces the likelihood of rapid nanoparticle formation that could occur. The larger particle sizes observed at higher silver precursor concentrations suggest that, while smaller sizes are initially favored under cold conditions, the increased concentration of silver ions encourages the further growth of the nanoparticles. The slow warming of the sample in the oven facilitates this growth, potentially intensified by Ostwald ripening, especially at higher silver precursor concentrations where a greater number of ions are available for redeposition onto larger particles due to their thermodynamically lower surface energy.
3.4. Oven Temperature Effect on AgNPs Average Size
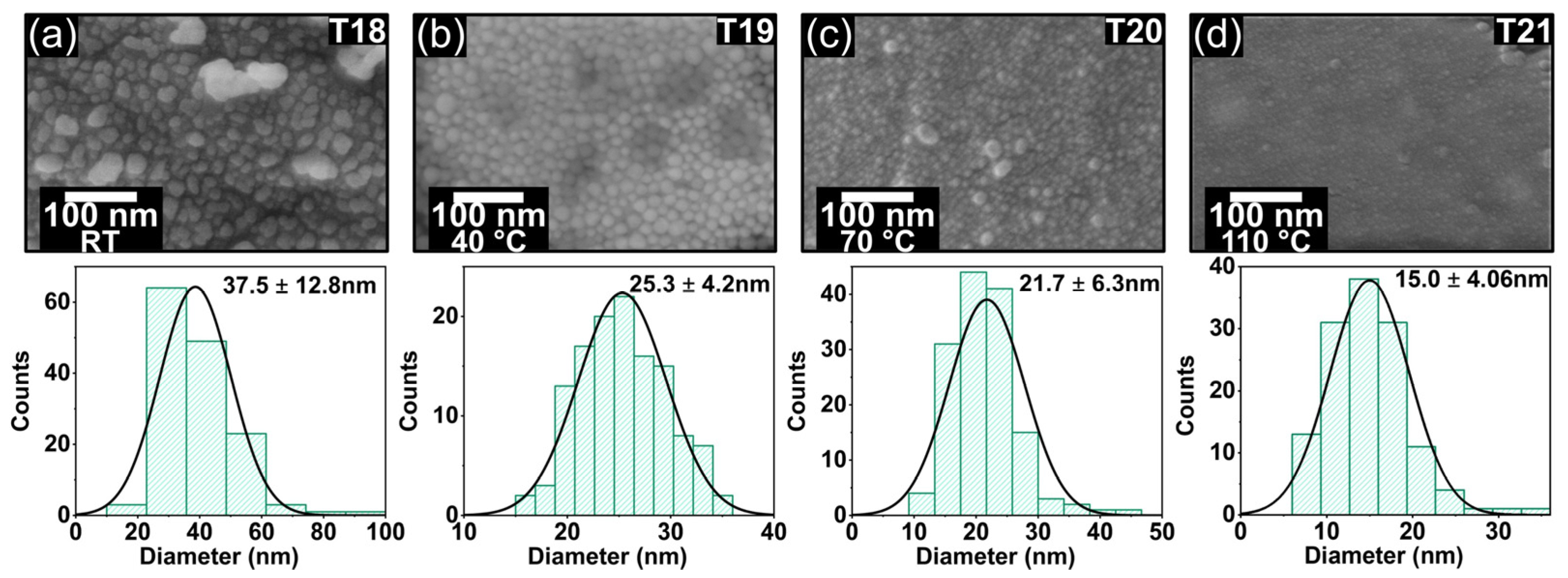
The relationship between oven temperature and the dimensions of self-assembled AgNPs was investigated using a vacuum environment set at 254 Torr (10 inches of mercury). Under these conditions, AgNPs were produced with a wide range of sizes, with diameters varying from 15 to 37.5 nm, as illustrated in
Figure 7. Four distinct temperatures were utilized for sample preparation: room temperature (T18), 40 °C (T19), 70 °C (T20), and 110 °C (T21). The solvents—EtOH (boiling point 78.37 °C) and IPA (boiling point 82.5 °C)—were heated to 70 °C. The concentration of the silver precursor was held constant at 6.66 g·L
−1, while the reducer concentration remained at 3.75 g·L
−1. As the temperature increased, a noticeable reduction in the average diameter of the AgNPs was observed. Specifically, the average size of the AgNPs decreased by 33% when the temperature was raised from room temperature to 40 °C, and by 40% when it increased from 40 °C to 110 °C. This trend is shown in
Figure 7a–d.
The reduction in AgNP size observed in this study can be attributed to several interrelated factors. As the temperature in the vacuum oven rises, the reduction of silver ions in AgClO
4 by NaBH
4 occurs more rapidly, leading to faster nucleation of AgNPs [
36]. Additionally, the elevated temperatures increase the evaporation rates of ethanol and isopropyl alcohol [
37], resulting in a higher concentration of reactants [
38], which promotes the formation of numerous nuclei and subsequently reduces the growth time of individual AgNPs. Moreover, the increase in temperature enhances reactant mobility and improves mixing efficiency [
39] due to a decrease in solvent viscosity, leading to a more uniform reaction throughout the liquid coating. Beyond a certain point, as the temperature increases, the enhanced movement of the involved species increases the aggregation rate in favored directions, leading to the formation of relatively nonuniform large NPs.
3.5. Curing Time Effect on AgNPs Average Size
The average size of AgNPs was influenced by the curing time in the oven, albeit to a lesser extent than the effects of silver precursor concentration and temperature. Nevertheless, this influence remains significant. For example, extending the curing time from 1 min to 3 min (samples T22 to T24) resulted in an average particle size increase of approximately 5 nm.
Figure 8 illustrates the effect of curing time on the average size of AgNPs for durations of 1 min (a), 3 min (b), and 6 min (c).
