
Interlaboratory Comparison of SpermX™ and Conventional Differential Extractions Indicated High Male DNA Recovery by the SpermX Method



Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and Pre-Quantification
2.2. Mixture Sample Preparation
2.3. SpermX Method for All Three Laboratories
2.4. Conventional Differential Extraction Method (DE)
2.5. DNA Quantification
2.6. STR Amplification and Typing
2.7. Method Comparison Studies
3. Results
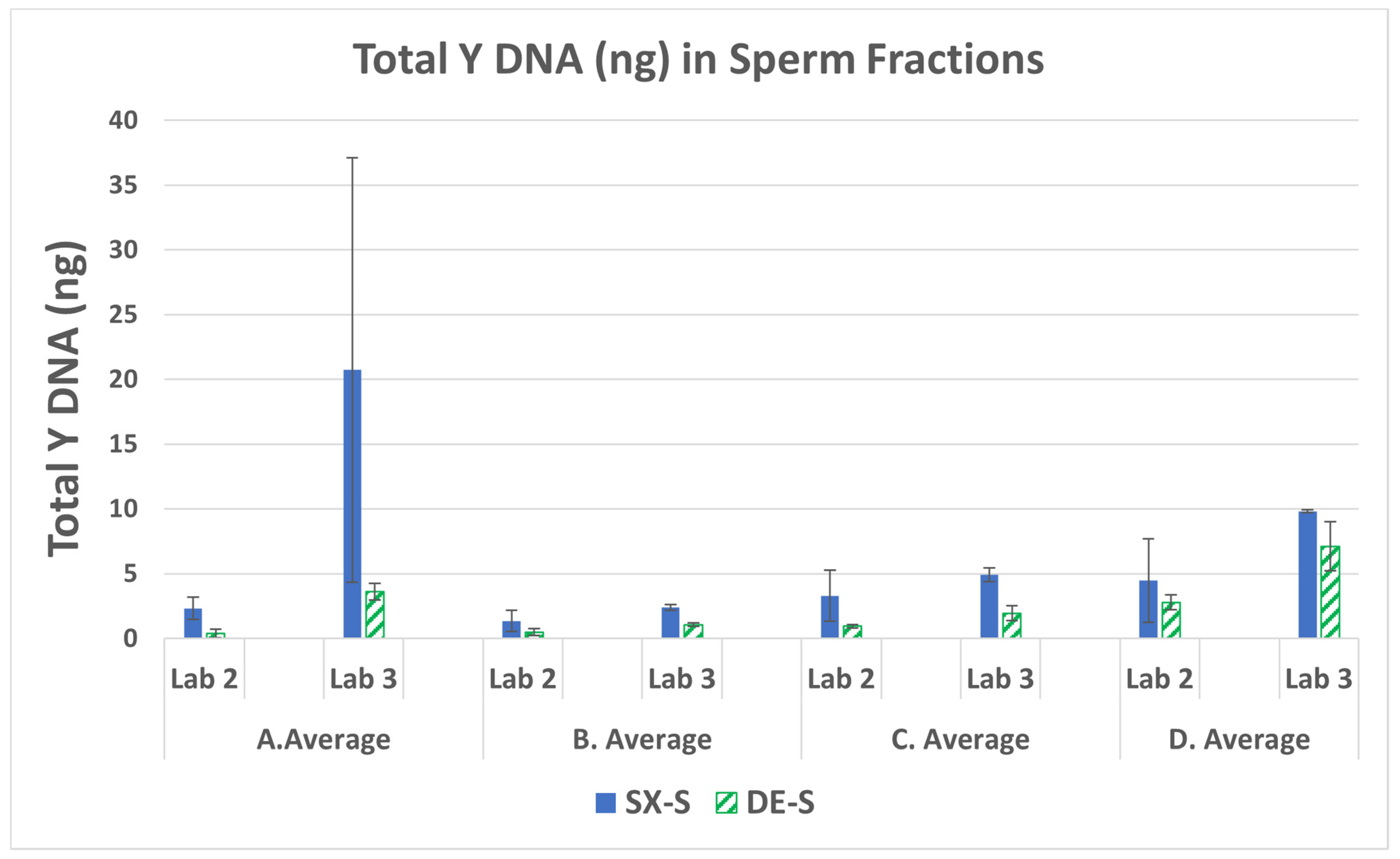
3.1. Male DNA in Sperm Fractions
3.2. Total Human to Male Ratios (S/Y) in the Sperm Fractions
