Transcriptomics of the Rooibos (Aspalathus linearis) Species Complex

 , , , , and
, , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Materials
2.2. Plant Sampling
2.3. Biochemical Analyses
2.4. RNA Extraction
2.5. Illumina Sequencing
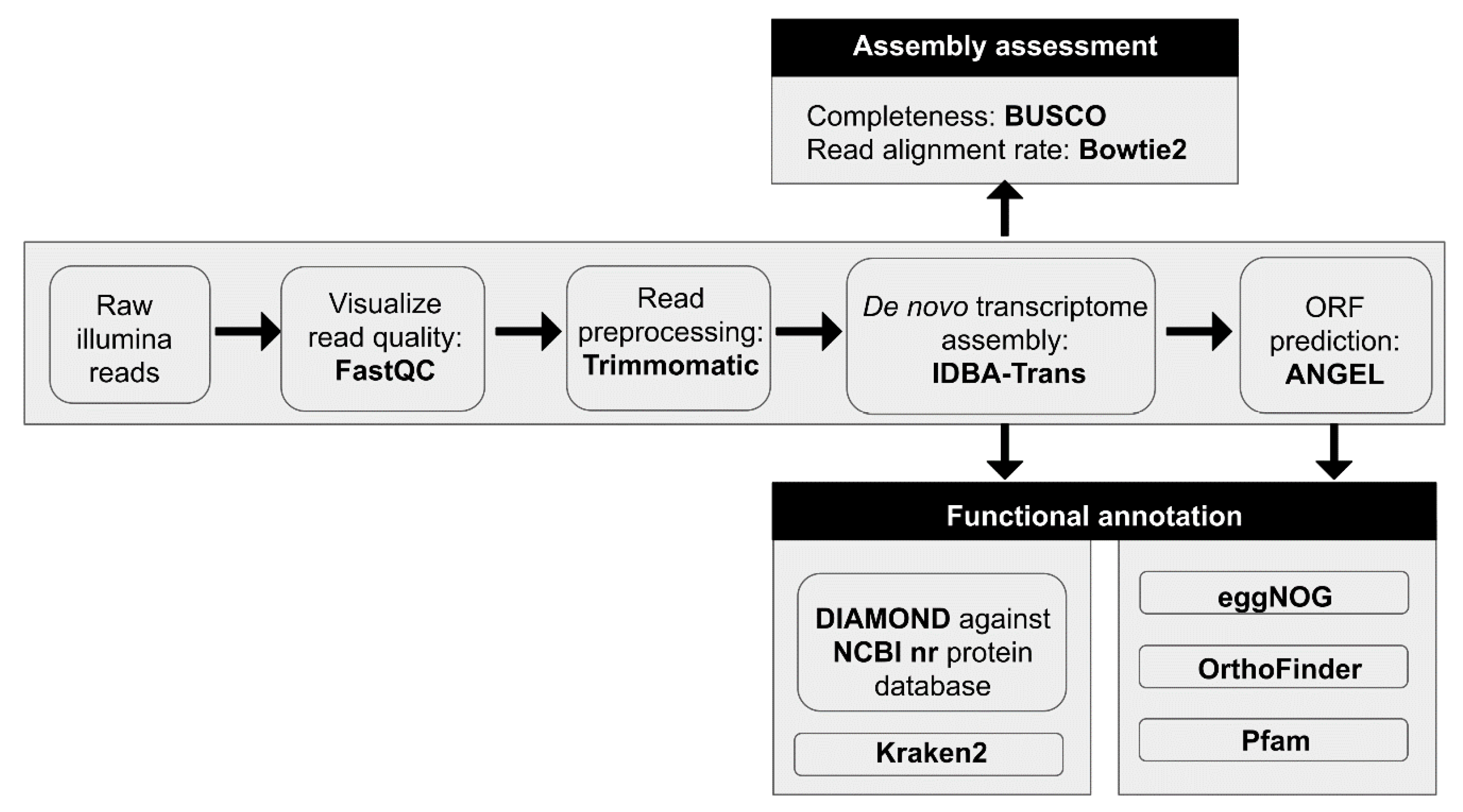
2.6. RNA-Seq Read Quality Control and Preprocessing
2.7. De novo Transcriptome Assemblies
2.8. Protein Prediction
2.9. Functional and Taxonomic Annotation
3. Results
3.1. Plant Sample Collection
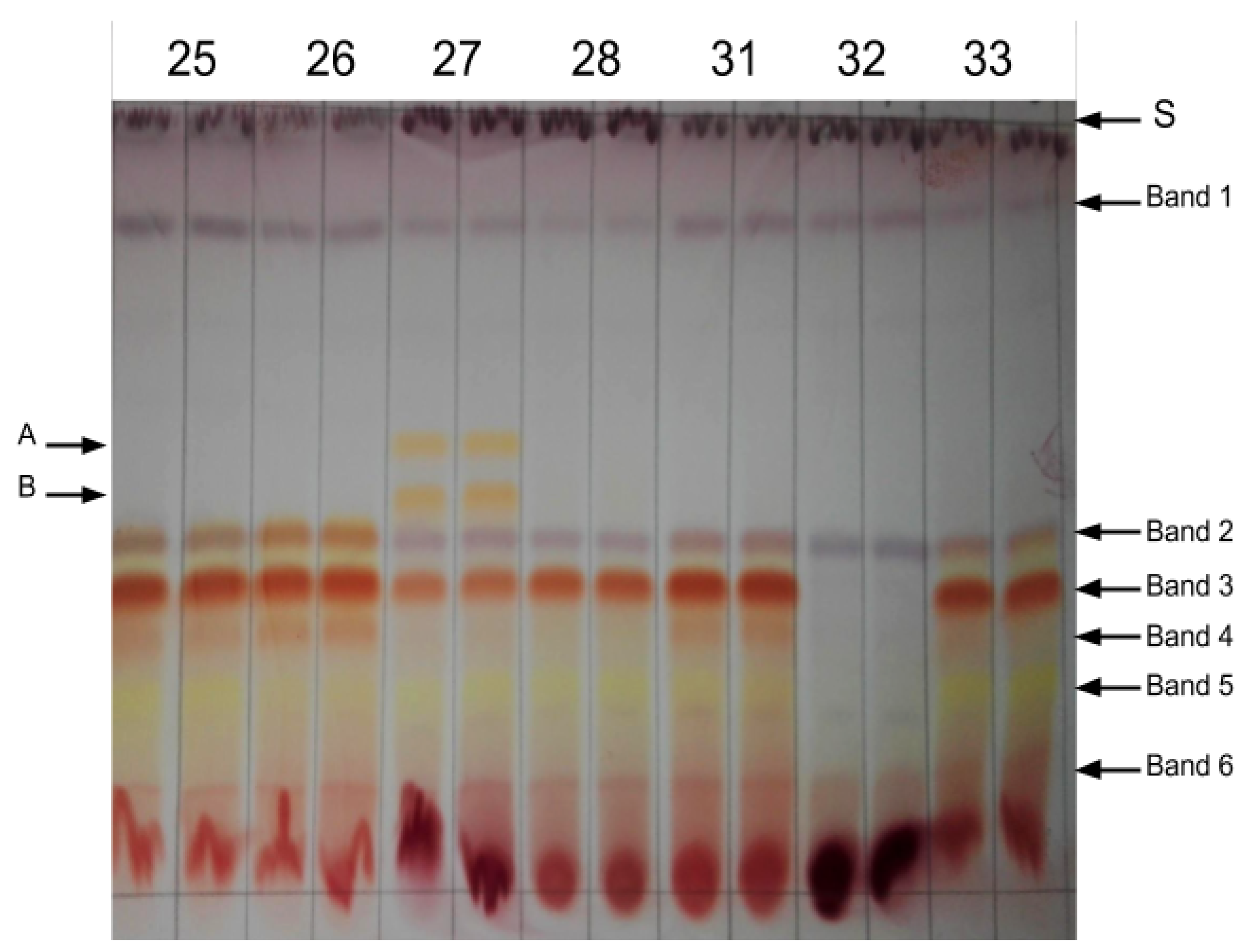
3.2. Biochemical Screening of Plant Material
3.3. Assessment of Transcriptome Assembly Programs
3.4. Assessment of ORF Prediction Tools
3.5. Rooibos Transcriptome Assemblies
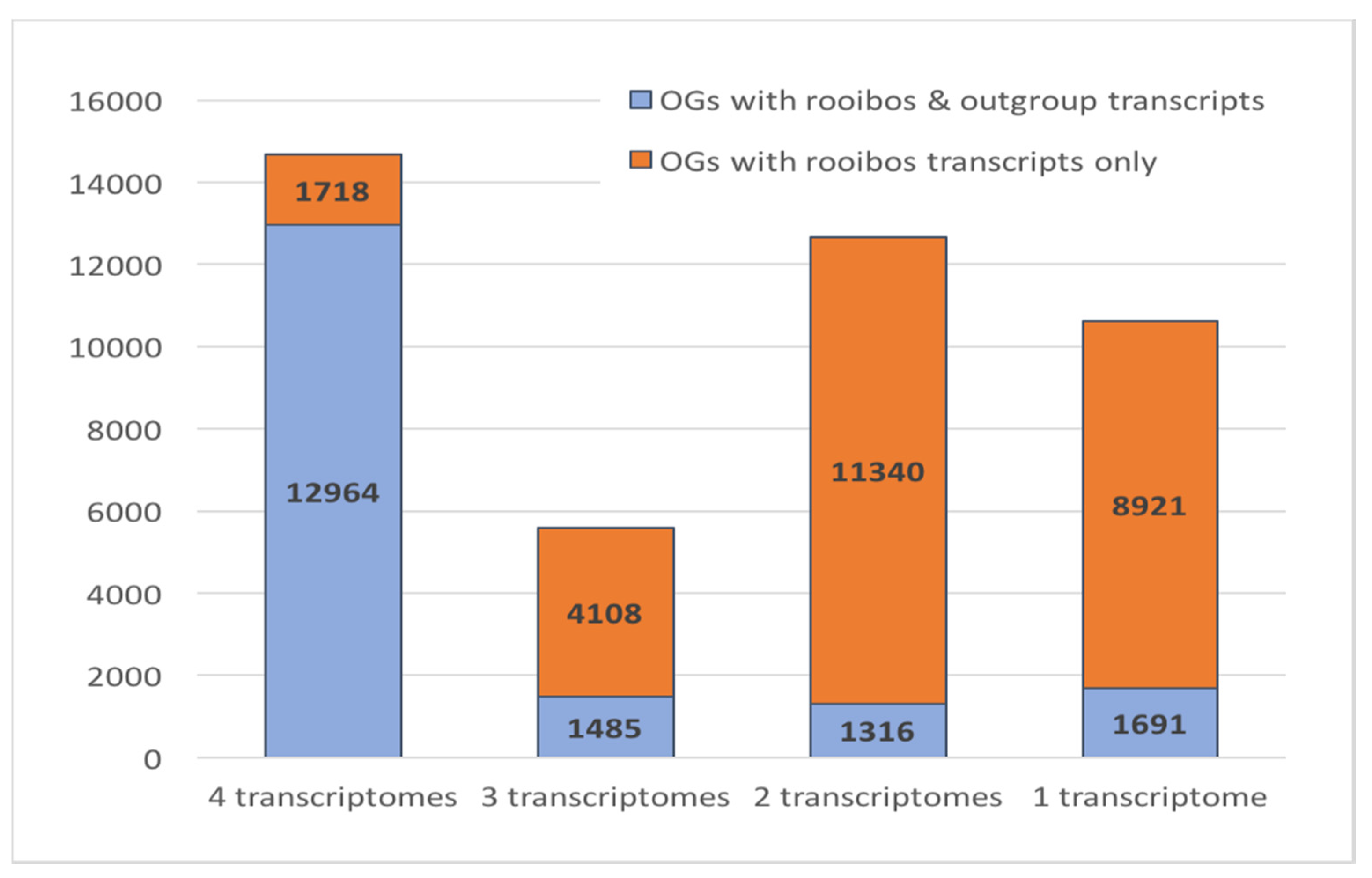
3.6. Rooibos Transcriptome Annotation: Comparative Genomics
3.7. Rooibos Transcriptome Annotation: Taxonomic Transcript Classification
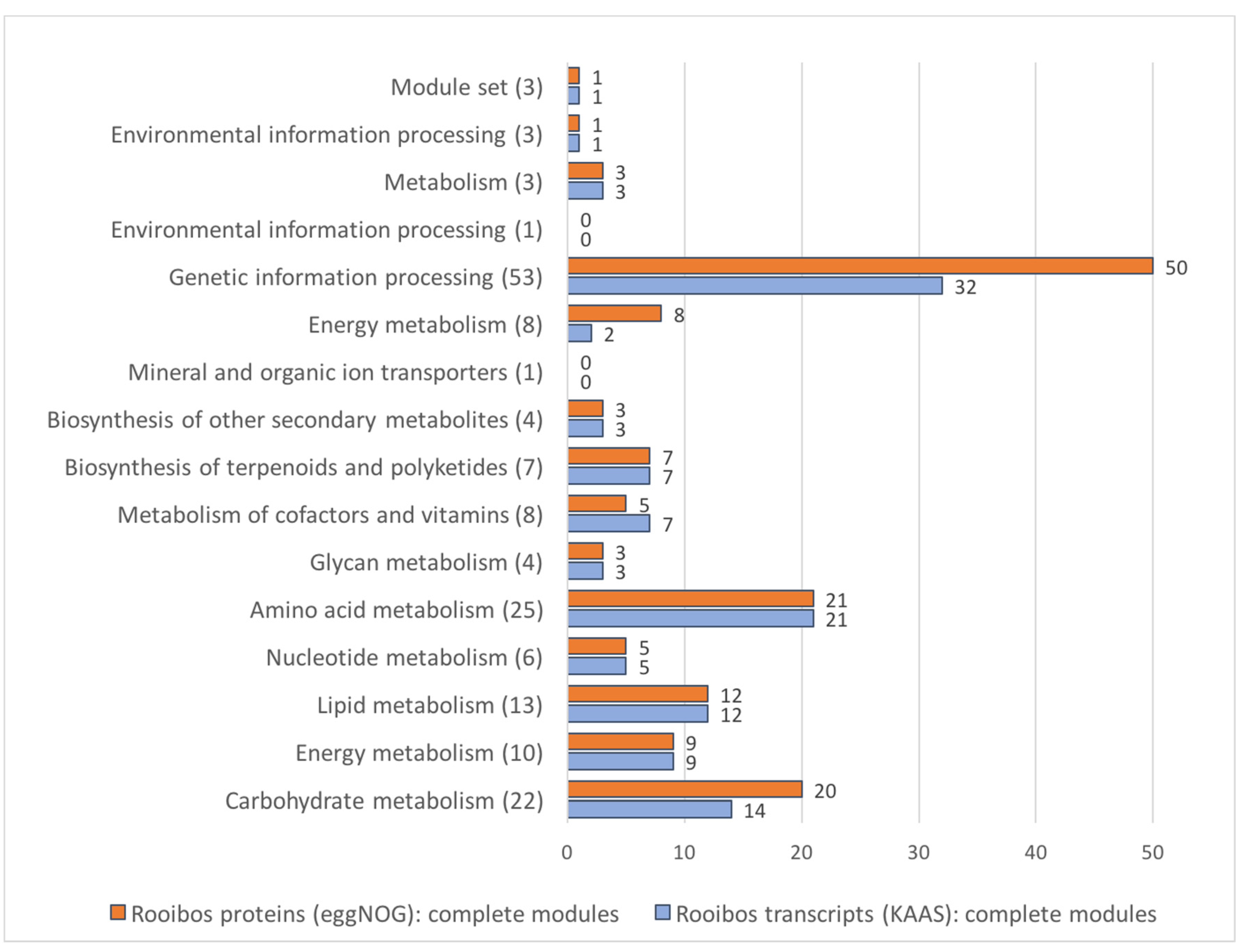
3.8. Rooibos Transcriptome Annotation: Functional Annotation
4. Discussion
4.1. High-Throughput Screening of Wild Plants for Transcriptome Analyses
4.2. Establishing Biocomputational Procedures for Non-Model Plant Transcriptome Analyses
4.3. The Rooibos Transcriptomes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Data Availability
References
- Van Wyk, B.-E.; Gorelik, B. The History and Ethnobotany of Cape Herbal Teas. South Afr. J. Bot. 2017, 110, 18–38. [Google Scholar] [CrossRef]
- Van Heerden, F.R.; van Wyk, B.-E.; Viljoen, A.M.; Steenkamp, P.A. Phenolic Variation in Wild Populations of Aspalathus linearis (Rooibos Tea). Biochem. Syst. Ecol. 2003, 31, 885–895. [Google Scholar] [CrossRef]
- Malgas, R.R.; Potts, A.J.; Oettlé, N.M.; Koelle, B.; Todd, S.W.; Verboom, G.A.; Hoffman, M.T. Distribution, Quantitative Morphological Variation and Preliminary Molecular Analysis of Different Growth Forms of Wild Rooibos (Aspalathus linearis) in the Northern Cederberg and on the Bokkeveld Plateau. South Afr. J. Bot. 2010, 76, 72–81. [Google Scholar] [CrossRef]
- Stander, M.A.; Van Wyk, B.E.; Taylor, M.J.C.; Long, H.S. Analysis of Phenolic Compounds in Rooibos Tea (Aspalathus linearis) with a Comparison of Flavonoid-Based Compounds in Natural Populations of Plants from Different Regions. J. Agric. Food Chem. 2017, 65, 10270–10281. [Google Scholar] [CrossRef] [PubMed]
- Van der Bank, M.; van der Bank, F.H.; van Wyk, B.-E. Evolution of Sprouting versus Seeding in Aspalathus linearis. Plant Syst. Evol. 1999, 219, 27–38. [Google Scholar] [CrossRef]
- Hawkins, H.-J.; Malgas, R.; Biénabe, E. Ecotypes of Wild Rooibos (Aspalathus linearis (Burm. F.) Dahlg., Fabaceae) Are Ecologically Distinct. South Afr. J. Bot. 2011, 77, 360–370. [Google Scholar] [CrossRef]
- Kunishiro, K.; Tai, A.; Yamamoto, I. Effects of Rooibos Tea Extract on Antigen-Specific Antibody Production and Cytokine Generation in vitro and in vivo. Biosci. Biotechnol. Biochem. 2001, 65, 2137–2145. [Google Scholar] [CrossRef] [PubMed]
- Hendricks, R.; Pool, E.J. The in vitro Effects of Rooibos and Black Tea on Immune Pathways. J. Immunoass. Immunochem. 2010, 31, 169–180. [Google Scholar] [CrossRef]
- Mueller, M.; Hobiger, S.; Jungbauer, A. Anti-Inflammatory Activity of Extracts from Fruits, Herbs and Spices. Food Chem. 2010, 122, 987–996. [Google Scholar] [CrossRef]
- Schloms, L.; Storbeck, K.-H.; Swart, P.; Gelderblom, W.C.A.; Swart, A.C. The Influence of Aspalathus linearis (Rooibos) and Dihydrochalcones on Adrenal Steroidogenesis: Quantification of Steroid Intermediates and End Products in H295R Cells. J. Steroid Biochem. Mol. Biol. 2012, 128, 128–138. [Google Scholar] [CrossRef]
