
Pagodane—Solution and Solid-State Vibrational Spectra


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Selection Rules
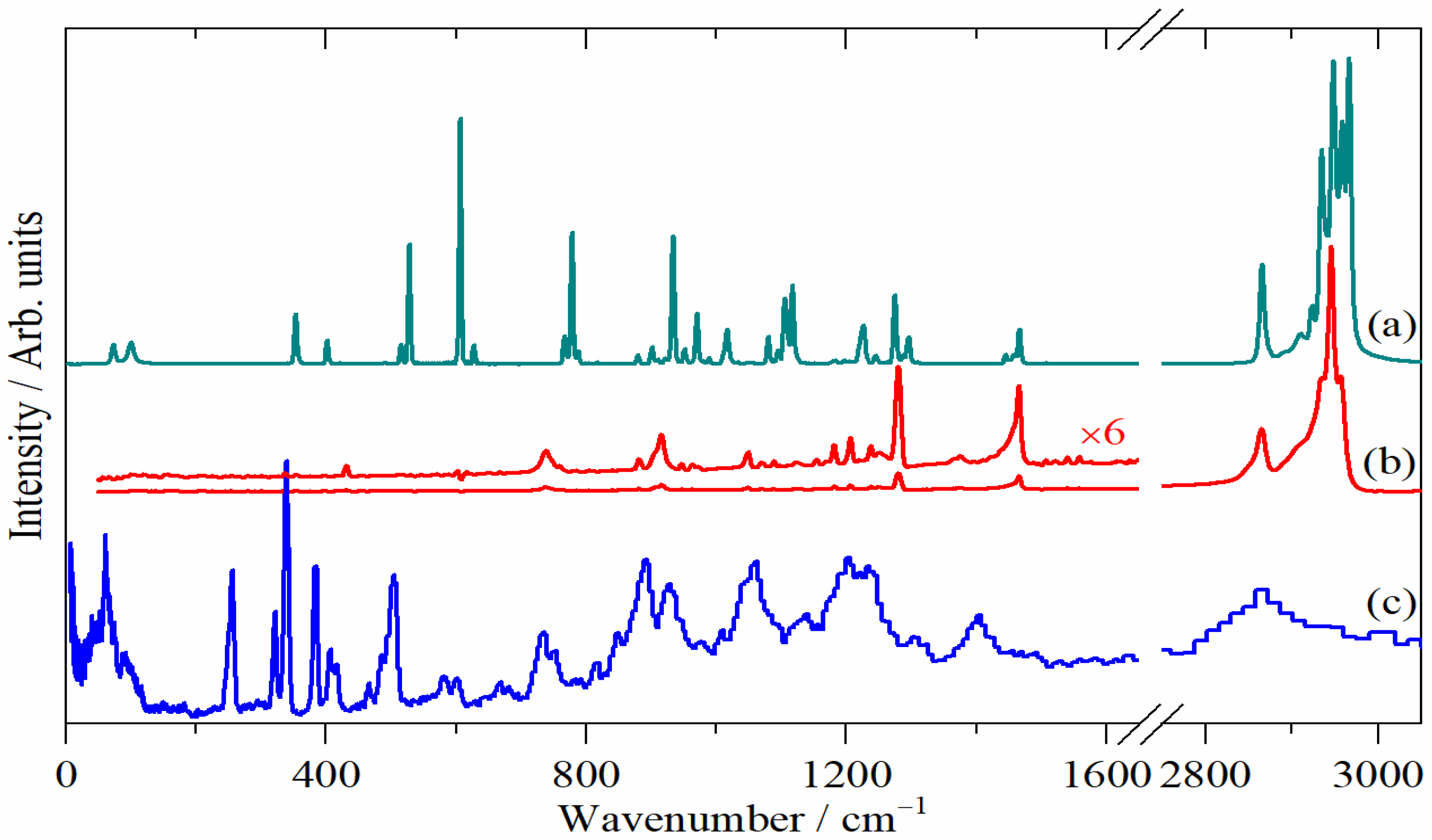
3.2. Pagodane in Solution
3.3. Pagodane in the Solid State
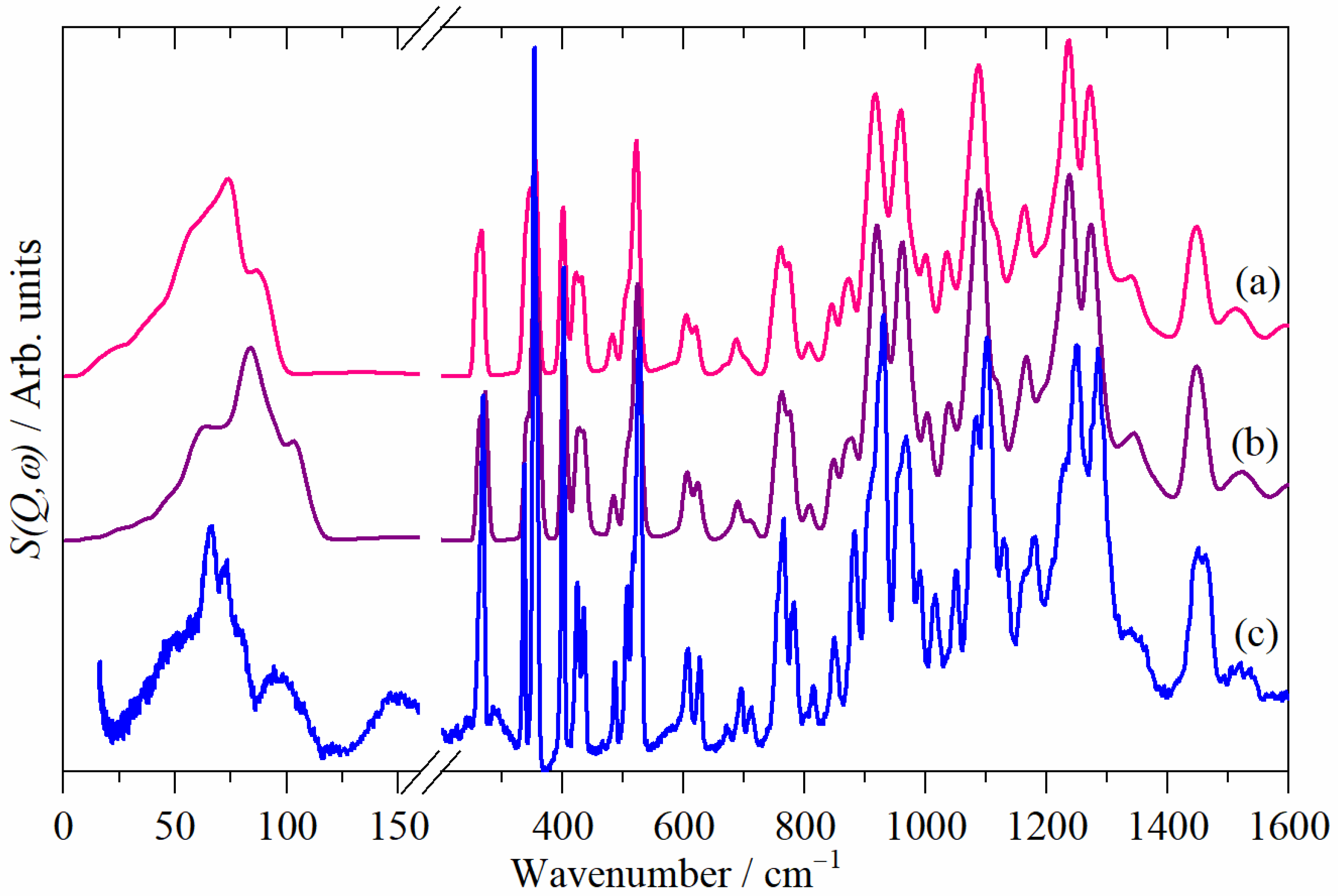
3.4. Computational Studies
3.4.1. The Lattice Mode Region (<200 cm−1)
3.4.2. The Fingerprint Region (200–1600 cm−1)
3.4.3. The C–H Stretch Region (>2800 cm−1)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fessner, W.-D.; Prinzbach, H.; Riho, C. Pagodane—An undecacyclic C20H20-polyquinane. Tetrahedron Lett. 1983, 24, 5857–5860. [Google Scholar] [CrossRef]
- Ternansky, R.J.; Balogh, D.W.; Paquette, L.A. Dodecahedrane. J. Am. Chem. Soc. 1982, 104, 4503–4504. [Google Scholar] [CrossRef]
- Prinzbach, H.; Weber, W. From an insecticide to Plato’s universe—The pagodane route to dodecahedranes: New pathways and new perspectives. Angew. Chem. Int. Ed. Engl. 1994, 33, 2239–2257. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Krishnamurthy, V.V.; Herges, R.; Bau, R.; Yuan, H.; Olah, G.A.; Fessner, W.-D.; Prinzbach, H. [1.1.1.1] and [2.2.1.1] Pagodane dications: Frozen two-electron Woodward-Hoffmann transition state models. J. Amer. Chem. Soc. 1988, 110, 7764–7772. [Google Scholar] [CrossRef]
- Allis, D.G.; Prinzbach, H.; Hudson, B.S. Inelastic neutron scattering spectra of pagodane: Experiment and DFT calculations. Chem. Phys. Lett. 2004, 386, 356–363. [Google Scholar] [CrossRef]
