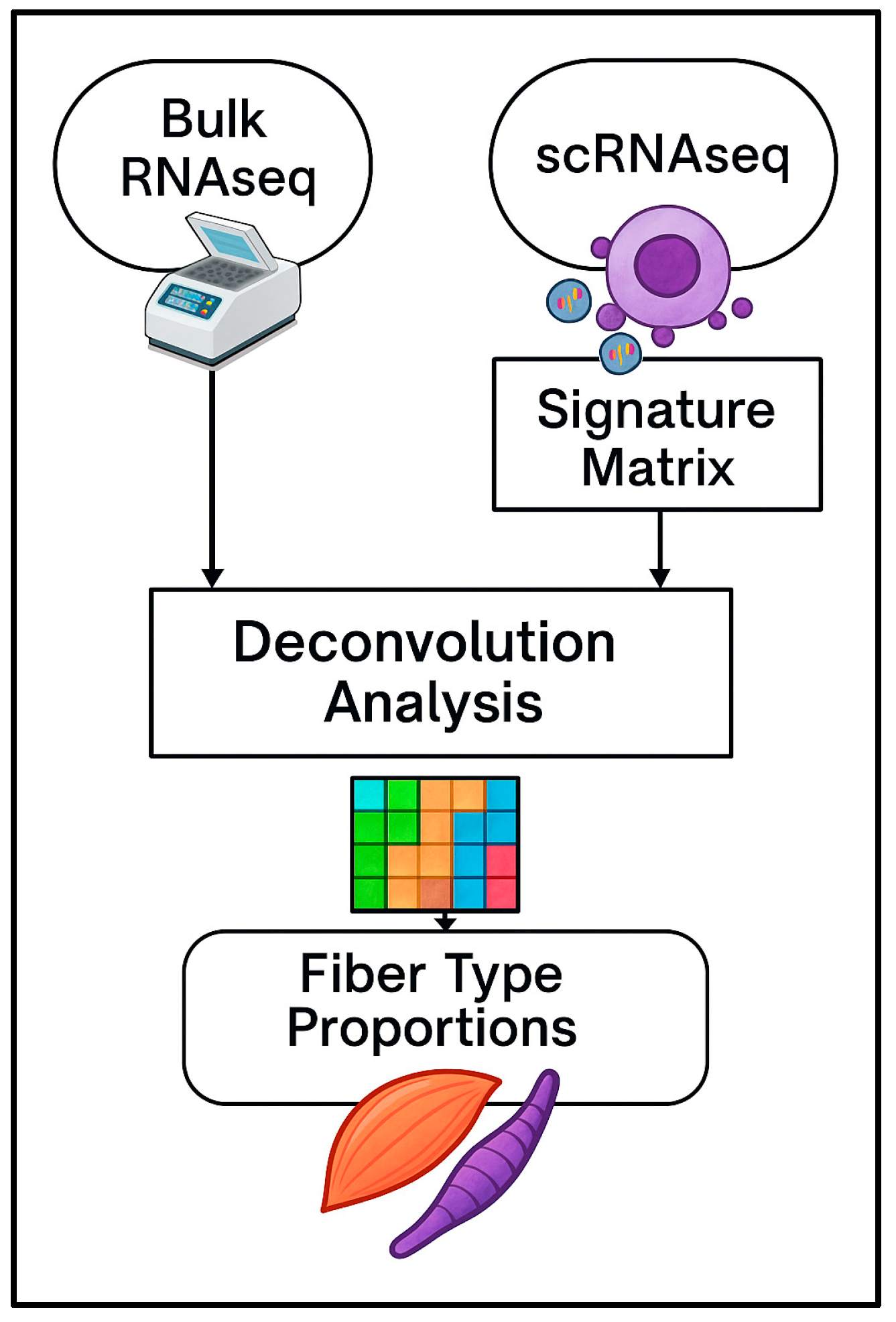
1. Introduction
RNA sequencing (RNA-seq) has revolutionized the analysis of transcriptomes, with bulk RNA-seq enabling analysis of differential gene expression at the global level. Although RNA-seq is a straightforward and cost-effective technique, it cannot fully capture the intrinsic heterogeneity of samples to reveal the contribution of different cell types and states of development to global gene expression [
1,
2,
3]. Single-cell RNA-seq (scRNA-seq), on the other hand, provides cell-type-specific transcriptome profiling, enabling the determination of cellular composition and proportions. However, the relatively high cost of scRNA-seq and technical challenges such as tissue dissociation and preparation of single-cell or nuclear suspensions limits its applicability, especially when working with archived or previously collected samples [
4,
5]. The recent emergence of computational deconvolution methods provides cost-effective alternatives to estimate the proportions of different cell types from bulk RNA-seq data [
2]. These approaches infer the cellular composition of heterogeneous tissues by mathematically decomposing bulk gene expression profiles, often using reference signatures derived from scRNA-seq data. Depending on the algorithm, deconvolution can be performed through linear regression, support vector regression, or probabilistic modeling, allowing researchers to estimate relative contributions of individual cell types without requiring single-cell data generation [
6].
