Abstract
This study introduces the intelligent learning engine (ILE) optimization technology, a novel approach designed to revolutionize screening processes in bioinformatics, cheminformatics, and a range of other scientific fields. By focusing on the efficient and precise identification of candidates with desirable characteristics, the ILE technology marks a significant leap forward in addressing the complexities of candidate selection in drug discovery, protein classification, and beyond. The study’s primary objective is to address the challenges associated with optimizing screening processes to efficiently select candidates across various fields, including drug discovery and protein classification. The methodology employed involves a detailed algorithmic process that includes dataset preparation, encoding of protein sequences, sensor nucleation, and optimization, culminating in the empirical evaluation of molecular activity indexing, homology-based modeling, and classification of proteins such as G-protein-coupled receptors. This process showcases the method’s success in multiple sequence alignment, protein identification, and classification. Key results demonstrate the ILE’s superior accuracy in protein classification and virtual high-throughput screening, with a notable breakthrough in drug development for assessing drug-induced long QT syndrome risks through hERG potassium channel interaction analysis. The technology showcased exceptional results in the formulation and evaluation of novel cancer drug candidates, highlighting its potential for significant advancements in pharmaceutical innovations. The findings underline the ILE optimization technology as a transformative tool in screening processes due to its proven effectiveness and broad applicability across various domains. This breakthrough contributes substantially to the fields of systems optimization and holds promise for diverse applications, enhancing the process of selecting candidate molecules with target properties and advancing drug discovery, protein classification, and modeling.
Key Contribution:
1. The Intelligent Learning Engine (ILE) optimization technology is a novel approach that revolutionizes screening processes in drug discovery and protein analysis. 2. ILE demonstrates superior accuracy in virtual high-throughput screening, protein classification and chemical indexing for their activities.
1. Introduction
In the ever-evolving landscape of science and technology, the critical task of accurately identifying and optimizing potential candidate molecules from a vast array of options is of the utmost importance [1]. This process is especially significant in the life sciences, where precise selection is key to advancing drug discovery [2], protein classification and modeling [3], and other areas in bioinformatics [4] and cheminformatics [5]. This paper introduces an innovative approach, “intelligent learning engine (ILE) optimization technology,” which is designed to revolutionize and optimize the screening process.
The primary aim of this paper is to address the challenges to effectively optimizing screening processes, focusing on the efficient selection of candidates with desirable characteristics. While the applications of this technology are broad, spanning various scientific and technological fields, this paper primarily underscores its utility in bioinformatics and cheminformatics. However, it is important to note the wide applicability of this advanced optimization technology in diverse domains, such as healthcare, finance, telecommunications, and so forth.
Across different scientific and technological areas, the recurring challenge is selecting the most appropriate candidate molecules from a vast array of possibilities. To tackle this challenge, various approaches have been developed, categorized primarily into optimization methods and modeling techniques. The key optimization methods include the following: 1—genetic algorithms (GAs): these methods generate and evaluate a population of potential solutions [6,7,8]; 2—simulated annealing (SA): a global optimization method utilizing a stochastic acceptance function [9,10]; 3—taboo searching (TBS): this approach combines extensive exploration with targeted local searches [11]; 4—stochastic elimination (ISE): this improves the efficiency of stochastic programming by eliminating suboptimal solutions early in the process [12,13,14]; 5—statistical methods (SMs): these methods optimize objective functions using Bayesian models [15,16]. The key modeling methods include the following: 1—neural networks (NNs): brain-inspired computational models [17,18,19]; 2—support vector machines (SVMs): these use statistical learning theory to classify data [20,21,22]; 3—Monte Carlo (MC) methods: these generate random samples for stochastic algorithms [23,24]; 4—Bayesian networks and Markov models: these offer probabilistic frameworks for optimization, aiding in decision making and learning from data [25,26]; 5—hidden Markov models (HMMs): these are effective for modeling time-series and linear sequences [27,28]; 6—discriminant analysis (DA): this uses statistical variables to classify objects based on their properties [29,30].