3.3. Male Recovery Percentage in the Sperm Fractions
3.4. Percent Female Carryover in the Sperm Fractions
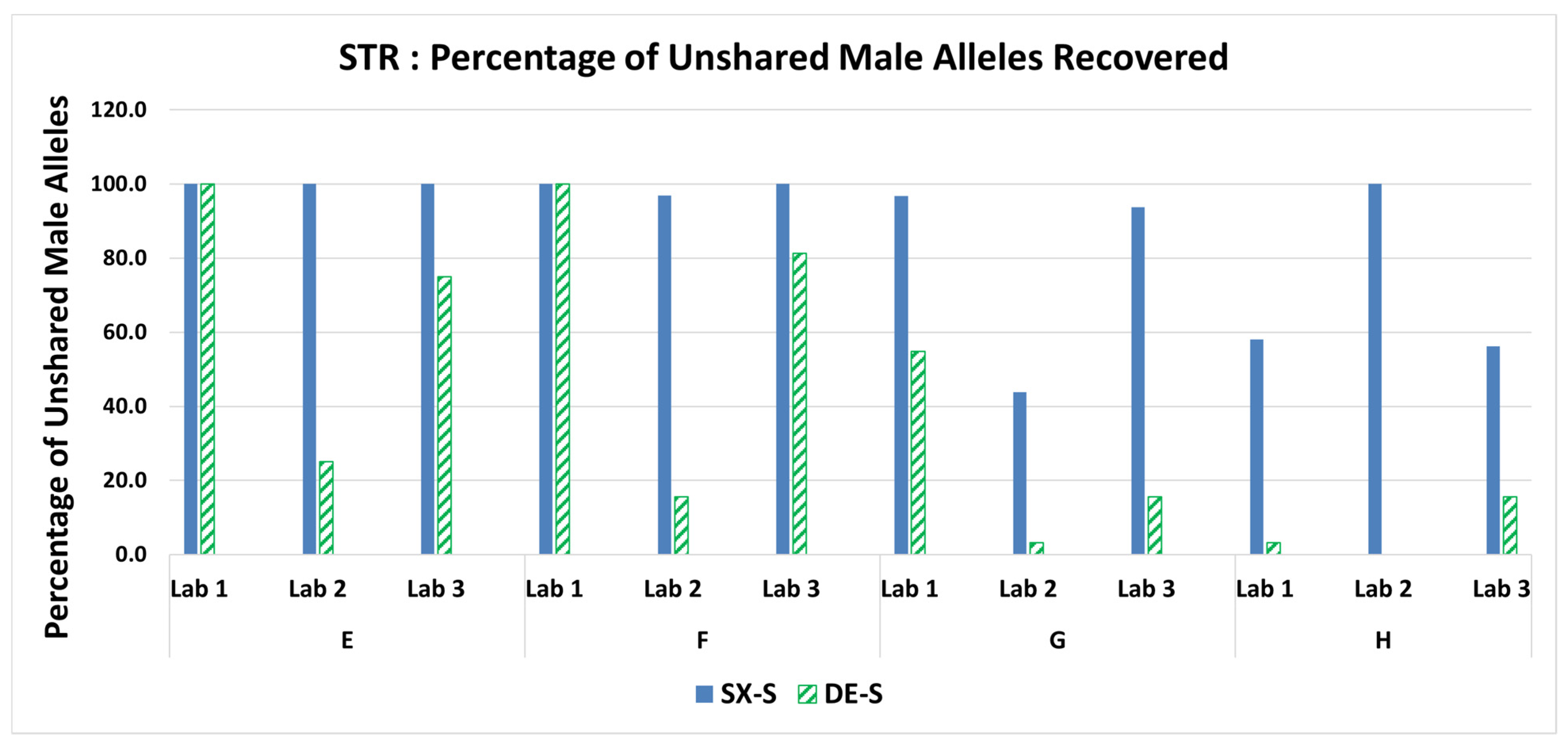
3.5. STR: Percentage of Unshared Male Alleles Recovered in the Sperm Fraction Profiles
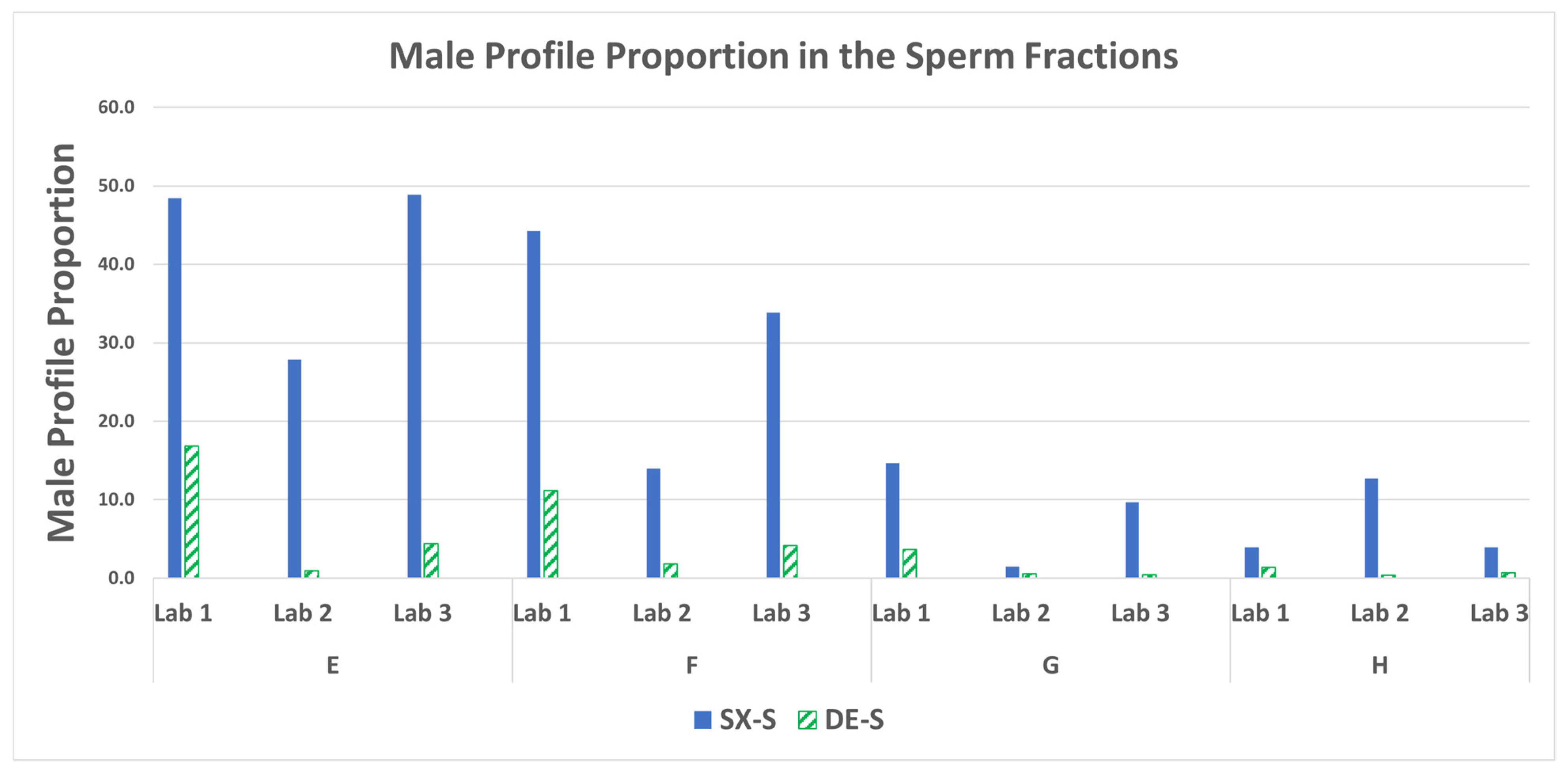
3.6. STR: Male Profile Proportion in the Sperm Fractions
3.7. STR Statistics: Probabilistic Genotyping
3.8. Variability in Recovery and Triplicate Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Prior Presentations
References
- National Sexual Assault Kit Initiative. The National Sexual Assault Kit Initiative (SAKI). 2023. Available online: https://sakitta.org/metrics/ (accessed on 10 April 2023).
- Peterson, J.; Johnson, D.; Herz, D.; Graziano, L.; Oehler, T. Sexual Assault Kit Backlog Study, NCJ Number 238500. 2012. Available online: https://nij.ojp.gov/library/publications/sexual-assault-kit-backlog-study (accessed on 3 January 2023).
- David, K. Sexual Assault Kit Processing at the Alaska Scientific Crime Detection Laboratory-Letter- Public Information. 1922. Available online: https://dps.alaska.gov/getmedia/ac3e3f7d-6812-45d3-a730-29e0eeb9527c/April-2022-Sexual-Assault-Kit-Processing-Workflow.pdf (accessed on 26 April 2023).