Various software tools are currently available for estimating cell proportions, each differing in reference dataset requirements and statistical methodologies used for deconvolution analysis. Tools that use a reference based on previously generated scRNA-seq data generally provide more reliable estimates of cell proportions compared to those that do not use a reference dataset [
3]. Among reference-based tools, CIBERSORTx [
7] is a commonly used deconvolution tool that applies a support vector regression algorithm to gene expression data for the estimation of cell type proportions. CIBERSORTx is particularly effective for analyzing complex heterogeneous samples [
3,
8]. Similarly, Cellanneal [
9] is a deconvolution method that uses simulated annealing to optimize Spearman’s rank correlation between experimental and computational gene expression vectors. By ranking gene expression values, Cellanneal reduces the influence of highly expressed genes and incorporates a broad set of informative genes, minimizing dependency on a specific gene list [
9]. DeconvR [
10] offers multiple methods for deconvolving bulk RNA-seq data, including non-negative least squares (NNLS) and robust linear regression (RLM). NNLS [
11] minimizes the sum of squared residuals while ensuring that all estimated proportions remain non-negative, making it suitable for datasets with non-negative constraints, such as gene expression levels or proportions of cell types. On the other hand, RLM is more resilient to outliers, as it minimizes the influence of extreme values, providing robust estimates when the data may contain noise or large variability [
10].
Despite its potential, deconvolution analyses remain underutilized in studies focused on livestock animals. A major challenge lies in the limited availability of reference data for specific tissues, compounded by the fact that existing scRNA-seq datasets are often derived from diverse breeds within the same species. Most available studies focus on humans and mice, leaving gaps in understanding how deconvolution performs across livestock breeds. In cattle, for instance, only one study reporting scRNA-seq analysis of bovine skeletal muscle tissue samples is currently publicly available [
12]. This study produced scRNA-seq data of skeletal muscle from Wagyu and Brahman cattle, and their crossbreed, and revealed mechanisms contributing to differential intramuscular fibrogenesis and adipogenesis between the breeds [
12]. However, there are over 800 recognized cattle breeds, categorized into
Bos taurus and
Bos indicus [
13]. Although significant opportunities exist in applying deconvolution analysis to bulk RNA-seq data to enhance our understanding of physiology of livestock in general, and cattle in particular, knowledge of how scRNA-seq reference data from different breeds may impact cell type identification is needed.
In dairy cattle, skeletal muscle plays an important role as a protein reserve to support milk production, particularly in early lactation [
14]. Mobilization of skeletal muscle in early lactation provides essential energy and amino acids to sustain high milk production [
14]. Skeletal muscle of mature cattle is primarily composed of slow myofibers (type I) and fast myofibers (types IIa and IIx) [
15]. We recently performed transcriptome analysis of
longissimus dorsi (LD) muscle biopsied from Holstein Friesian (
Bos taurus) cows in late gestation and found expression levels of genes that encode for type I, IIa, and IIx myofibers varied between cattle with high LD muscle depth versus low depth at three weeks before expected calving [
16]. However, since these studies were conducted using bulk RNA-seq and not scRNA-seq analysis, the proportion of specific cell fractions and their functional implications could not be surmised. A precise characterization of muscle fiber types via deconvolution may offer valuable insights into muscle physiology and its influence on key productive and reproductive traits in dairy cattle.