The diversity of these methods reflects the complexity of optimization challenges across various fields. It highlights the ongoing need for innovative approaches to select candidates with target properties, a gap this paper aims to fill with the introduction of ILE optimization technology. In the realm of pharmaceutical development, indexing, the efficient cataloging and analysis of molecules, is a pivotal process [2]. It organizes molecules based on specific characteristics and streamlines the retrieval, analysis, and comparison of molecular data [31]. A pressing concern in this area is the increasing use of non-antiarrhythmic drugs, such as antihistamines [32,33,34] and antipsychotics [35,36], which are implicated in prolonging the QT interval [37,38] and thereby increasing the risk of drug-induced long QT syndrome (LQTS)—a condition marked by potentially fatal cardiac arrhythmias.
Central to understanding LQTS is the human ether-à-go-go-related gene (hERG) potassium channel [39,40], instrumental in cardiac excitability and rhythm regulation. The hERG channel’s unique structure makes it susceptible to interactions with a diverse array of drugs, leading to QT interval prolongation—a phenomenon termed “acquired LQTS”. Over 80% of drugs that prolong the QT interval are known to inhibit the hERG K+ channel [12]. This binding, often weak and reversible, poses a significant risk, as it can escalate to torsades de pointes (TdP), a severe form of ventricular tachycardia, even in patients with otherwise normal hearts.
Given these risks, assessing TdP liability is a critical step in drug development, typically evaluated through the drug’s binding affinity to the hERG channel. The advent of ILE technology marks a transformative step in this regard. This chemoinformatics approach, utilizing descriptors such as molecular weight, logP, and the number of rotatable bonds, makes it possible to differentiate between hERG potassium channel blockers and non-blockers. The ILE model assigns a hERG liability index (ELI) to each molecule, estimating its potential as a hERG channel blocker. Such advanced in silico methods are invaluable for early-stage toxicity assessment, enhancing the safety and efficacy of drug development. ILE technology, therefore, represents a significant leap forward in mitigating hERG-related liabilities, paving the way for safer pharmaceutical innovations.
ILE optimization technology, a cornerstone of this study, has already demonstrated groundbreaking potential in drug development. Notably, it has played a pivotal role in the formulation and evaluation of several cancer treatment candidates. For instance, IDD-1040, a paclitaxel–lipoate conjugate, exhibited remarkable anti-tumor efficacy in non-small-cell lung cancer models, outperforming conventional treatments in both tumor inhibition and toxicity profiles [41]. Similarly, IDD-1010, a novel docetaxel–biotin conjugate, proved to be superior to traditional drugs in treating prostate cancer and demonstrated enhanced therapeutic effectiveness and safety [42]. These instances underscore the transformative impact of ILE optimization technology on pharmaceutical innovation, setting a promising precedent for its application in protein analysis and other scientific domains.
Previous studies on protein identification and classification have employed a variety of techniques, including support vector machines (SVMs) [43], hidden Markov models (HMMs) [44], neural networks [45], and probabilistic classifiers [46]. These foundational methods have paved the way for more advanced approaches, such as the ILE technology presented in this study, which notably enhances the accuracy and efficiency of protein analysis.
To summarize, this study introduces an advanced technique designed to significantly enhance screening processes, which has demonstrated its versatility across a wide spectrum of applications, ranging from the selection of drug candidates to the categorization of protein families and beyond. ILE optimization technology transcends the traditional boundaries of bioinformatics and cheminformatics and shows immense potential in various fields. Its capabilities include identifying individuals with a high risk of disease, optimizing investment strategies, improving resource allocation in communication networks, and navigating complex transportation systems, among other uses.
While the primary focus of this study was the application of this technology in bioinformatics, our empirical evaluations centered on several key areas. These included molecular activity indexing; the classification of proteins, such as G-protein-coupled receptors; and the homology-based modeling of serine proteases. These examples highlight the broad impact of ILE optimization technology and illustrate its potential to revolutionize screening processes across diverse scientific and technological fields.