- Khan, A.U.; Gilani, A.H. Selective Bronchodilatory Effect of Rooibos Tea (Aspalathus linearis) and Its Flavonoid, Chrysoeriol. Eur. J. Nutr. 2006, 45, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Persson, I.A.-L.; Persson, K.; Hägg, S.; Andersson, R.G. Effects of Green Tea, Black Tea and Rooibos Tea on Angiotensin-Converting Enzyme and Nitric Oxide in Healthy Volunteers. Public Health Nutr. 2010, 13, 730. [Google Scholar] [CrossRef] [PubMed]
- Persson, I.A.-L.; Josefsson, M.; Persson, K.; Andersson, R.G.G. Tea Flavanols Inhibit Angiotensin-Converting Enzyme Activity and Increase Nitric Oxide Production in Human Endothelial Cells. J. Pharm. Pharmacol. 2010, 58, 1139–1144. [Google Scholar] [CrossRef] [PubMed]
- Persson, I.A.-L. The Pharmacological Mechanism of Angiotensin-Converting Enzyme Inhibition by Green Tea, Rooibos and Enalaprila–A Study on Enzyme Kinetics. Phyther. Res. 2011, 26, 517–521. [Google Scholar] [CrossRef]
- Marnewick, J.L.; Rautenbach, F.; Venter, I.; Neethling, H.; Blackhurst, D.M.; Wolmarans, P.; Macharia, M. Effects of Rooibos (Aspalathus linearis) on Oxidative Stress and Biochemical Parameters in Adults at Risk for Cardiovascular Disease. J. Ethnopharmacol. 2011, 133, 46–52. [Google Scholar] [CrossRef]
- Dludla, P.V.; Muller, C.J.F.; Louw, J.; Joubert, E.; Salie, R.; Opoku, A.R.; Johnson, R. The Cardioprotective Effect of an Aqueous Extract of Fermented Rooibos (Aspalathus linearis) on Cultured Cardiomyocytes Derived from Diabetic Rats. Phytomedicine 2014, 21, 595–601. [Google Scholar] [CrossRef]
- Uličná, O.; Vančová, O.; Božek, P.; Čársky, J.; Šebeková, K.; Boor, P.; Nakano, M.; Greksák, M. Rooibos Tea (Aspalathus linearis) Partially Prevents Oxidative Stress in Streptozotocin-Induced Diabetic Rats. Physiol. Res. 2006, 55, 157–164. [Google Scholar]
- Muller, C.J.F.; Joubert, E.; de Beer, D.; Sanderson, M.; Malherbe, C.J.; Fey, S.J.; Louw, J. Acute Assessment of an Aspalathin-Enriched Green Rooibos (Aspalathus linearis) Extract with Hypoglycemic Potential. Phytomedicine 2012, 20, 32–39. [Google Scholar] [CrossRef]
- Mazibuko, S.E.; Muller, C.J.F.; Joubert, E.; de Beer, D.; Johnson, R.; Opoku, A.R.; Louw, J. Amelioration of Palmitate-Induced Insulin Resistance in C2C12 Muscle Cells by Rooibos (Aspalathus linearis). Phytomedicine 2013, 20, 813–819. [Google Scholar] [CrossRef]
- Kamakura, R.; Son, M.J.; de Beer, D.; Joubert, E.; Miura, Y.; Yagasaki, K. Antidiabetic Effect of Green Rooibos (Aspalathus linearis) Extract in Cultured Cells and Type 2 Diabetic Model KK-A(y) Mice. Cytotechnology 2015, 67, 699–710. [Google Scholar] [CrossRef]
- Mueller, M.; Jungbauer, A. Culinary Plants, Herbs and Spices—A Rich Source of PPARγ Ligands. Food Chem. 2009, 117, 660–667. [Google Scholar] [CrossRef]
- Beltrán-Debón, R.; Rull, A.; Rodríguez-Sanabria, F.; Iswaldi, I.; Herranz-López, M.; Aragonès, G.; Camps, J.; Alonso-Villaverde, C.; Menéndez, J.A.; Micol, V.; et al. Continuous Administration of Polyphenols from Aqueous Rooibos (Aspalathus Linearis) Extract Ameliorates Dietary-Induced Metabolic Disturbances in Hyperlipidemic Mice. Phytomedicine 2011, 18, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, M.; Mazibuko, S.E.; Joubert, E.; de Beer, D.; Johnson, R.; Pheiffer, C.; Louw, J.; Muller, C.J.F. Effects of Fermented Rooibos (Aspalathus inearis) on Adipocyte Differentiation. Phytomedicine 2014, 21, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Ajuwon, O.R.; Gowder, L.M.D.E.-S.J.T. Rooibos (Aspalathus linearis) and Its Major Flavonoids—Potential Against Oxidative Stress-Induced Conditions. In Basic Principles and Clinical Significance of Oxidative Stress; Marnewick, J.L., Ed.; IntechOpen: Rijeka, Croatia, 2015; Chapter 7. [Google Scholar]