To address these challenges, the present study aimed to assess the performance of four deconvolution methods—CIBERSORTx (v. 1.05), Cellanneal (v. 1.1.0), DeconvR-NNLS (v. 1.15.0), and DeconvR-RLM (v. 1.15.0) —in estimating the proportions of type I, IIa, and IIx myofibers. This assessment was performed using bulk RNA-seq data from Casey et al. [
16], in combination with what is, to the best of our knowledge, the only currently available single-cell RNA-seq reference dataset, derived from two genetically distinct bovine breeds: Wagyu (
Bos taurus) and Brahman (
Bos indicus) [
12].
Despite the increasing availability of deconvolution tools, it remains unclear how breed-specific differences in single-cell reference data influence the accuracy and consistency of muscle fiber type estimation from bulk RNA-seq in cattle. We hypothesize that using genetically distinct scRNA-seq references may lead to significant differences in the inferred myofiber proportions. Therefore, our objective was to systematically evaluate the consistency and biological relevance of deconvolution outputs generated by each method across different reference panels, aiming to identify the most robust approach for studying muscle composition in dairy cattle.
4. Discussion
4.1. Novel Breed-Specific Effects in Deconvolution Analysis
The deconvolution analysis of bulk RNA-seq data from Holstein cows found that the breed of cattle used in the reference scRNA-seq data and the software tools applied significantly influenced the predicted myofiber proportions in LD muscle. According to Im and Kim [
2], benchmarking is essential for validating deconvolution methods. This is the first study to show that the cattle breed used in single-cell RNA-seq reference data significantly impacts the outcomes of deconvolution analyses for muscle fiber type estimation. Our comprehensive comparison of four deconvolution methods using reference data from two genetically distinct cattle breeds revealed consistent breed-specific effects on the estimation of type IIa and IIx myofiber proportions, while type I fiber estimation remained relatively stable across reference breeds.
The systematic nature of the breed effects observed across multiple deconvolution algorithms suggests that these differences reflect genuine biological variations in gene expression patterns between cattle breeds rather than methodological artifacts. However, without true scRNA-seq data from Holstein cows as ground truth, we cannot definitively determine which breed reference or deconvolution method provides the most accurate estimates of myofiber proportions. This limitation underscores the need for future studies to generate breed-matched single-cell reference data to validate deconvolution approaches and establish definitive benchmarks for accuracy assessment. Interestingly, the consistency of breed-specific effects across CIBERSORTx, DeconvR-NNLS, and DeconvR-RLM for type IIa and IIx fibers, despite their different algorithmic approaches, suggests that breed compatibility between reference and target datasets is a critical factor for accurate deconvolution results. These findings highlight two potential strategies for improving deconvolution accuracy: either using breed-matched reference datasets when analyzing specific populations, or alternatively, developing comprehensive multi-breed reference datasets that capture the full spectrum of genetic diversity within and across species. The latter approach could support OneHealth initiatives by enabling cross-species comparative analyses and leveraging the broader biological knowledge available across different breeds and species to enhance deconvolution robustness.
In domestic animals, the existence of multiple breeds or lineages within a species—each selected for specific traits such as intramuscular fat, growth rate, milk yield, muscle fiber composition, or heat tolerance—leads to distinct gene expression profiles across tissues. These breed-specific transcriptomic differences help explain the systematic effects observed in our deconvolution results, reinforcing the importance of using representative or breed-matched reference datasets for accurate cell type proportion estimation.
4.2. Performance Comparison of Deconvolution Methods
CIBERSORTx and DeconvR-RLM demonstrated similar estimates of fiber type distribution across both reference breeds. Additionally, when analyzing segmented myofibers, both methods calculated similar proportions of type I fibers using the different breed reference datasets. In contrast, the estimated proportions of type IIa and IIx myofibers varied depending on the reference breed, suggesting these fiber types may be more sensitive to breed-specific transcriptional differences. CIBERSORTx and DeconvR-RLM were considered the most consistent methods due to their low inter-sample variability and agreement across the reference datasets. Their estimates also aligned with established physiological expectations in bovine skeletal muscle, particularly the predominance of type II fibers and a total myofiber content of approximately 90–95% in muscle samples, which supports the biological plausibility of their outputs. On the other hand, DeconvR-NNLS showed notably poor performance by consistently estimating unrealistic total myofiber proportions near 100%, which contradicts the known cellular heterogeneity in muscle tissue that includes fibro-adipogenic progenitors, satellite cells, and immune cells. This limitation likely stems from the non-negative least squares constraint, which forces the algorithm to assign all transcriptomic signal to the available myofiber types rather than recognizing the presence of other cell populations, thereby undermining the method’s ability to provide biologically accurate estimates of muscle composition.