2. Materials and Methods
This study utilizes the intelligent learning engine (ILE), a novel optimization algorithm for classifying objects and indexing chemicals for their activity against biological targets.
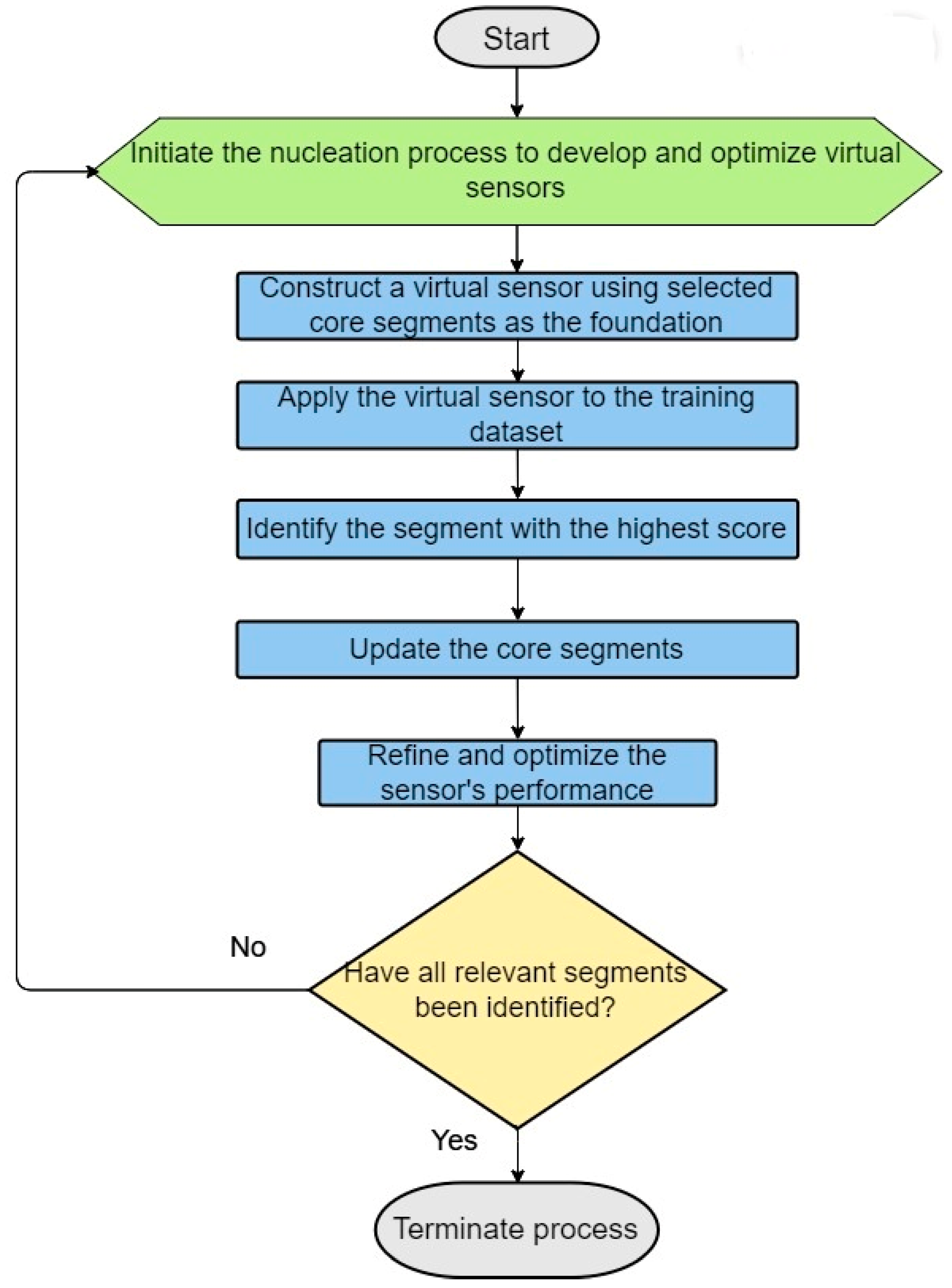
The following are the main steps in the process (see the algorithm flowchart in Figure 1):
- (1)
- Dataset Preparation: Two distinct datasets are prepared—one containing true positive (TP) matches and the other true negative (TN) matches. These datasets should then be split into training and testing sets, with a typical allocation of two-thirds for training and one-third for testing (see Figure 2). This division is crucial for facilitating effective model training and validation.
