2. Model
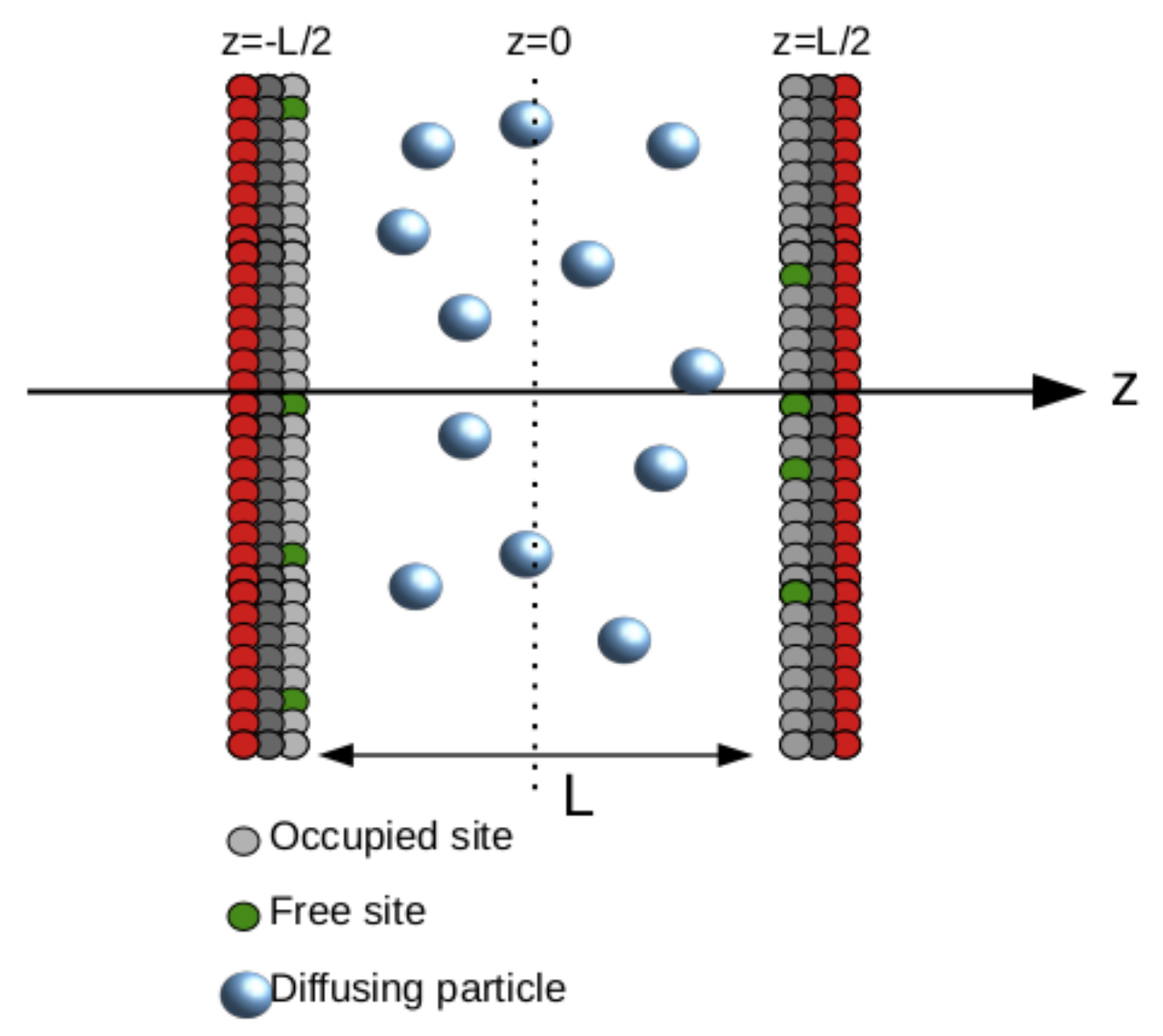
Since we want to study diffusing particles diluted in a liquid in contact with adsorbing (desorbing) walls, we assume a sample in the shape of a slab where the only relevant direction is the
z−axis. Such geometry is similar to experimental situations such as those found in liquid crystal displays, for example, where the adsorption and desorption of particles is known to play critical roles in the organization of the material [
3]. The cell has thickness
L, and the substrates are located at
(left side) and
(right side), as shown in
Figure 1. Particles in bulk obey the diffusion equation, here assumed to be diluted so the Fickian approach can be used, that is
where
D is the diffusion coefficient, here assumed to be constant in space, and
is the time- and space-dependent bulk density of particles. At the walls limiting the sample, we assume that for
, all the particles are in the bulk (all adsorption sites are vacant), and that particles may be adsorbed–desorbed according to the well-known Langmuir’s kinetic equation:
In Equation (
2), the subscript
represents the set of parameters characterizing either the surface on the left side or the surface on the right side of the sample. Moreover,
is the density of adsorbed particles and
is a parameter representing the rate of adsorption, while
represents the rate (or time) of desorption. Furthermore,
is the number of adsorption sites available at surface
i. The Langmuir kinetics assumes that the adsorption rate
) is proportional to the difference between adsorption and desorption rates and that both rates are the same at equilibrium.
Figure 1 shows the system studied here, including the occupied and free sites at the walls.
Notice that if we consider an infinity number of sites available for adsorption (
), we recover the linearized version previously used in different works [
21]. Moreover, if the total density of particles is
, and are initially in the bulk, i.e.,
, we can introduce the normalized quantities
and
, and, at equilibrium, arrive at the Langmuir isotherm:
where
is a parameter governing the steady-state equation for each substrate. The set of Equations (
1) and (
2) are solved with the aid of the current density at the walls, that is,
which implies that the number of particles must be conserved, that is
To account for memory effects, we modify Equation (
2). We introduce a kernel dependence as follows:
where
is a kernel introduced to represent distinct scenarios related to the adsorption-desorption phenomena. An equation such as Equation (
6) has been widely used to describe memory effects and non-Debye relaxations [
21,
22,
27,
28]. It has been used to describe chemisorption, physisorption, or a combination of both depending on the choice of
[
21,