Performance differences between CIBERSORTx and DeconvR-NNLS may also reflect their underlying algorithmic assumptions, with CIBERSORTx’s support vector regression framework providing greater noise tolerance and adaptive feature selection compared to DeconvR-NNLS’s strict non-negative constraints that limit flexibility in handling crossbreed transcriptomic variability. For instance, Jin and Liu [
30], in their evaluation of 11 deconvolution methods across 1766 different conditions in
Homo sapiens, also demonstrated that CIBERSORTx achieved the best performance. Muscle samples from cows exhibit significant cellular heterogeneity, and during late lactation, gene expression profiles may shift due to physiological changes [
16]. Moreover, immune cell populations can increase as part of an inflammatory process before parturition [
31]. CIBERSORTx and DeconvR-RLM, which rely on linear regression for deconvolution, seem to perform effectively with heterogeneous tissue samples and manage outliers through Bayesian frameworks or M-estimators [
3,
7,
32]. These factors can make CIBERSORTx and DeconvR-RLM particularly well-suited for accurately predicting transcriptomic signals and identifying myofiber fractions in dynamic biological contexts.
DeconvR-NNLS consistently estimated total myofiber proportions near 100%. However, this assumption is likely incorrect. Wang et al. [
12] demonstrated the presence of various cell types, including fibro-adipogenic progenitors (FAPs), satellite cells (SCs), monocytes/macrophages, neutrophils, and lymphocytes, in muscle samples of Wagyu and Brahman cattle. Similarly, Xiao et al. [
33], analyzing scRNA-seq data from the LD muscle in pigs, identified endothelial cells, myotubes, FAPs, satellite cells, myoblasts, myocytes, Schwann cells, smooth muscle cells, dendritic cells, pericytes, and neutrophils. Zhu et al. [
34] reported similar findings in the LD muscle of goats, describing not only myofibers but also fibroblasts, endothelial cells, satellite cells, and smooth muscle cells. The limitation of DeconvR-NNLS likely arises from a methodological constraint, wherein the algorithm enforces non-negative estimates for proportions to provide a biologically interpretable output. While this constraint avoids negative proportions, it undermines the method’s ability to detect cellular heterogeneity, particularly for cell populations present at low abundance within the analyzed tissue. This methodological bias hampers the accurate representation of the diverse cellular composition typically observed in muscle tissues.
Wang et al. [
4] benchmarked four deconvolution methods, including CIBERSORTx and DeconvR-NNLS, in pancreatic islet cells from humans under two distinct scenarios. In the first, both scRNA-seq and bulk RNA-seq data were derived from the same individuals. In this case, DeconvR-NNLS and CIBERSORTx performed comparably, with correlation values of 0.85 and 0.89, respectively. In the second scenario, where scRNA-seq and bulk RNA-seq data came from different individuals, the performance of both methods declined. The correlation for CIBERSORTx dropped more significantly to 0.76, compared to 0.82 for DeconvR-NNLS, indicating a greater decrease in its performance under this condition. However, in our study, despite using a different breed reference to deconvolute the Holstein muscle transcriptome, CIBERSORTx demonstrated more consistent results across different reference breeds compared to DeconvR-NNLS.