- (2)
- Encoding of the Molecules/Protein Sequences into Binary Vectors: The molecules’ protein sequences are encoded into binary vectors, such that each position along the vector is marked as 1 if it has the specific characteristic (for example if its molecular weight falls in a range of 155 to 220 daltons or if the amino acid type at a certain position is aspartic acid), and otherwise as 0.
- (3)
- Virtual Sensor Construction through Nucleation: Virtual sensors are defined by their sensor weight scores (SWSs), which are determined for specific segments of the binary vector. To calculate a sensor’s overall SWS, logical operations such as exclusive OR (XOR) and exclusive NOR (XNOR) are utilized to integrate the sensors with segments of the vector. This facilitates the dynamic generation of features capable of identifying distinct patterns in the binary vectors, especially patterns that mirror the intrinsic biological or chemical attributes of the molecules or proteins involved.
- (4)
- Sensor Optimization: The configuration of sensors is optimized using scoring functions such as specificity, sensitivity, and the Matthews correlation coefficient (MCC). The optimized sensors are then evaluated against the test set to verify their accuracy and reliability; this is aimed at minimizing false positives and false negatives.
- (5)
- Maximization of Virtual Sensor Efficiency: Factors are applied to the weights of the virtual sensors to boost their effectiveness and to augment the model’s capability to distinguish between true positive (TP) and true negative (TN) cases.
- (6)
- Usage of the Optimized Selected Sensors for Modeling Purposes: The refined model, which includes the selected optimized virtual sensors, is applied to specific tasks, such as indexing molecular activity, identifying and classifying proteins, modeling homology, and so forth.
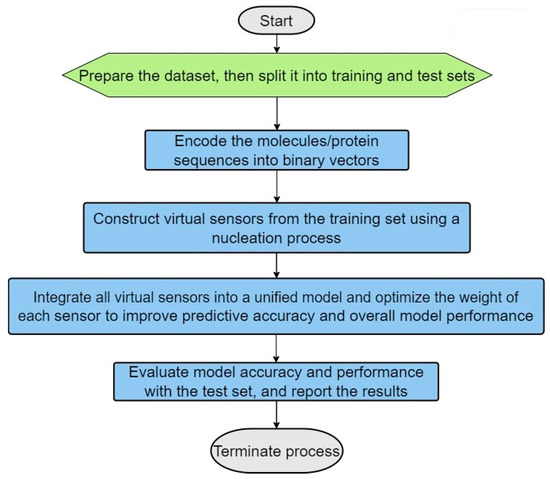
Figure 1.
Primary steps involved in the ILE (iterative learning estimation) algorithm.
Figure 1.
Primary steps involved in the ILE (iterative learning estimation) algorithm.