22]. Although we introduced the kernel in the desorption term of Equation (
2), it is important to stress that a memory effect represents that the previous state of the particle is important for the next state, so the kernel modifies the whole dynamics of the surface. Unfortunately, the process of solving Equations (
2), (
5) and (
6) is difficult and does not have an analytical solution, so we employ a numerical method to solve it (see, for example, refs. [
29,
30]). We first introduce reduced quantities in such a way that Equation (
1) becomes
where
,
,
, and
is the diffusion time. Regarding Equation (
2), we first notice that the parameter
has the dimension of time; thus, we write it as
(now
, which we call either
l or
r, meaning left or right side, respectively) from now on. Second, we have to choose the form of the kernel
to proceed with the solution procedure. As proposed in [
21], the form
, which is a non-localized function of time, is an excellent choice to represent memory effects, such as may occur during multiple collisions of particles with the surfaces, in which energy is lost after each collision, and therefore the previous state of the particle is important in determining the next state. This memory time, represented by
, reproduces the adsorption–desorption phenomena often found in physisorption or mixed processes [
21]. If
, we recover the original kinetic equation, Equation (
2), which is better suited to describe chemisorption processes. Numerically, it is more convenient to work with differential equations than with integral equations. We therefore take the time derivative on both sides of Equation (
6) and apply the kernel
to arrive at:
where
is the adsorption time and
is a parameter that relates the number of particles available in bulk to be adsorbed to the number of sites available at the surfaces. Thus, if
, the specific surface has more sites than particles to be adsorbed, whereas if
, there are not enough sites at the surface for all the particles in bulk. The conservation of the number of particles, Equation (
5), now becomes
which also implies:
A summary of the main parameters and a brief description is given in
Appendix A. Now, in order to solve the set of Equations (
7)–(
9) (or (
10)), we employ a numerical method based on finite differences [
31]. We use a mesh with
points separated by a fixed distance
. All derivatives were taken using a central difference with a second-order approximation, using ghost points at the borders. Thus, including the two walls, it results in a system of ordinary equations in
, with
equations. The time integration was performed with the 8th-order Dormand–Prince method implemented by the GSL library [
32]. To ensure that the simulations were performed without instabilities, we monitored the conservation of the number of particles during each small increment in time by checking if Equation (