- Smith, C.; Swart, A. Aspalathus linearis (Rooibos)–A Functional Food Targeting Cardiovascular Disease. Food Funct. 2018, 9, 5041–5058. [Google Scholar] [CrossRef]
- Sasaki, M.; Nishida, N.; Shimada, M. A Beneficial Role of Rooibos in Diabetes Mellitus: A Systematic Review and Meta-Analysis. Molecules 2018, 23, 839. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; De Beer, D.; Dludla, P.V.; Ferreira, D.; Muller, C.J.; Joubert, E. Aspalathin from Rooibos (Aspalathus linearis): A Bioactive C-Glucosyl Dihydrochalcone with Potential to Target the Metabolic Syndrome. Planta Med. 2018, 84, 568–583. [Google Scholar] [CrossRef]
- Joubert, E.; Gelderblom, W.C.A.; Louw, A.; de Beer, D. South African Herbal Teas: Aspalathus linearis, Cyclopia Spp. and Athrixia phylicoides—A Review. J. Ethnopharmacol. 2008, 119, 376–412. [Google Scholar] [CrossRef]
- Rojas-Garbanzo, C.; Zimmermann, B.F.; Schulze-Kaysers, N.; Schieber, A. Characterization of Phenolic and Other Polar Compounds in Peel and Flesh of Pink Guava (Psidium guajava L. Cv. ‘Criolla’) by Ultra-High Performance Liquid Chromatography with Diode Array and Mass Spectrometric Detection. Food Res. Int. 2017, 100, 445–453. [Google Scholar] [CrossRef]
- Kreuz, S.; Joubert, E.; Waldmann, K.-H.; Ternes, W. Aspalathin, a Flavonoid in Aspalathus linearis (Rooibos), Is Absorbed by Pig Intestine as a C-Glycoside. Nutr. Res. 2008, 28, 690–701. [Google Scholar] [CrossRef]
- McKay, D.L.; Blumberg, J.B. A Review of the Bioactivity of South African Herbal Teas: Rooibos (Aspalathus linearis) and Honeybush (Cyclopia intermedia). Phyther. Res. 2007, 21, 1–16. [Google Scholar] [CrossRef]
- Joubert, E.; de Beer, D. Rooibos (Aspalathus linearis) beyond the Farm Gate: From Herbal Tea to Potential Phytopharmaceutical. South Afr. J. Bot. 2011, 77, 869–886. [Google Scholar] [CrossRef]
- Joubert, E.; de Beer, D.; Malherbe, C.J.; Muller, N.; Bonnet, S.L.; van der Westhuizen, J.H.; Ferreira, D. Occurrence and Sensory Perception of Z-2-(β-d-Glucopyranosyloxy)-3-Phenylpropenoic Acid in Rooibos (Aspalathus linearis). Food Chem. 2013, 136, 1078–1085. [Google Scholar] [CrossRef] [PubMed]
- Muller, C.J.F.; Malherbe, C.J.; Chellan, N.; Yagasaki, K.; Miura, Y.; Joubert, E. Potential of Rooibos, Its Major C-Glucosyl Flavonoids, and Z-2-(β-D-Glucopyranosyloxy)-3-Phenylpropenoic Acid in Prevention of Metabolic Syndrome. Crit. Rev. Food Sci. Nutr. 2018, 58, 227–246. [Google Scholar] [CrossRef]
- Le Nguyen, K.; Grondin, A.; Courtois, B.; Gantet, P. Next-Generation Sequencing Accelerates Crop Gene Discovery. Trends Plant Sci. 2019, 24, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Volpicella, M.; Leoni, C.; Costanza, A.; Fanizza, I.; Placido, A.; Ceci, L.R. Genome walking by next generation sequencing approaches. Biology 2012, 1, 495–507. [Google Scholar] [CrossRef]
- Góngora-Castillo, E.; Fedewa, G.; Yeo, Y.; Chappell, J.; DellaPenna, D.; Buell, C.R. Genomic Approaches for Interrogating the Biochemistry of Medicinal Plant Species. Methods Enzym. 2012, 517, 139–159. [Google Scholar]
- Tripathi, S.; Jadaun, J.S.; Chandra, M.; Sangwan, N.S. Medicinal Plant Transcriptomes: The New Gateways for Accelerated Understanding of Plant Secondary Metabolism. Plant Genet. Resour. 2016, 14, 256–269. [Google Scholar] [CrossRef]