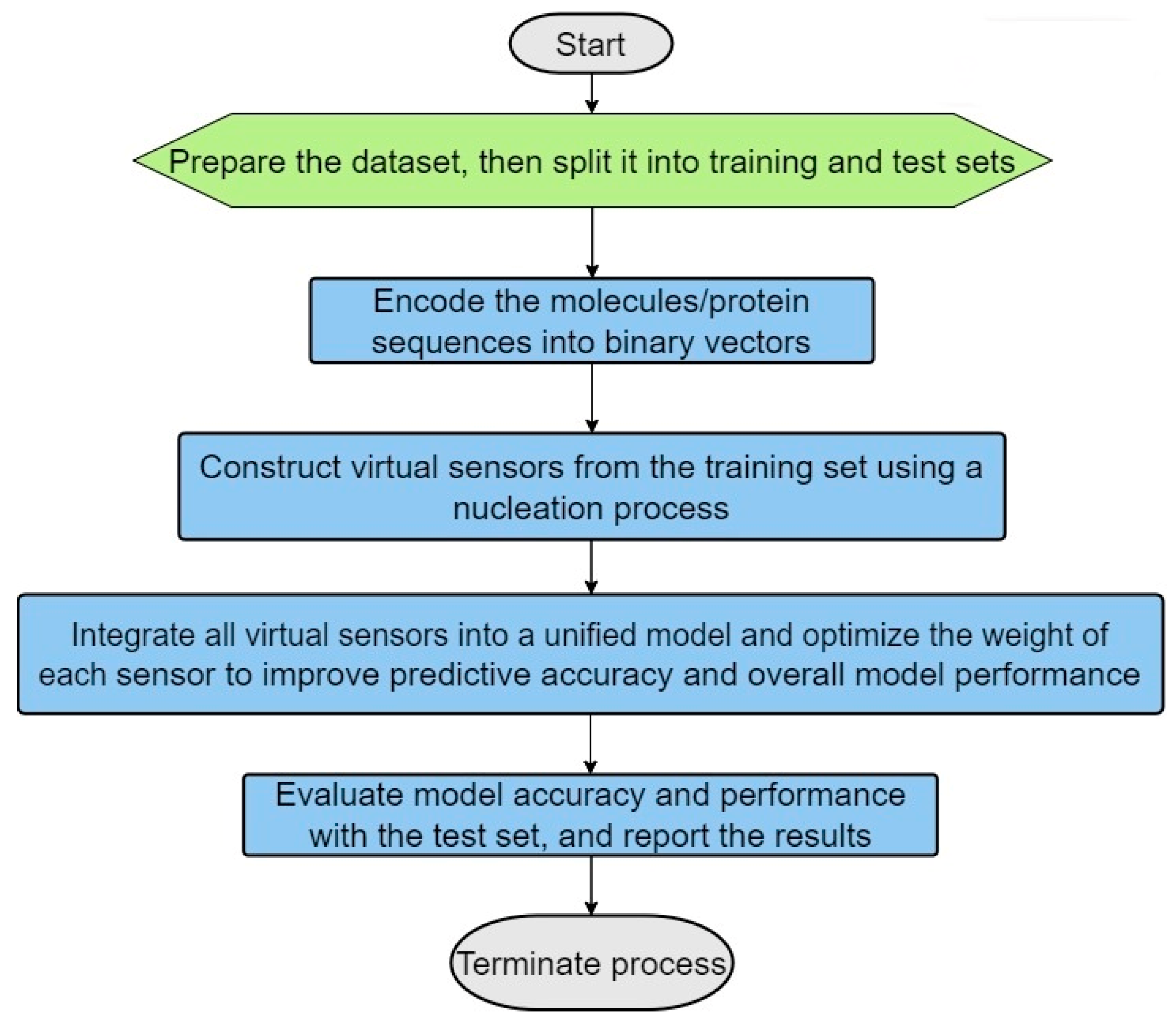
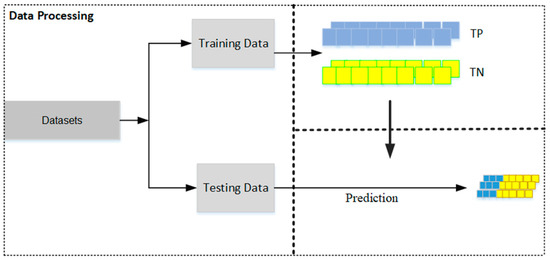
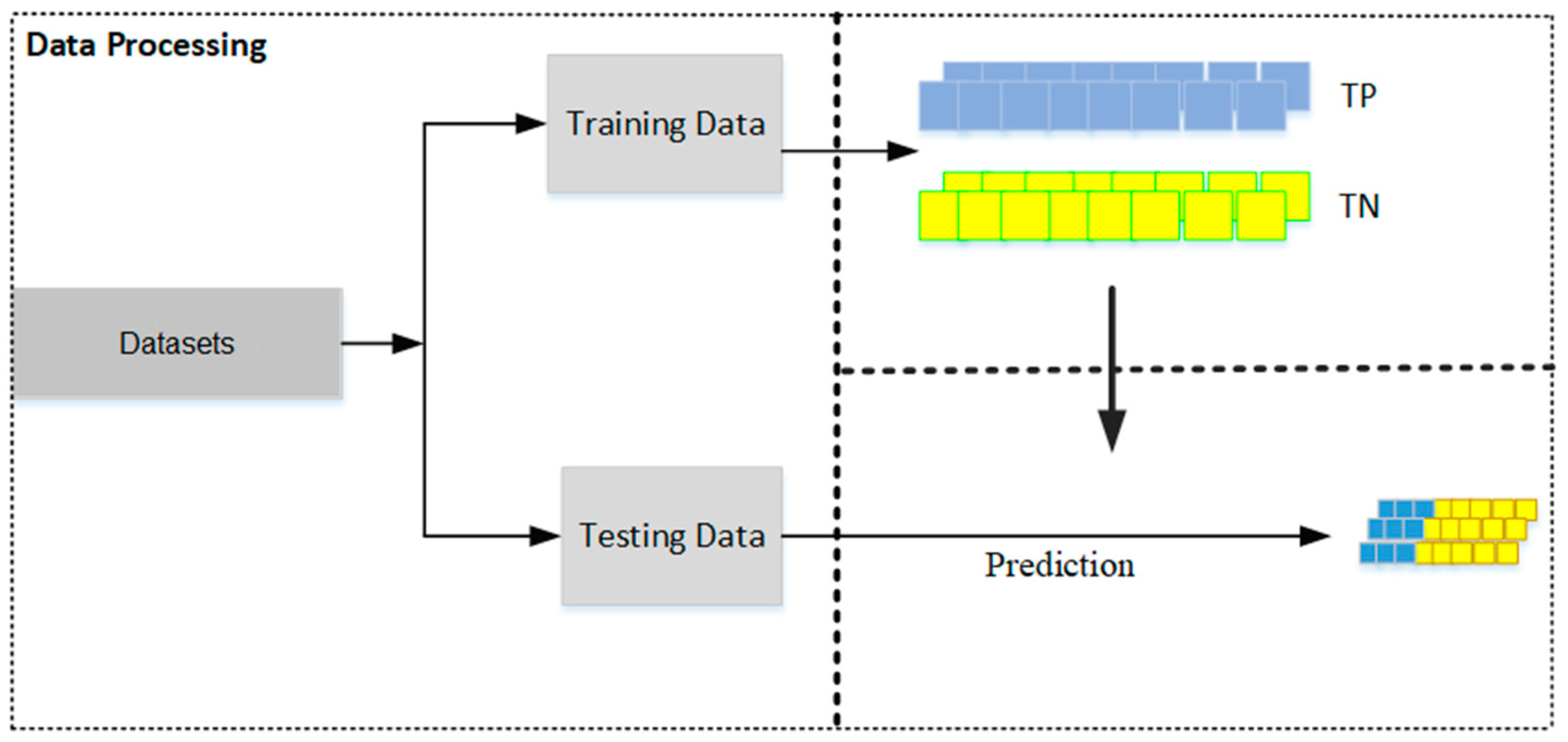
Figure 2.
The dataset is partitioned into a training set, used to construct prediction models, and a testing set, used to evaluate the performance of the models.