- Gill, P.; Jeffreys, A.J.; Werrett, D.J. Forensic application of DNA ‘fingerprints’. Nature 1985, 318, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.B.; Buoncristiani, M.R. Evaluating the efficacy of DNA differential extraction methods for sexual assault evidence. Forensic. Sci. Int. Genet. 2017, 29, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Bogas, V.; Bento, A.; Serra, A.; Brito, P.; Lopes, V.; Sampaio, L.; Gouveia, N.; Cunha, P.; Balsa, F.; São-Bento, M.; et al. Validation of sampletype I-sep DL for differential extraction and purification with prepfiler express in the automate express DNA extraction system. Forensic. Sci. Int. Genet. Suppl. Ser. 2017, 6, e353–e354. [Google Scholar] [CrossRef][Green Version]
- Paul, J. Project FORESIGHT Annual Report, 2020–2021. Faculty & Staff Scholarship. 3093. 2022. Available online: https://researchrepository.wvu.edu/faculty_publications/3093. (accessed on 26 April 2023).
- Volk, P.; Holt, A.; Chen, A.; Hanson, E.; Ballantyne, J. Enhancing the sexual assault workflow: Development of a rapid male screening assay incorporating molecular non-microscopic sperm identification. Forensic. Sci. Int. Genet. Suppl. Ser. 2019, 7, 21–22. [Google Scholar] [CrossRef]
- Timken, M.D.; Klein, S.B.; Kubala, S.; Scharnhorst, G.; Buoncristiani, M.R.; Miller, K.W.P. Automation of the standard DNA differential extraction on the Hamilton AutoLys STAR system: A proof-of-concept study. Forensic. Sci. Int. Genet. 2019, 40, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, M.C.; Cox, J.O.; Seman, L.B.; Cruz, T.D. Improved resolution of mixed STR profiles using a fully automated differential cell lysis/DNA extraction method. Forensic. Sci. Res. 2020, 5, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Nittis, M.; Franco, M.; Cochrane, C. New oral cut-off time limits in NSW. J. Forensic. Leg. Med. 2016, 44, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Spivak, H. National Best Practices for Sexual Assault Kits: A Multidisciplinary Approach. Available online: https://nij.ojp.gov/topics/articles/national-best-practices-sexual-assault-kits-multidisciplinary-approach (accessed on 3 January 2023).
- Sinha, S.K.; Brown, H.; Holt, H.; Khan, M.; Brown, R.; Sgueglia, J.B.; Loftus, A.; Murphy, G.; Montgomery, A. Development and validation of a novel method “SpermXTM” for high throughput differential extraction processing of sexual assault kits (SAKs) for DNA analysis. Forensic. Sci. Int. Genet. 2022, 59, 102690. [Google Scholar] [CrossRef] [PubMed]
- Casey, D.G.; Domijan, K.; MacNeill, S.; Rizet, D.; O’Connell, D.; Ryan, J. The Persistence of Sperm and the Development of Time Since Intercourse (TSI) Guidelines in Sexual Assault Cases at Forensic Science Ireland, Dublin, Ireland. J. Forensic. Sci. 2017, 62, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Loftus, A.; Murphy, G.; Brown, H.; Montgomery, A.; Tabak, J.; Baus, J.; Carroll, M.; Green, A.; Sikka, S.; Sinha, S. Development and validation of InnoQuant® HY, a system for quantitation and quality assessment of total human and male DNA using high copy targets. Forensic. Sci. Int. Genet. 2017, 29, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Fedder, J. Nonsperm Cells in Human Semen: With Special Reference to Seminal Leukocytes and their Possible Influence on Fertility. Arch. Androl. 1996, 36, 41–65. [Google Scholar] [CrossRef] [PubMed]
- Shewale, J.G.; Sikka, S.C.; Schneida, E.; Sinha, S.K. DNA profiling of azoospermic semen samples from vasectomized males by using Y-PLEXTM6 amplification kit. J. Forensic. Sci. 2003, 48, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Gillooly, J.F.; Hein, A.; Damiani, R. Nuclear DNA content varies with cell size across human cell types. Cold Spring Harb. Perspect. Biol. 2015, 7, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Qiagen. Available online: https://www.qiagen.com/us/products/human-id-and-forensics/investigator-solutions/qiaamp-dna-investigator-kit/ (accessed on 7 November 2022).