9) is still satisfied. Through all simulations, we used
, and set the absolute error to be smaller than
, allowing a free time step size, and retrieved data at every
. Moreover, we assumed the initial surface density for both walls was zero (
. For the initial distribution in bulk, we used two different initial configurations: 1—particles are uniformly distributed across the cell (
), or 2—particles are initially concentrated in a plane set in the center of the sample, that is, the initial bulk distribution obeys a Dirac delta configuration. Finally, the characteristic times used in this work come from experimental data of similarly confined systems [
3]. The adsorption parameter,
, is usually in the order of
ms
[
3,
33]. At the same time, the desorption time was estimated to be nearly 0.01 s for liquid crystal samples and close to 1 s for other isotropic samples [
3,
33]. A typical slab sample such as the one studied here is around 10 μm thick, while the diffusion coefficient is in the order of
m
2s
−1, so
s. Last, the memory time was estimated from experimental results to be
s [
21]. It is important to notice that, as long as the system obeys Fick’s law and Langmuir’s kinetic, it in principle can be investigated within the scope of this model.
3. Results and Discussion
We start by analyzing how bulk and surface distributions change over time as the characteristic parameters of the system change. In particular, we want to understand how the parameter
, which gives the ratio between particles available to be adsorbed and the number of sites available for adsorption, affects the bulk distribution of particles. Indeed, previous attempts to model adsorption–desorption with diffusing particles in a limited system [
21] have used a kinetic equation that considers the number of sites to be infinity, so we can check here the importance of having a limited number of sites on the dynamics of the system. Furthermore, in our model, we can treat both surfaces as being non-identical; that is, each surface has its dynamics, so the model becomes more closely related to experimental situations (where it is difficult to assure both surfaces are completely identical) and to other cases where adsorption is present and substrates are non-identical, such as in fuel cells (hybrid microfluidic fuel cells, for example) [
34], hybrid aligned liquid crystal cells [
35], wetting layers [
36], liquid crystals doped with dyes [
37], and polymer adsorption in confined regions [
38].
We start by showing a simple example where the role played by the parameter
can be clearly understood. For this first case, we consider both surfaces to be identical (so
,
and so on) and that the initial bulk distribution is uniform, i.e.,
.