- Mitchell, P.C.H.; Parker, S.F.; Ramirez-Cuesta, A.J.; Tomkinson, J. Vibrational Spectroscopy with Neutrons, with Applications in Chemistry, Biology, Materials Science and Catalysis; World Scientific: Singapore, 2005. [Google Scholar]
- Parker, S.F.; Lennon, D.; Albers, P.W. Vibrational spectroscopy with neutrons—A review of new directions. Appl. Spec. 2011, 65, 1325–1341. [Google Scholar] [CrossRef]
- Parker, S.F.; Fernandez-Alonso, F.; Ramirez-Cuesta, A.J.; Tomkinson, J.; Rudić, S.; Pinna, R.S.; Gorini, G.; Fernández Castañon, J. Recent and future developments on TOSCA at ISIS. J. Phys. Conf. Ser. 2014, 554, 12003. [Google Scholar] [CrossRef]
- Pinna, R.S.; Rudić, S.; Parker, S.F.; Armstrong, J.; Zanetti, M.; Škoro, G.; Waller, S.P.; Zacek, D.; Smith, C.A.; Capstick, M.J.; et al. The neutron guide upgrade of the TOSCA spectrometer. Nucl. Instrum. Methods Phys. Res. Sect. A 2018, 896, 68–74. [Google Scholar] [CrossRef]
- The ISIS Neutron and Muon Source. Available online: https://www.isis.stfc.ac.uk/Pages/About.aspx (accessed on 31 July 2024).
- Griffiths, P.R.; de Haseth, J.A. Fourier Transform Infrared Spectrometry; Wiley-Interscience: New York, NY, USA, 1986. [Google Scholar]