- Romsos, E.L.; Kline, M.C.; Farkas, N.; Duewer, D.L.; Toman, B. Certification of Standard Reference Material® 2372a Human DNA Quantitation Standard; Special Publication (NIST SP); National Institute of Standards and Technology: Gaithersburg, MD, USA, 2018. [Google Scholar] [CrossRef]
- Buckleton, J.S.; Bright, J.; Gittelson, S.; Moretti, T.R.; Onorato, A.J.; Bieber, F.R.; Budowle, B.; Taylor, D.A. The Probabilistic Genotyping Software STRmix: Utility and Evidence for its Validity. J. Forensic. Sci. 2019, 64, 393–405. [Google Scholar] [CrossRef] [PubMed]
- Simek, E.; Janssens, L. Assessment of an Automated Differential Separation Utilizing a Novel Nanofiber Filter for Sexual Assault Cases. In Proceedings of the International Symposium of Human Identification, Orlando, FL, USA, 13–16 September 2021; Available online: https://www.ishinews.com/events/assessment-of-an-automated-differential-separation-utilizing-a-novel-nanofiber-filter-for-sexual-assault-cases/ (accessed on 30 November 2023).












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Swab Set | F:M Ratio | Female (ng) | Male (ng) |
|---|---|---|---|
| A | 100:1 | 1000 | 10 |
| B | 1000:1 | 2500 | 2.5 |
| C | 1000:1 | 5000 | 5 |
| D | 750:1 | 7500 | 10 |
| E | 600:1 | 1500 | 2.5 |
| F | 1200:1 | 1500 | 1.25 |
| G | 4545:1 | 1500 | 0.33 |
| H | 18,182:1 | 1500 | 0.0825 |
| Mixture | Lab | Fold Difference | Average |
|---|---|---|---|
| A | LAB 1 | Not Performed | 5.9 |
| LAB 2 | 6.1 | ||
| LAB 3 | 5.8 | ||
| B | LAB 1 | Not Performed | 9.9 |
| LAB 2 | 10.8 | ||
| LAB 3 | 8.9 | ||
| C | LAB 1 | Not Performed | 6.0 |
| LAB 2 | 6.9 | ||
| LAB 3 | 5.0 | ||
| D | LAB 1 | Not Performed | 1.5 |
| LAB 2 | 1.6 | ||
| LAB 3 | 1.4 | ||
| E | LAB 1 | 15.3 | 20.1 |
| LAB 2 | 37.6 | ||
| LAB 3 | 7.2 | ||
| F | LAB 1 | 23.0 | 22.4 |
| LAB 2 | 30.1 | ||
| LAB 3 | 14.2 | ||
| G | LAB 1 | 167.8 | 124.3 |
| LAB 2 | 165.3 | ||
| LAB 3 | 39.9 | ||
| H | LAB 1 | 878.2 | 1533.0 |
| LAB 2 | 2787.9 | ||
| LAB 3 | 932.9 |
| Mixture | Lab | SX | DE |
|---|---|---|---|
| G | LAB 1 | 30 | 17 |
| LAB 2 | 14 | 1 | |
| LAB 3 | 30 | 5 | |
| H | LAB 1 | 18 | 1 |
| LAB 2 | 32 | 0 | |
| LAB 3 | 18 | 5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sgueglia, J.B.; Holt, H.; Hanson, E.; Nichols, J.; Kalafut, T.; Khan, M.-r.; Walsh, T.; Foley, M.; Brown, H.; Ballantyne, J.; et al. Interlaboratory Comparison of SpermX™ and Conventional Differential Extractions Indicated High Male DNA Recovery by the SpermX Method. Forensic Sci. 2023, 3, 592-610. https://doi.org/10.3390/forensicsci3040043
Sgueglia JB, Holt H, Hanson E, Nichols J, Kalafut T, Khan M-r, Walsh T, Foley M, Brown H, Ballantyne J, et al. Interlaboratory Comparison of SpermX™ and Conventional Differential Extractions Indicated High Male DNA Recovery by the SpermX Method. Forensic Sciences. 2023; 3(4):592-610. https://doi.org/10.3390/forensicsci3040043
Chicago/Turabian StyleSgueglia, Joanne B., Hailey Holt, Erin Hanson, Justina Nichols, Tim Kalafut, Mah-ro Khan, Thomas Walsh, Megan Foley, Hiromi Brown, Jack Ballantyne, and et al. 2023. "Interlaboratory Comparison of SpermX™ and Conventional Differential Extractions Indicated High Male DNA Recovery by the SpermX Method" Forensic Sciences 3, no. 4: 592-610. https://doi.org/10.3390/forensicsci3040043
APA StyleSgueglia, J. B., Holt, H., Hanson, E., Nichols, J., Kalafut, T., Khan, M.-r., Walsh, T., Foley, M., Brown, H., Ballantyne, J., & Sinha, S. K. (2023). Interlaboratory Comparison of SpermX™ and Conventional Differential Extractions Indicated High Male DNA Recovery by the SpermX Method. Forensic Sciences, 3(4), 592-610. https://doi.org/10.3390/forensicsci3040043