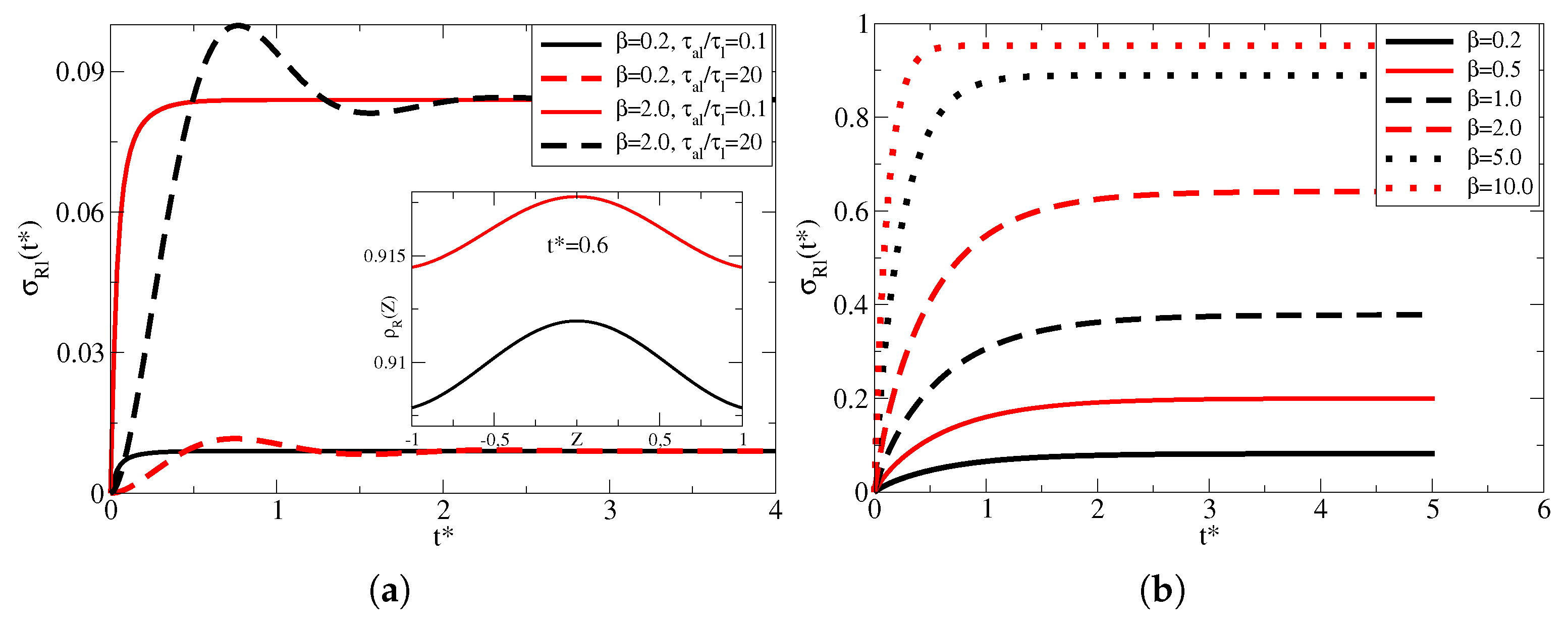
Figure 2a reproduces the surface density (in
) vs.
for
,
and for two values of
, that is, 0.1 or 20, meaning in the first case very short memory time and in the second case a long memory time, which results in the oscillations seen during the adsorption phenomena [
21]. As discussed elsewhere [
21], memory effects occur when the adsorption process depends on the previous state of the particle being adsorbed. For example, if a particle falls into an adsorbing well, it may be desorbed and eventually adsorbed again. Since the energy landscape changes after each process, the next phenomenon depends on the previous state, which in such a dynamic model it is represented by the parameter
[
21,
22]. We used two different values of
, 0.2 and 2, meaning in the first case there are five times more sites on the surfaces to adsorb than particles to be adsorbed, while in the second case, there are two times more particles in bulk to be adsorbed than available sites. Since
, the desorption rate is larger than adsorption, so the equilibrium density is fairly low. However, in the case of
, the surfaces reach a larger value, as expected (see Equation (
3)). It is also interesting to note that the adsorption peak position (
), related to memory effects, also changes depending on
, which is indicative that memory effects are also sensitive to the ratio between the number of particles to the available number of adsorbing sites. To the best of our knowledge, this is the first time memory effects are considered in Langmuir kinetics. The inset shows the bulk density for
, indicating that for larger
there is a tendency of larger bulk density at all times.
Figure 2b shows the left-side surface density for
,
and
and several values of
. Since the adsorption and desorption rates are the same, the surface coverage is more extensive and determined by the parameter
. For example, if the number of particles is ten times larger than the number of sites, the surfaces are nearly filled to maximum once the equilibrium is achieved. Clearly, the surface density for
reaches saturation quicker when compared with
.