- Rossi Albertini, V.; Colognesi, D.; Tomkinson, J. A study on the calibration of a time-focused inelastic neutron scattering spectrometer. J. Neutron. Res. 2000, 8, 245–259. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Krist. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Matthias Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Tkatchenko, A.; Scheffler, M. Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data. Phys. Rev. Lett. 2009, 102, 073005. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Pack, J.D.; Monkhorst, H.J. “Special points for Brillouin-zone integrations”—A reply. Phys. Rev. B 1977, 16, 1748–1749. [Google Scholar] [CrossRef]
- Refson, K.; Tulip, P.R.; Clark, S.J. Variational density-functional perturbation theory for dielectrics and lattice dynamics. Phys. Rev. B 2006, 73, 155114. [Google Scholar] [CrossRef]
- Gonze, X.; Lee, C. Dynamical matrices, Born effective charges, dielectric permittivity tensors, and interatomic force constants from density-functional perturbation theory. Phys. Rev. B 1997, 55, 10355–10368. [Google Scholar] [CrossRef]
- Milman, V.; Perlov, A.; Refson, K.; Clark, S.J.; Gavartin, J.; Winkler, B. Structural, electronic and vibrational properties of tetragonal zirconia under pressure: A density functional theory study. J. Phys. Condens. Matter 2009, 21, 485404. [Google Scholar] [CrossRef]
- Biovia Materials Studio. Available online: https://www.3ds.com/products-services/biovia/products/molecular-modeling-simulation/biovia-materials-studio (accessed on 12 July 2024).