Several interrelated processes likely contribute to the observed increase in the average size of AgNPs with longer curing times. Prolonged heat treatment allows for additional growth as more silver ions are reduced and incorporated into the existing nanoparticles. During this period, increased diffusion enhances the likelihood of nanoparticle collisions, which can lead to their coalescence into larger entities [
40,
41]. Additionally, the sustained application of heat during the extended curing time promotes the mobility of silver atoms on the nanoparticle surfaces, facilitating their rearrangement into more energetically stable, larger crystalline structures.
We used EDS and FESEM to analyze the nanoparticles grown on the cotton yarn surface. Before FESEM imaging, sample surfaces were coated with a 5 nm gold layer.
Figure S1 presents FESEM images of the uncoated cotton yarn at various magnifications, revealing the surface morphology. The EDS analysis confirmed the presence of AgNPs on the cotton yarn, evidenced by the characteristic Ag peak observed at 3 keV in the EDS spectra [
42]. Additionally, EDS allowed for the determination of the various elements present on the cotton yarn surface, including silver, gold, carbon, and oxygen. The EDS spectra are detailed in
Figures S2 and S3. These results provide clear evidence of AgNP formation and offer insights into the elemental composition of the coated yarn surface.
3.6. Fourier-Transformed Infrared Spectroscopy and X-Ray Diffraction
Fourier-transform infrared spectroscopy (FTIR) analysis revealed the characteristic peaks of the cellulosic structure of cotton within specific wavenumber ranges. These peaks include 3322–3334 cm⁻
1, 2890–2947 cm⁻
1, 1141–1167 cm⁻
1, and 1017–1044 cm⁻
1 [
43,
44,
45]. By comparing these detected peaks with an infrared spectroscopy correlation chart, the FTIR data allow for the identification of specific functional groups present in the sample. The peak observed at 1022 cm⁻
1 can be attributed to the stretching vibrations of the C-O-C group, indicating the presence of ether functional groups. The peak at 1159 cm⁻
1 corresponds to C-O stretching bonds, typically associated with alcohols, esters, or ethers. The presence of alkanes is inferred from the distinctive C-H stretching vibrations, signified by the absorption peak at 2890 cm⁻
1. Lastly, the peak at 3328 cm⁻
1 is characteristic of -OH stretching, commonly observed in alcohols and phenols. This spectroscopic fingerprint is crucial for identifying the molecular components and understanding the chemical composition of the cotton yarn surface.
The surface of the cotton yarn is characterized by a range of organic functional groups, as shown in
Figure 9a. FTIR analysis reveals that the growth of AgNPs did not induce any significant changes in the chemical structure of the cotton fibers. The absence of new peaks or notable shifts in the existing peaks within the FTIR spectrum is consistent with other findings and suggests no chemical bonding between the cellulosic structure of the cotton and the AgNPs [
43]. This result implies that the AgNPs are physically adsorbed onto the surface of the cotton yarn rather than forming chemical bonds with the cellulose fibers [
44].
A confirmation of the crystalline nature of the AgNPs deposited on the cotton yarn was obtained through X-ray diffraction (XRD) analysis.
Figure 9b presents the XRD pattern of the AgNP-coated cotton sample, where the characteristic peaks corresponding to the (111), (200), and (220) planes indicate the face-centered cubic (FCC) crystal structure of silver [
46]. For comparison, the XRD spectrum of the uncoated cotton yarn displays a peak at a 2θ angle of 54°, which remains present in the AgNP-coated samples. In the coated samples, a slight shift in the XRD peaks associated with the silver lattice was observed, primarily attributed to the variation in the size of the AgNPs. Additionally, the XRD patterns exhibit peak broadening, a common feature in nanoparticles that reflects their small size. This broadening can also be influenced by factors such as structural imperfections within the nanoparticles and the resulting strain, which further contribute to the observed shifts in the XRD peaks.
3.7. Antiviral Effectivity of Different Sizes of AgNPs
AgNPs have been shown to be effective antibacterial and antiviral agents. Previous research has extensively proved AgNPs’ efficiency against a broad spectrum of bacteria and viruses. The size of AgNPs is an important element in antimicrobial activity since not all sizes are effective against certain bacteria or viruses. The possibility of synthesizing AgNPs of various sizes provides a versatile approach toward targeting a wide range of life-threatening pathogens.
Table 2 describes antiviral efficacy of AgNPs against a variety of virus types and addresses the possibility for size-variable AgNPs that allow tailored antimicrobial applications.
4. Conclusions
We have successfully developed a scalable method for impregnating cotton yarn with silver nanoparticles (AgNPs), presenting a cost-effective approach with potential applications in functional coatings, wearable electronics, biomedical applications, and smart textiles. Our study highlights several critical parameters that influence the growth and average size of AgNPs during their in situ synthesis on cotton yarn. Key findings include the observation that operating under vacuum conditions (254 Torr) promotes the formation of larger AgNPs. Similarly, increasing the concentrations of the silver precursor and reducing agents results in larger nanoparticles. In contrast, raising the curing temperature from room temperature to 110 °C reduces the average size of the AgNPs. Prolonged curing times, however, lead to an increase in particle size. The use of an ice bath during synthesis effectively slows down the reaction kinetics, producing smaller nanoparticles. These insights provide a framework for controlling AgNP synthesis on cotton yarn, enabling the customization of nanoparticle size and properties to meet specific application requirements. Looking forward, further research could explore the integration of these AgNP-coated yarns into advanced textile manufacturing processes. Additionally, investigating the long-term stability and functional performance of the AgNPs under real-world conditions would be valuable. Such studies could pave the way for the development of next-generation smart textiles with enhanced functionalities tailored for diverse industrial and medical applications.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}