We now start investigating the cases in which the surfaces are not identical. This is particularly interesting because one can see how one surface affects the other and the bulk distribution. Moreover, it is helpful to understand the effect of on such distributions if the substrates are seen individually. For our study, we fixed the left substrate () with the following parameters: , and . Furthermore, we kept the diffusion time the same for all the analyses, that is, .
Figure 3a shows a case in which all the parameters are the same for both substrates, except the parameter
. We chose
,
,
,
, and varied
. The substrate on the left side (parameters are always the same) is shown in the main figure, while the inset shows the substrate in
. As
gets larger, the coverage in
grows, which is expected since larger
represents more particles to be adsorbed compared with the number of sites on that specific surface. The left-side surface also saturates at higher concentration values when the parameter
of the right-side surface increases. Since the right side has fewer sites for adsorption (as
increases), the left-side surface has more particles to adsorb, since
. This trend can also be understood by Equation (
9). If
,
, whereas for
,
, where “bulk” stands for the final concentration in the bulk. Since the number of particles in bulk is never larger than 1,
saturates at higher values as
increases. In
Figure 3b, we keep the surface in
with the same parameters as in
Figure 3a but used the parameters
,
and
and
. Notice that the adsorption time is ten times larger than the desorption time, so the overall coverage is low for the right surface. In this case, changing
does not affect the left surface. Thus, for
, the surface has five times more sites than particles available, and the resulting coverage is small. If
, the right surface has room for only half of the particles, so the coverage is higher. Moreover, since the memory time is longer, the right surface displays oscillations in the adsorption–desorption process. The inset shows the bulk density for three different times, where the black curves represent
and the red curves
. In the initial moments, both cases are identical. As time passes, for
, the bulk density near the right surface for
is slightly higher because the right surface has fewer sites to adsorb, so there is an excess of particles when compared with the case where
. As time increases, the bulk densities for the two values of