- Sun, L.; Rai, A.; Rai, M.; Nakamura, M.; Kawano, N.; Yoshimatsu, K.; Suzuki, H.; Kawahara, N.; Saito, K.; Yamazaki, M. Comparative Transcriptome Analyses of Three Medicinal Forsythia Species and Prediction of Candidate Genes Involved in Secondary Metabolisms. J. Nat. Med. 2018, 72, 867–881. [Google Scholar] [CrossRef]
- Cao, X.; Zhang, F.; Yuan, B.; Meng, L.; Yang, X.; Jiang, J. De Novo Transcriptome Sequencing and Analysis of Euphorbia pekinensis Rupr. and Identification of Genes Involved in Diterpenoid Biosynthesis. Plant Gene. 2017, 12, 33–42. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Guo, F.; Zhan, L.; Mohr, T.; Cheng, P.; Huo, N.; Gu, R.; Pei, D.; Sun, J.; et al. Deep Sequencing and Transcriptome Analyses to Identify Genes Involved in Secoiridoid Biosynthesis in the Tibetan Medicinal Plant Swertia Mussotii. Sci. Rep. 2017, 7, 43108. [Google Scholar] [CrossRef]
- Evangelistella, C.; Valentini, A.; Ludovisi, R.; Firrincieli, A.; Fabbrini, F.; Scalabrin, S.; Cattonaro, F.; Morgante, M.; Mugnozza, G.S.; Keurentjes, J.J.B. De Novo Assembly, Functional Annotation, and Analysis of the Giant Reed (Arundo donax L.) Leaf Transcriptome Provide Tools for the Development of a Biofuel Feedstock. Biotechnol. Biofuels 2017, 10, 138. [Google Scholar] [CrossRef]
- Lin, Z.; An, J.; Wang, J.; Niu, J.; Ma, C.; Wang, L.; Yuan, G.; Shi, L.; Liu, L.; Zhang, J.; et al. Integrated Analysis of 454 and Illumina Transcriptomic Sequencing Characterizes Carbon Flux and Energy Source for Fatty Acid Synthesis in Developing Lindera glauca Fruits for Woody Biodiesel. Biotechnol. Biofuels 2017, 10, 134. [Google Scholar] [CrossRef] [PubMed]
- Sainger, M.; Jaiwal, A.; Sainger, P.A.; Chaudhary, D.; Jaiwal, R.; Jaiwal, P.K. Advances in Genetic Improvement of Camelina Sativa for Biofuel and Industrial Bio-Products. Renew. Sustain. Energy Rev. 2017, 68, 623–637. [Google Scholar] [CrossRef]
- Chen, C.; Zheng, Y.; Zhong, Y.; Wu, Y.; Li, Z.; Xu, L.-A.; Xu, M. Transcriptome Analysis and Identification of Genes Related to Terpenoid Biosynthesis in Cinnamomum Camphora. BMC Genom. 2018, 19, 550. [Google Scholar] [CrossRef]
- Ali, M.; Li, P.; She, G.; Chen, D.; Wan, X.; Zhao, J. Transcriptome and Metabolite Analyses Reveal the Complex Metabolic Genes Involved in Volatile Terpenoid Biosynthesis in Garden Sage (Salvia Officinalis). Sci. Rep. 2017, 7, 16074. [Google Scholar] [CrossRef] [PubMed]
- Narnoliya, L.K.; Kaushal, G.; Singh, S.P.; Sangwan, R.S. De Novo Transcriptome Analysis of Rose-Scented Geranium Provides Insights into the Metabolic Specificity of Terpene and Tartaric Acid Biosynthesis. BMC Genom. 2017, 18, 74. [Google Scholar] [CrossRef]
- Goldblatt, P.; Manning, J. Cape plants: A conspectus of the Cape flora of South Africa; South African National Biodiversity Institute Libraries: Pretoria, South Africa, 2000; Volume 9. [Google Scholar]
- Van Wyk, B.-E.; Van Oudtshoorn, B.; Gericke, N. Medicinal Plants of South Africa; Briza Publications: Pretoria, South Africa, 2014. [Google Scholar]
- Stander, E.A.; Williams, W.; Rautenbach, F.; Le Roes-Hill, M.; Mgwatyu, Y.; Marnewick, J.; Hesse, U. Visualization of Aspalathin in Rooibos (Aspalathus linearis) Plant and Herbal Tea Extracts Using Thin-Layer Chromatography. Molecules 2019, 24, 938. [Google Scholar] [CrossRef]
- Bramati, L.; Minoggio, M.; Gardana, C.; Simonetti, P.; Mauri, P.; Pietta, P. Quantitative Characterization of Flavonoid Compounds in Rooibos Tea (Aspalathus linearis) by LC−UV/DAD. J. Agric. Food Chem. 2002, 50, 5513–5519. [Google Scholar] [CrossRef]