- Dymkowski, K.; Parker, S.F.; Fernandez-Alonso, F.; Mukhopadhyay, S. AbINS: The modern software for INS interpretation. Phys. B 2018, 551, 443–448. [Google Scholar] [CrossRef]
- Fair, R.L.; Jackson, A.J.; Voneshen, D.J.; Jochym, D.B.; Le, M.D.; Refson, K.; Perring, T.G. Euphonic: Inelastic neutron scattering simulations from force constants and visualization tools for phonon properties. J. Appl. Cryst. 2022, 55, 1689–1703. [Google Scholar] [CrossRef]
- Fateley, W.G.; Dollish, F.R.; McDevitt, N.T.; Bentley, F.F. Infrared and Raman Selection Rules for Molecular and Lattice Vibrations: The Correlation Method; Wiley-Interscience: New York, NY, USA, 1972. [Google Scholar]
- Lin-Vien, D.; Colthup, N.B.; Fateley, W.G.; Grasselli, J.G. The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules; Academic Press: Boston, MA, USA, 1991. [Google Scholar]
- Perrichon, A. On the spectral resolution of the broad-band indirect-geometry time-of-flight neutron spectrometer TOSCA. Nucl. Instrum. Methods Phys. Res. A 2022, 1041, 167401. [Google Scholar] [CrossRef]
- Bellamy, L.J. The Infrared Spectra of Complex Molecules, 3rd ed.; Chapman and Hall: London, UK, 1975; p. 16. [Google Scholar]
- Hudson, B.S.; Allis, D.G.; Parker, S.F.; Ramirez-Cuesta, A.J.; Braden, D.A.; Herman, H.; Prinzbach, H. The infrared, Raman and inelastic neutron scattering spectra of dodecahedrane: An Ih molecule in Th site symmetry. J. Phys. Chem. A 2005, 109, 3418–3424. [Google Scholar] [CrossRef] [PubMed]
- Parker, S.F.; Fortes, A.D.; Nye, D.W.; Day, S.J.; Kuck, D. Centrohexaindane, a unique polyaromatic hydrocarbon bearing the rare Cq(Cq)4 core: Inelastic neutron scattering, infrared and Raman spectroscopy. Chem. Eur. J. 2023, 29, e202302057. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, S.F.; Mason, H.E.; Wilson, C.T.; Jackson, A.J. Pagodane—Solution and Solid-State Vibrational Spectra. Physchem 2024, 4, 524-535. https://doi.org/10.3390/physchem4040036
Parker SF, Mason HE, Wilson CT, Jackson AJ. Pagodane—Solution and Solid-State Vibrational Spectra. Physchem. 2024; 4(4):524-535. https://doi.org/10.3390/physchem4040036
Chicago/Turabian StyleParker, Stewart F., Hannah E. Mason, Campbell T. Wilson, and Adam J. Jackson. 2024. "Pagodane—Solution and Solid-State Vibrational Spectra" Physchem 4, no. 4: 524-535. https://doi.org/10.3390/physchem4040036
APA StyleParker, S. F., Mason, H. E., Wilson, C. T., & Jackson, A. J. (2024). Pagodane—Solution and Solid-State Vibrational Spectra. Physchem, 4(4), 524-535. https://doi.org/10.3390/physchem4040036