become very similar, being only slightly higher for
due to the limited capability of the right surface to adsorb the particles.
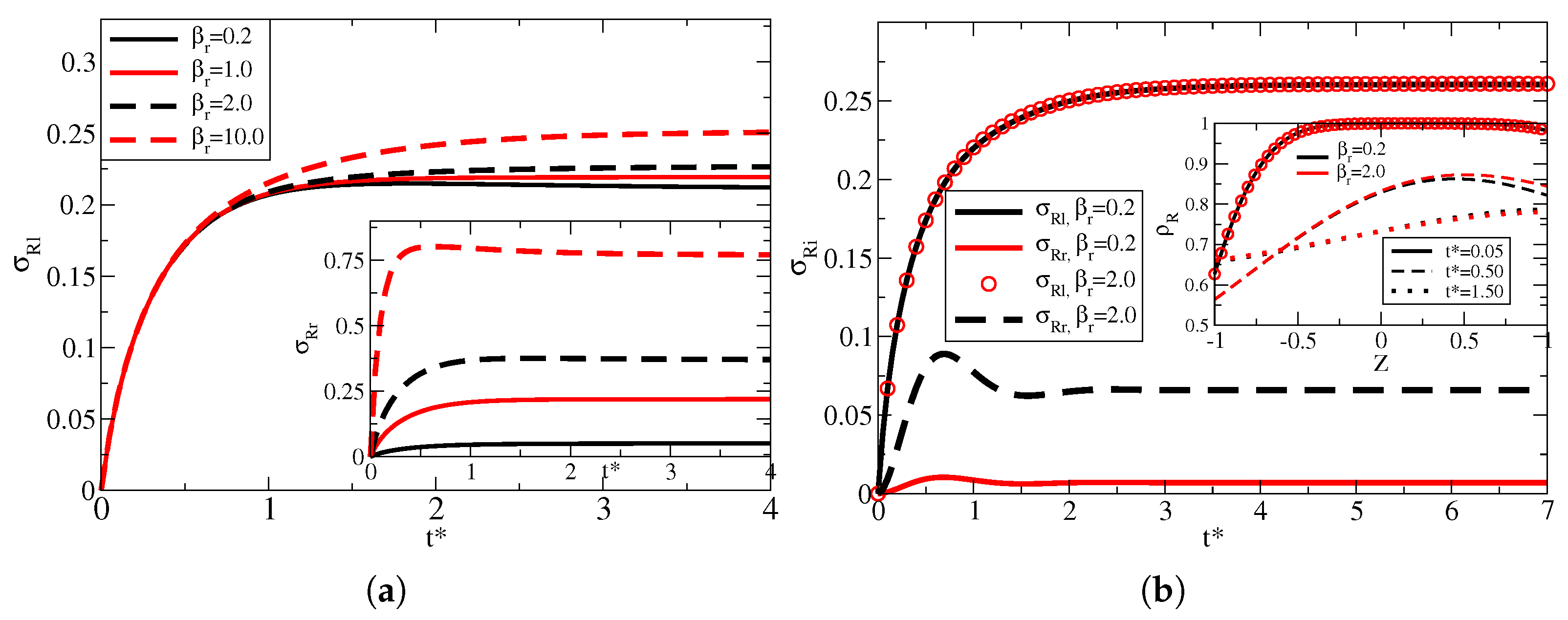
To demonstrate that a larger
represents a lower overall density of particles compared with lower
cases, in
Figure 4, we show the density of adsorbed particles for both surfaces normalized by their respective
. For this figure, we used the same parameters for the left surface as in
Figure 3. For the right surface, we used
and
, so the right surface has a high adsorption rate and neglectable memory effects. As expected, for both
and
, the right surface reaches higher coverage values when compared with the left side, which is a consequence of the high adsorption rate. However, when
, both surfaces reach the equilibrium (the left coverage is still slowly decreasing for
) much more quickly when compared with
, which is due to the quick filling of the available sites when
. On the other hand, for
, the system is still evolving (filling the adsorption sites) when
.
Figure 4 clearly shows that the equilibrium point depends on the number of available sites rather than just adsorption and desorption rates. The inset of
Figure 4 shows the bulk dynamics three different times for both values of
. Initially, when most particles are still in bulk, the distribution for both cases is the same. For
, the distribution near the left side is the same, whereas, near the right surface, the bulk distribution is lower for
when compared with
. As the system continues evolving, the case for
quickly reaches an equilibrium value, which is larger when compared with
, where even for
, the bulk distribution is still not at equilibrium (which is also seen from the surface curves).
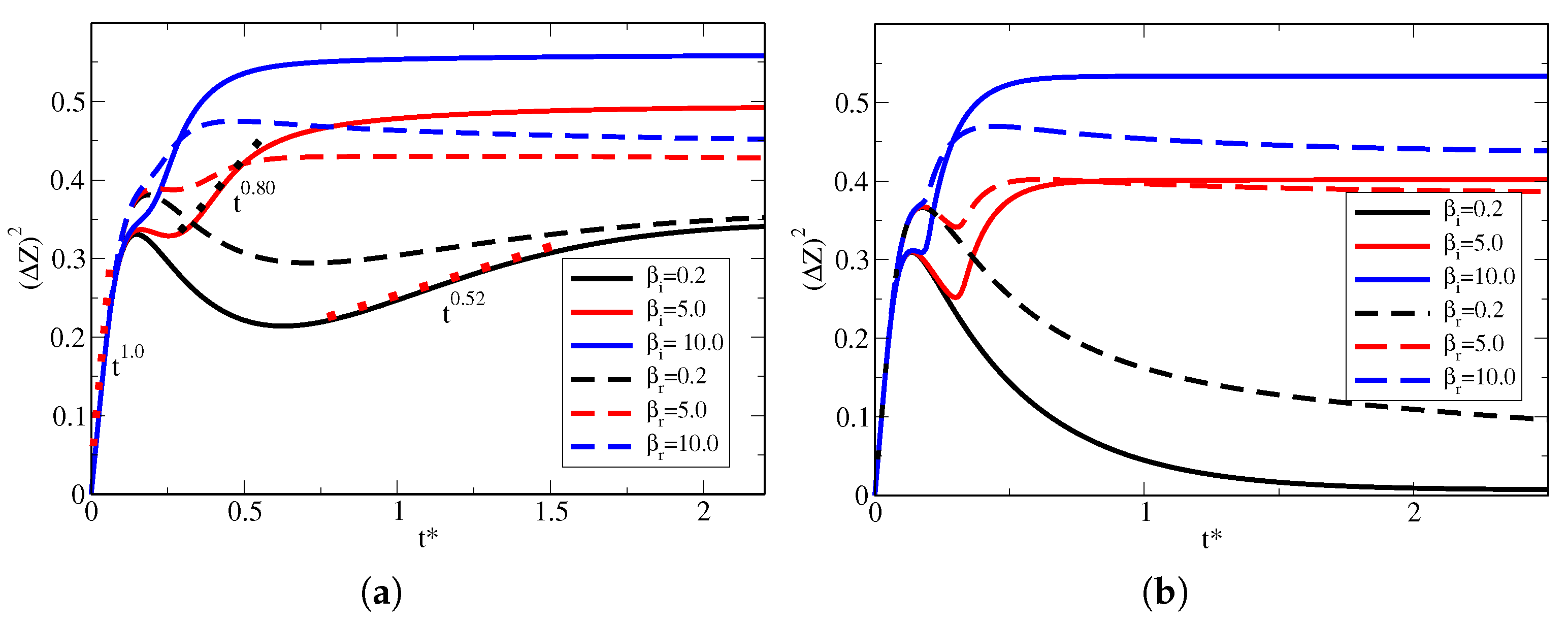
Now, we use the mean square displacement (MSD) to probe how the diffusive regimes affect the number of sites at the surfaces. This is an important parameter because it is related to the spreading of particles across the cell, which characterizes the time-dependent diffusion coefficient,
. Thus, we are now interested in understanding how the number of sites, hence the parameter
, changes the way particles diffuse in bulk. Indeed, it is known that limiting surfaces may affect how particles diffuse in the bulk [
24], which is of particular importance mainly for transport in living cells [
39,