- Smart, M.; Roden, L.C. A Small-Scale RNA Isolation Protocol Useful for High-Throughput Extractions from Recalcitrant Plants. South Afr. J. Bot. 2010, 76, 375–379. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhuru, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De Novo Transcriptome Assembly with Short RNA-Seq Reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Chin, F.Y.L.; Leung, H.C.M.; Lv, M.-J.; Yiu, S.-M.; Zhu, X.-G. IDBA-Tran: A More Robust de Novo de Bruijn Graph Assembler for Transcriptomes with Uneven Expression Levels. Bioinformatics 2013, 29, i326–i334. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing Genome Assembly and Annotation Completeness with Single-Copy Orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO Applications from Quality Assessments to Gene Prediction and Phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357. [Google Scholar] [CrossRef]
- Tang, S.; Lomsadze, A.; Borodovsky, M. Identification of Protein Coding Regions in RNA Transcripts. Nucleic Acids Res. 2015, 43, e78. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving Fundamental Biases in Whole Genome Comparisons Dramatically Improves Orthogroup Inference Accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef]
- Tang, H.; Krishnakumar, V.; Bidwell, S.; Rosen, B.; Chan, A.; Zhou, S.; Gentzbittel, L.; Childs, K.L.; Yandell, M.; Gundlach, H.; et al. An Improved Genome Release (Version Mt4.0) for the Model Legume Medicago truncatula. BMC Genom. 2014, 15, 312. [Google Scholar] [CrossRef]
- Kawahara, Y.; de la Bastide, M.; Hamilton, J.P.; Kanamori, H.; McCombie, W.R.; Ouyang, S.; Schwartz, D.C.; Tanaka, T.; Wu, J.; Zhou, S.; et al. Improvement of the Oryza Sativa Nipponbare Reference Genome Using Next Generation Sequence and Optical Map Data. Rice 2013, 6, 4. [Google Scholar] [CrossRef]
- Sato, S.; Nakamura, Y.; Kaneko, T.; Asamizu, E.; Kato, T.; Nakao, M.; Sasamoto, S.; Watanabe, A.; Ono, A.; Kawashima, K.; et al. Genome Structure of the Legume, Lotus Japonicus. DNA Res. 2008, 15, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Hane, J.K.; Ming, Y.; Kamphuis, L.; Nelson, M.N.; Garg, G.; Atkins, C.; Bayer, P.E.; Bravo, A.; Bringans, S.; Cannon, S.; et al. A Comprehensive Draft Genome Sequence for Lupin (Lupinus angustifolius), an Emerging Health Food: Insights into Plant-Microbe Interactions and Legume Evolution. Plant Biotechnol. J. 2017, 15, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2014, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An Automatic Genome Annotation and Pathway Reconstruction Server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Bork, P.; von Mering, C.; Szklarczyk, D.; Jensen, L.J. Fast Genome-Wide Functional Annotation through Orthology Assignment by EggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef]
- Fin, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Quareshi, M.; Vegas, A.S.; et al. The Pfam Protein Families Database: Towards a More Sustainable Future. Nucleic Acids Res. 2016, 44, 222–230. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast Metagenomic Sequence Classification Using Exact Alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Mohammad, A.; Bhawani, S.A.; Sharma, S. Analysis of Herbal Products by Thin-Layer Chromatography: A Review. Int. J. Pharma Bio Sci. 2010, 1, PS16. [Google Scholar]
- Sherma, J.; Rabel, F. A Review of Thin Layer Chromatography Methods for Determination of Authenticity of Foods and Dietary Supplements. J. Liq. Chromatogr. Relat. Technol. 2018, 41, 645–657. [Google Scholar] [CrossRef]