4.3. Biological Basis for Breed-Specific Expression Patterns
The importance of reference breeds became particularly evident for certain myofiber types (
Figure 2b,c). Despite no differences being found between breeds for type I fibers, we observed variability across all methods for type IIa fibers. For type IIx fibers, CIBERSORTx, DeconvR-NNLS, and RLM analyses showed clear discrepancies between reference breeds. Wagyu is well known for its high intramuscular fat (IMF) content [
35]. Gotoh et al. [
36] reported significantly higher IMF levels in Wagyu compared to Holstein and found a positive correlation between IMF and type I fiber abundance, in contrast to a negative correlation for type II fibers. When using Wagyu as the reference, estimates for type IIa fibers were generally higher and more consistent across methods, suggesting that the transcriptomic profile of Wagyu muscle may enhance the detection or resolution of type-IIa-specific gene signatures. However, for type IIx fibers, Wagyu-based references produced the lowest estimated proportions in CIBERSORTx, DeconvR-NNLS, and RLM analyses. In contrast, Wang et al. (2023) [
12] showed that Brahman has a greater fibrogenesis capacity than Wagyu, which may amplify muscle-specific gene expression signals and improve the accuracy of myofiber type deconvolution. Consequently, Brahman outperformed Wagyu as a reference breed when deconvoluting Holstein muscle tissue, particularly for type IIx fibers.
4.4. Comparison with the Literature and Validation Context
There is a notable lack of scRNA-seq or single nuclear RNA-seq (snRNA-seq) studies of the fractions of cell types in the LD muscle of cattle. Notably, our findings indicate a higher proportion of muscle cells compared to those documented in the existing literature in the LD of other species such as swine. Yi et al. [
37] found that the total muscle myofiber proportions ranged from 64.12% to 73.60% of the cells using snRNA-seq of the LD muscle of Dahe pigs at 194 days of age. Similarly, Wang et al. [
25], using snRNA-seq analysis of LD muscle in Laiwu pigs, reported total muscle fiber values of 73.74% and 82.59% of cells. In contrast, Xiao et al. [
33], using scRNA-seq of LD muscle from a 1-day-old newborn Suhuai boar, found a lower percentage of total muscle fibers, approximately 28.5%.
Mammalian skeletal muscle typically comprises myofiber isoform types I, IIa, IIb, and IIx [
38]. In Casey et al. [
16], our bulk RNA-seq dataset, normalized read counts were shown for MYH4, the gene encoding type IIb fibers. However, in our deconvolution study, we identified proportions only for type I, IIa, and IIx myofibers in Holstein cows. This is likely because the reference data used here did not detect type IIb myofibers in Wagyu or Brahman breeds [
12], preventing their detection in our results. Other studies have also reported the absence of type IIb myofibers in bovine species, including Holstein [
39] and Simmental cattle [
15], particularly in the LD muscle.
According to the literature, the proportion of slow myofibers in bovine LD muscle is consistently lower, ranging from 25% to 32.5%, compared to fast myofibers, which range from 67.5% to 75% [
40,
41]. Similarly, Casey et al. [
16], who analyzed bulk RNA-seq data from the same Holstein cows used in our study, reported that expression of MYH7 (which encodes the type I myosin heavy chain) was substantially lower (~13%) compared to the combined expression of MYH1 and MYH2 (which encode fast-twitch fiber types, ~87%). These expression levels were observed in LD muscle sampled three weeks before expected calving, with differences noted between high and low muscle depth groups. However, because bulk RNA-seq does not provide cell type resolution, these findings could not determine the precise proportions of myofiber subtypes or their functional implications.
In the study performed by Wang et al. [
12], from which we sourced the scRNA-seq data as a reference for our study, the fractions of myofibers identified through scRNA-seq in the LD muscle were 57.21% for type I, 33.81% for type IIa, and 3.07% for type IIx. The higher proportions of type I myofiber in their study, compared to the proportion identified in this study is likely related to the age of the animals (relatively younger in Wang et al.’s [
12] paper, ~4 months old). As demonstrated by Zhang et al. [
15], the expression of type I in bovines reduces significantly between 3 to 36 months of age. In the bulk RNA-seq used in the deconvolution, the Holstein cows were at least 36 months of age. Also, Wang et al. [
12] reported that the lower abundance of type IIx fibers was likely due to their larger size, as larger fragments were more prone to removal during sample processing.