40], but in general, the number of sites is neglected during the modeling. The MSD, which takes into account the conservation of particles (bulk distribution does not remain normalized at all times as particles leave the volume to be adsorbed at the walls), as in Equation (
9), is given by
. In other words, the MSD is calculated as:
In
Figure 5, shows the MSD vs. time for different values of the parameter
for the situations in which the surfaces are identical and the case where the surfaces are non-identical. For all cases, we used
. For the case of identical surfaces, depicted by the solid lines, we used
and
. Notice that when the MSD increases with time, the bulk distribution is spreading up, while MSD decreasing with time means that the distribution is shrinking; that is, particles are returning to the bulk. We first notice that for all the curves, the initial behavior of the MSD is equal, which corresponds to the initial spreading of particles from the center of the cell toward the surfaces. This initial diffusing process is not affected by the surfaces and corresponds to the usual diffusion that occurs for boundless samples. The black solid line represents
, while the solid red line shows
, and the solid blue curve depicts the case
. For
, after the initial free diffusion, the MSD decreases until
. This is a consequence of the desorption process that takes place together with the adsorption process (
), which is favored by the small number of particles remaining in bulk. After this minimum occurs for the MSD, the adsorption process continues, favored by the larger number of sites compared with particles in bulk, until an equilibrium is reached. For
, this minimum happens much faster and is much less pronounced. At the same time, for
, it does not appear at all, which is a consequence of a large number of particles compared with the available sites, resulting in a much weaker desorption process of the high particle concentration in bulk at all times. The dashed lines in
Figure 5a represent the same values of the parameter
but for non-identical walls. Here, the right surface is the same as used to produce the solid curves, only
is changed, but the left surface is fixed at
,
and
. Interestingly, the bulk dynamic is also affected by the non-identical surface, not only by changing how the spreading and shrinking of the distribution occurs but also by changing the overall amount of particles to reach equilibrium in bulk, a direct consequence of the ratio between the number of particles and number of sites.