- Bräutigam, A.; Kajala, K.; Wullenweber, J.; Sommer, M.; Gagneul, D.; Weber, K.L.; Carr, K.M.; Gowik, U.; Maß, J.; Lercher, M.J.; et al. An MRNA Blueprint for C4 Photosynthesis Derived from Comparative Transcriptomics of Closely Related C3 and C4 Species. Plant Physiol. 2011, 155, 142–156. [Google Scholar] [CrossRef]
- Gowik, U.; Bräutigam, A.; Weber, K.L.; Weber, A.P.M.; Westhoff, P. Evolution of C4 Photosynthesis in the Genus Flaveria: How Many and Which Genes Does It Take to Make C4? Plant Cell 2011, 23, 2087–2105. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gribskov, M. Comprehensive Evaluation of de Novo Transcriptome Assembly Programs and Their Effects on Differential Gene Expression Analysis. Bioinformatics 2017, 33, 327–333. [Google Scholar] [PubMed]
- Simsek, O.; Donmez, D.; Kacar, Y.A. RNA-Seq Analysis in Fruit Science: A Review. Am. J. Plant Biol. 2017, 2, 1–7. [Google Scholar]
- Bolger, M.E.; Arsova, B.; Usadel, B. Plant Genome and Transcriptome Annotations: From Misconceptions to Simple Solutions. Brief. Bioinform. 2018, 19, 437–449. [Google Scholar] [CrossRef]
- Martin, J.A.; Wang, Z. Next-Generation Transcriptome Assembly. Nat. Rev. Genet. 2011, 12, 671. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.T.J.; Carpenter, E.J.; Tian, Z.; Bruskiewich, R.; Burris, J.N.; Carrigan, C.T.; Chase, M.W.; Clarke, N.D.; Covshoff, S.; Edgar, P.P.; et al. Evaluating Methods for Isolating Total RNA and Predicting the Success of Sequencing Phylogenetically Diverse Plant Transcriptomes. PLoS ONE 2012, 7, e50226. [Google Scholar] [CrossRef]
- Syed, N.H.; Kalyna, M.; Marquez, Y.; Barta, A.; Brown, J.W.S. Alternative splicing in plants—Coming of age. Trends Plant Sci. 2012, 17, 616–623. [Google Scholar] [CrossRef]
- Almeida, A.M.R.; Piñeyro-Nelson, A.; Yockteng, R.B.; Specht, C.D. Comparative Analysis of Whole Flower Transcriptomes in the Zingiberales. PEER J. 2018, 6, e5490. [Google Scholar] [CrossRef]
- Moreno-Pachon, N.M.; Leeggangers, H.A.C.F.; Nijveen, H.; Severing, E.; Hilhorst, H.; Immink, R.G.H. Elucidating and Mining the Tulipa and Lilium Transcriptomes. Plant Mol. Biol. 2016, 92, 249–261. [Google Scholar] [CrossRef]
- Bazinet, A.L.; Ondov, B.D.; Sommer, D.D.; Ratnayake, S. BLAST-Based Validation of Metagenomic Sequence Assignments. PEER J. 2018, 6, e4892. [Google Scholar] [CrossRef]
- Barnes, I.; Crous, P.W.; Wingfield, B.D.; Wingfield, M.J. Multigene Phylogenies Reveal That Red Band Needle Blight of Pinus Is Caused by Two Distinct Species of Dothistroma, D. Septosporum and D. Pini. Stud. Mycol. 2004, 50, 551–565. [Google Scholar]
- Lichtenzveig, J.; Bonfil, D.J.; Zhang, H.-B.; Shtienberg, D.; Abbo, S. Mapping Quantitative Trait Loci in Chickpea Associated with Time to Flowering and Resistance to Didymella rabiei the Causal Agent of Ascochyta Blight. Appl. Genet. 2006, 113, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.A.; Ewaze, J.O.; Summerbell, R.C.; Arocha-Rosete, Y.; Maharaj, A.; Guardiola, Y.; Saleh, M.; Wong, B.; Bogale, M.; O’Hara, M.J.; et al. Multilocus DNA Sequencing of the Whiskey Fungus Reveals a Continental-Scale Speciation Pattern. Personia 2016, 37, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Xing, X.-K.; Zhang, L.-C.; Xing, Y.-M.; Guo, S.-X. Identification of Hortaea Werneckii Isolated from Mangrove Plant Aegiceras comiculatum Based on Morphology and rDNA Sequences. Mycopathologia 2012, 174, 457–466. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plant | Growth Type | Seeder/Sprouter | Growth Form | Location |