4.5. Study Limitations and Technical Considerations
The discrepancies in muscle cell proportions may be partly attributed to methodological differences in the preparation of input samples, as scRNA-seq tends to exclude some myofibers due to their larger diameter, whereas snRNA-seq captures nuclei from all cell types, including those from larger myofibers, leading to a more comprehensive representation of muscle composition and potentially different estimated cell proportions. This underscores a key technical limitation of using scRNA-seq for analyzing myofibers. Large myofibers are frequently filtered out during the tissue dissociation and cell preparation steps required for scRNA-seq, which may compromise the results by underestimating certain fiber types, particularly the larger type IIx fibers. This technical bias could lead to skewed reference datasets that do not accurately represent the true cellular composition of muscle tissue. Future studies should consider employing snRNA-seq approaches, which can better capture nuclei from large myofibers and provide a more comprehensive representation of muscle fiber composition. Additionally, standardization of scRNA-seq preprocessing protocols (e.g., digestion steps) might help reduce technical biases during cell capture, thereby improving consistency across breeds and enabling more accurate comparisons of muscle composition.
The limited number of type IIx cells (10 cells) in the reference dataset represents a known limitation for deconvolution analysis, as rare cell types with insufficient reference cells often exhibit reduced signature matrix reliability and increased estimation variability across methods [
42,
43,
44]. Recent methodological advances, such as hierarchical deconvolution [
43], were specifically developed to address this challenge by employing hierarchical approaches to improve rare cell type estimation under limited reference conditions and should be explored in future studies. Another fundamental limitation of this study is the absence of ground truth single-cell RNA-seq data from Holstein cows to validate our deconvolution results. Without breed-matched reference data, we cannot definitively determine which deconvolution method or reference breed provides the most accurate estimates of myofiber proportions in Holstein muscle tissue. This limitation is critical for interpreting our findings and underscores the need for future studies to generate Holstein-specific single-cell reference data to establish definitive benchmarks for deconvolution accuracy assessment. Similarly, the age disparity between the reference animals (4-month-old calves in the Wang et al. [
12] study) and the bulk RNA-seq samples (multiparous cows) is also a potential limitation of this study, as gene expression patterns can vary significantly between developmental stages. Another technical consideration that may have influenced our results was the use of log normalization for both input datasets. Since the reference data were in a log-normalized scale, we transformed the bulk RNA-seq data to the same scale to perform the deconvolution analysis. However, this transformation could introduce biases that impact the results. Specifically, log transformation compresses the range of high-expression values while amplifying lower expression values, which could affect the accuracy of the estimated cell proportions. Future studies should investigate the impact of different normalization strategies on deconvolution outcomes and develop standardized protocols for data preprocessing that minimize technical biases while preserving biological signals.
6. Conclusions
To the best of our knowledge, this is the first study to show that the cattle breed used in single-cell RNA-seq reference data significantly impacts deconvolution analyses for muscle fiber type estimation. In this context, CIBERSORTx and DeconvR-RLM seemed to demonstrate superior performance, providing biologically plausible estimates of muscle fiber composition (90–95% total myofiber content) with appropriate sensitivity to breed-specific variations. Notably, both methods also produced estimates that are consistent with known muscle fiber biology, further supporting their reliability and relevance for muscle deconvolution analyses. Given the complexity of breed-specific effects and current limitations in available reference datasets, we conclude that cell type proportion estimates should be determined using breed-specific references for accuracy in deconvolution approaches. Despite these promising findings, some limitations must be acknowledged. The reference dataset likely underrepresents large myofibers due to technical constraints of scRNA-seq, such as tissue dissociation bias. The small number of type IIx cells in the reference reduces estimation reliability for this fiber type. Moreover, the absence of breed- and age-matched single-cell data from Holstein cows, along with the use of log normalization, may have introduced biases in the estimated cell proportions. Future studies should prioritize the generation of single-cell RNA-seq data from the same populations used for bulk RNA-seq analysis to establish robust, population-specific reference datasets. Alternatively, developing comprehensive multiple-breed/inter-species reference datasets that capture the full spectrum of genetic diversity could support OneHealth initiatives by enabling cross-species comparative analyses and leveraging broader biological knowledge. Moreover, these advancements have important implications for government agencies and policymakers, as more precise muscle composition profiling can inform breeding programs, livestock management strategies, and sustainable agricultural policies aimed at improving animal health and productivity.


 ,
, 


{kind=link}
{kind=link}