Figure 5b shows the cases in which
, when the surfaces are identical, and for the right surface (
) in the case of non-identical surfaces. The left surface for non-identical surfaces remains the same as
Figure 5a. Here, since the rate of adsorption is much larger than the rate of desorption, it is expected that the surface quickly adsorbs most of the particles, leaving the bulk with a low concentration. This is true for
, where the MSD is nearly zero for
, meaning that the distribution continuously decreases where most of the particles are trapped at the surfaces. However, as the number of particles increases when compared with the number of available sites, this is no longer true, which significantly affects how diffusion takes place in the bulk, indicating that indeed the parameter
is essential, rather than just looking at the rates of adsorption and desorption as usually performed. This is true for both identical and non-identical cases.
Figure 5b shows the cases in which
, when the surfaces are identical, and for the right surface (
) in the case of non-identical surfaces. The left surface for non-identical surfaces remains the same as
Figure 5a. Here, since the rate of adsorption is much larger than the rate of desorption, it is expected that the surface quickly adsorbs most of the particles, leaving the bulk with a low concentration. This is true for
, where the MSD is nearly zero for
, meaning that the distribution continuously decreases, where most of the particles are trapped at the surfaces. However, as the number of particles increases when compared with the number of available sites, this is no longer true, which significantly affects how diffusion takes place in the bulk, indicating that indeed the parameter
is essential, rather than just looking at the rates of adsorption and desorption as usually performed. This is true for both identical and non-identical cases. Notice how the dynamics of the MSD closely resemble the data observed for the diffusive characteristics observed in some systems that are essentially anomalous and with limited amount of adsorption sites to diffusing particles, such as in living cells [
41] and in the diffusion of gold-labeled dioleoylPE in the plasma membrane of fetal rat skin keratinocyte cells [
39].
Finally, noticing how the diffusive regimes change with the parameter
is interesting. Accordingly, (
)
, where the power
a is related to how particles diffuse, that is, if
, the diffusive regime is said to be subdiffusive, if
, it is called usual, whereas for
, the diffusive regime is called superdiffusive. Usually, confined systems are subjected to adsorption–desorption phenomena. However, the ratio
is not considered when studying the diffusive regime. We observe that in the initial moments before particles reach the surfaces, the diffusion is usual, that is,
. Nonetheless, after interacting with the surfaces, the diffusion becomes essentially subdiffusive and is heavily affected by the parameter
. In
Figure 5a, we show, as dotted lines, a few examples of exponents
a for certain time intervals in which the distribution spreads after a quick desorption process. For example, for
,
, while for
,
, indicating that
changes the diffusion regime. As it is well-known, molecular crowding is one source of anomalous diffusion, especially in biological fluids [
42], which, within this model, may be altered by changing the parameter
. This fact shall be further explored in future studies.
In conclusion, we modeled a system composed of an isotropic liquid limited by adsorbing surfaces where particles diffuse and may be adsorbed/desorbed following the complete Langmuir’s kinetic equation. In the modeling process, we incorporated, in addition to the natural parameters arising from the Langmuir kinetic equation such as the ratios of adsorption, desorption, and number of available sites, memory effects that may occur depending on the inherent nature of adsorbing walls, such as occurring during chemisorption, physisorption, or a mixed process. We scaled all the variables in terms of characteristic times of the system and kept as main parameter the ratio between the available sites to the amount of particles in the bulk. It turns out that not only the dynamics but the diffusive regimes are heavily affected by such ratio, which is often neglected during adsorption dynamics. We hope our results may be helpful in describing separation processes and other systems, such as in living mater, where the limited amount of adsorption sites plays a crucial role.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