|---|---|---|---|---|
| A | Red type (commercial) | seeder | upright, densely branched bush | S 031° 43′ 18′′ E 019° 07′ 32′′ |
| B | Nieuwoudtville sprouter | sprouter | low-growing, densely branched bush | S 031° 45′ 48′′ E 019° 07′ 54′′ |
| C | Black type | seeder | tall, slender shrub | S 031° 59′ 21′′ E 018° 50′ 35′′ |
| D | Grey sprouter | sprouter | upright, sparsely branched bush | S 032° 37′ 17′′ E 019° 03′ 24′′ |
| Plant | Aspalathin | Orientin | Iso-Orientin | Iso-Vitexin | Vitexin | Hyperoside | Luteolin | Quercetin |
|---|---|---|---|---|---|---|---|---|
| A | 66.52 ± 0.68 a | 4.70 ±0.19 a | 6.94 ± 0.05 a | 2.18 ± 0.07 a | 1.98 ± 0.10 a | 2.06 ± 0.09 a | 0.01 ± 0.00 a | 0.01 ± 0.00 a |
| B | 9.24 ± 0.17 b | 1.56 ± 0.04 c | 1.50 ± 0.01 c | 0.17 ± 0.00 c | 0.49 ± 0.01 b | 0.41 ± 0.01 b | 0.21 ± 0.00 b | 0.01 ± 0.00 a |
| C | 0.00 ± 0.00 c | 1.82 ± 0.08 c | 1.71 ± 0.03 c | 0.17 ± 0.00 c | 0.23 ± 0.01 c | 0.32 ± 0.01 b,c | 0.59 ± 0.01 c | 0.01 ± 0.00 a |
| D | 0.00 ± 0.00 c | 2.29 ± 0.06 b | 2.85 ± 0.08 b | 0.69 ± 0.01 b | 0.25 ± 0.00 c | 0.17 ± 0.00 c | 0.58 ± 0.01 c | 0.05 ± 0.00 b |
| Plant | A | B | C | D |
|---|---|---|---|---|
| RNA yield (ng/µL) | 53.0 | 59.0 | 58.0 | 46.0 |
| RNA integrity number (RIN) | 8.3 | 7.1 | 7.9 | 8.1 |
| Library insert size (bp) | 437 | 448 | 479 | 452 |
| # read pairs (in Mio) | 54.7 | 66.5 | 26.7 | 44.1 |
| # read pairs after quality processing (in Mio) | 54.6 | 65.8 | 26.5 | 43.8 |
| % read pairs remained after quality processing | 99.9 | 99.1 | 99.2 | 99.2 |
| % bases remained after quality processing | 83.6 | 83.7 | 83.8 | 83.7 |
| Length after trimming (bp) | 30–127 | 30–127 | 30–127 | 30–127 |
| Trinity_all | Trinity_longest | IDBA_Trans | SOAPdenovo_Trans | CLC | |
|---|---|---|---|---|---|
| Assembler running time (h) | 22 | 22 | 4 | 1 | 4 |
| # of Transcripts (≥300 nt) | 100,778 | 53,363 | 76,784 | 50,503 | 59,716 |
| 300–500 bp: | 28,701 | 22,049 | 15,941 | 11,203 | 27,416 |
| 501–1000 bp: | 27,747 | 14,995 | 19,885 | 13,139 | 18,131 |
| 1001–5000 bp: | 43,922 | 16,145 | 40,701 | 25,795 | 14,046 |
| 5001–10,000 bp: | 408 | 174 | 257 | 364 | 123 |
| >10,000 bp: | 0 | 0 | 0 | 2 | 0 |
| Overall read alignment rate (%) | 97.0 | 82.9 | 89.1 | 76.8 | 78.7 |
| Read pairs aligned concordantly (%) | 82.0 | 66.0 | 75.9 | 58.1 | 59.8 |
| Complete BUSCOs (C) | 1258 | 1092 | 1230 | 1019 | 923 |
| Single-copy BUSCOs (S) | 721 | 1061 | 374 | 825 | 870 |
| Duplicated BUSCOs (D) | 537 | 31 | 856 | 194 | 53 |
| Fragmented BUSCOs (F) | 73 | 152 | 59 | 229 | 244 |
| Missing BUSCOs (M) | 109 | 196 | 151 | 192 | 273 |
| # of transcripts that hit a BUSCO | 2065 | 1278 | 2561 | 1477 | 1223 |
| % of transcripts that hit a BUSCO | 2.0 | 2.4 | 3.3 | 2.9 | 2.0 |
| Transcriptome (76,784) | Angel | GenemarkS-T | TransDecoder | |
|---|---|---|---|---|
| # of predicted proteins | - | 74,767 | 58,284 | 54,205 |
| # of transcripts with ORFs | - | 71,791 | 54,754 | 54,205 |
| ORFs/transcript (mean) | - | 1.04 ± 0.21 | 1.06 ± 0.26 | 1.00 ± 0.00 |
| Complete BUSCOs | 1230 | 1211 | 1202 | 1200 |
| Single-copy BUSCOs | 374 | 396 | 394 | 390 |
| Duplicated BUSCOs | 856 | 815 | 808 | 810 |
| Fragmented BUSCOs | 59 | 71 | 68 | 74 |
| Missing BUSCOs | 151 | 158 | 170 | 166 |
| A | B | C | D | |
|---|---|---|---|---|
| Total Mbps | 121.87 | 122.11 | 98.02 | 102.83 |
| Transcripts | 91,171 | 96,865 | 76,784 | 80,456 |
| 300–500 bp | 20,986 | 24,955 | 15,941 | 18,460 |
| 501–1000 bp | 22,161 | 24,463 | 19,885 | 20,767 |
| 1001–5000 bp | 47,231 | 46,736 | 40,701 | 40,674 |
| 5001–10 000 bp | 793 | 707 | 257 | 547 |
| >10,000 bp | 0 | 4 | 0 | 8 |
| Predicted ORFs | 85,234 | 91,301 | 75,426 | 79,234 |
| Overall read alignment rate (%) | 77.11 | 66.75 | 89.05 | 81.11 |
| Read pairs aligned concordantly ≥ 1× (%) | 60.63 | 51.94 | 75.86 | 63.66 |
| Read pairs aligned discordantly (%) | 7.63 | 5.60 | 8.74 | 8.57 |
| RIN | 8.3 | 7.1 | 7.9 | 8.1 |
| Complete BUSCOs (C) | 1291 | 1221 | 1230 | 1242 |
| Complete and single-copy BUSCOs (S) | 383 | 335 | 374 | 391 |
| Complete and duplicated BUSCOs (D) | 908 | 886 | 856 | 851 |
| Fragmented BUSCOs (F) | 48 | 65 | 59 | 62 |
| Missing BUSCOs (M) | 101 | 154 | 151 | 136 |
| A (91,171) | B (96,865) | C (76,784) | D (80,456) | |||||
|---|---|---|---|---|---|---|---|---|
| NCBI Nr Taxonomic category | Transcripts | % | Transcripts | % | Transcripts | % | Transcripts | % |
| Fabaceae | 62,368 | 68.41 | 63,097 | 65.14 | 57,558 | 74.96 | 57,878 | 71.94 |
| Other Plants | 2905 | 3.19 | 3036 | 3.13 | 2324 | 3.03 | 2566 | 3.19 |
| Fungi | 4 | 0 | 3140 | 3.24 | 661 | 0.86 | 664 | 0.83 |
| Bacteria | 28 | 0.03 | 38 | 0.04 | 24 | 0.03 | 82 | 0.1 |
| Viruses | 15 | 0.02 | 14 | 0.01 | 4 | 0.01 | 8 | 0.01 |
| Other Eukaryotes | 16 | 0.02 | 333 | 0.34 | 199 | 0.26 | 0 | 0 |
| Total: | 65,336 | 71.66 | 69,658 | 71.91 | 60,770 | 79.14 | 61,198 | 76.06 |
| Kraken2 classification | transcripts | % | transcripts | % | transcripts | % | transcripts | % |
| Plant | 72,016 | 78.99 | 72,771 | 75.13 | 62,359 | 81.21 | 63,673 | 79.14 |
| Bacteria | 387 | 0.42 | 493 | 0.51 | 340 | 0.44 | 344 | 0.43 |
| Fungi | 78 | 0.09 | 1467 | 1.51 | 345 | 0.45 | 293 | 0.36 |
| Total: | 72,481 | 79.5 | 74,731 | 77.15 | 63,044 | 82.11 | 64,310 | 79.93 |
| A | B | C | D | |
|---|---|---|---|---|
| Total transcripts (>300 bp) | 91,171 | 96,865 | 76,784 | 80,456 |
| Transcripts > 300 bp annotated using NCBI(NR) | 65,336 | 69,658 | 60,770 | 61,198 |
| Total transcripts (>1000 bp) | 48,024 | 47,447 | 40,958 | 40,958 |
| Transcripts > 1000 bp annotated using KEGG | 21,040 | 21,126 | 19,210 | 18,646 |
| Total protein sequences | 85,234 | 91,301 | 75,426 | 79,234 |
| Proteins annotated using eggNOG: eggNOG annotations | 52,688 | 56,090 | 50,413 | 50,338 |
| Proteins annotated using eggNOG: KO annotations | 26,011 | 28,187 | 25,244 | 24,894 |
| Proteins annotated using Pfam-A | 44,390 | 46,868 | 42,244 | 42,258 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stander, E.A.; Williams, W.; Mgwatyu, Y.; Heusden, P.v.; Rautenbach, F.; Marnewick, J.; Roes-Hill, M.L.; Hesse, U. Transcriptomics of the Rooibos (Aspalathus linearis) Species Complex. BioTech 2020, 9, 19. https://doi.org/10.3390/biotech9040019
Stander EA, Williams W, Mgwatyu Y, Heusden Pv, Rautenbach F, Marnewick J, Roes-Hill ML, Hesse U. Transcriptomics of the Rooibos (Aspalathus linearis) Species Complex. BioTech. 2020; 9(4):19. https://doi.org/10.3390/biotech9040019
Chicago/Turabian StyleStander, Emily Amor, Wesley Williams, Yamkela Mgwatyu, Peter van Heusden, Fanie Rautenbach, Jeanine Marnewick, Marilize Le Roes-Hill, and Uljana Hesse. 2020. "Transcriptomics of the Rooibos (Aspalathus linearis) Species Complex" BioTech 9, no. 4: 19. https://doi.org/10.3390/biotech9040019
APA StyleStander, E. A., Williams, W., Mgwatyu, Y., Heusden, P. v., Rautenbach, F., Marnewick, J., Roes-Hill, M. L., & Hesse, U. (2020). Transcriptomics of the Rooibos (Aspalathus linearis) Species Complex. BioTech, 9(4), 19. https://doi.org/10.3390/biotech9040019