Figure 2.
The dataset is partitioned into a training set, used to construct prediction models, and a testing set, used to evaluate the performance of the models.
3. Building Better Virtual Sensors: An Iterative Strategy of Nucleation and Updates
Virtual sensors are integral to our algorithm, leveraging sensor weight scores (SWSs) to discern the discriminative power of binary vector segments in distinguishing between positives and negatives. These portions correspond to fragments within dataset items or queries, with SWSs being computed based on sensor scoring rules (SSRs), which are adapted to suit true positive (TP) and true negative (TN) items differently. For protein sequences, this process involves segmenting the sequences into frames, each frame representing a distinct subset of amino acids. The SWSs for these frames are then calculated within the training set, facilitating the creation of virtual sensors. The process of sensor selection and replacement (SSR) employs logical operations such as XOR (for TN items) and XNOR (for TP items), which are used to integrate sensors with vector segments, thereby computing the SWSs for these segments. The aggregated SWSs of these segments contribute to the overall sensor score.
Building on this foundation, this article delves into the iterative strategy behind constructing more effective virtual sensors, as outlined in our case study for step 2.3 of the ILE algorithm (see Figure 3). This begins with developing a virtual sensor specifically designed to predict the likelihood of a segment’s being the initial transmembrane helix, based on a comprehensive analysis rule set derived from core segments identified in the training dataset. Selection of the initial core segments is based on experimental data and/or expert insights, with a focus on the physicochemical and relational attributes of segment positions.
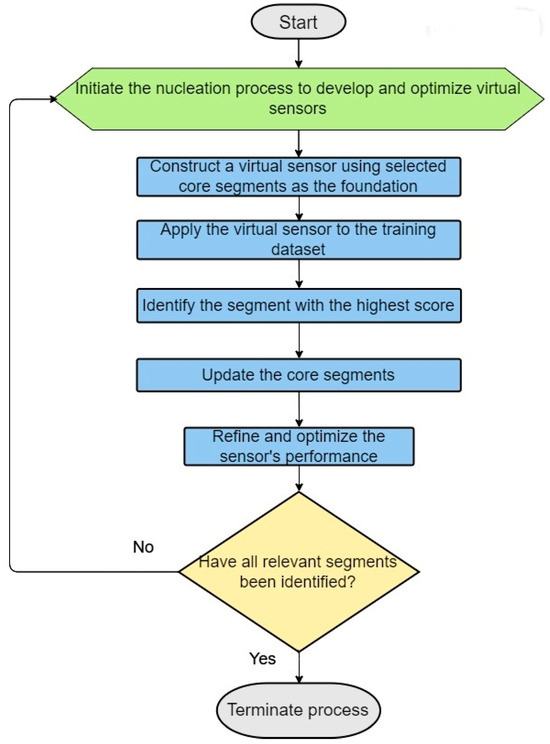
Figure 3.
Steps in the dynamic construction of virtual sensors employing nucleation and iterative updates.
This virtual sensor is then iteratively applied across the training set to pinpoint likely segments that represent the first transmembrane helix, incorporating the highest scoring segment into the core for subsequent virtual sensor development. This cyclical enhancement continues until a refined sensor emerges, capable of accurately detecting the first transmembrane helix.
Critical to our methodology is the development of virtual sensors for all seven transmembrane helices, ensuring that each G protein-coupled receptor (GPCR) in the positive set is thoroughly screened for the presence and proper sequence of these helices. This guarantees a robust dataset. We calculate an overall score for each GPCR based on the combined scores from the seven virtual sensors, fine-tuning the weight of each sensor to maximize predictive efficiency and performance.
Through this systematic and iterative approach, we significantly advance the development of virtual sensors, enhancing their accuracy and efficiency at identifying essential transmembrane helix segments. Such progress is crucial for protein classification, homology-based modeling, and deepening our understanding of GPCR structures and functions.
The molecular descriptors used to model hERG liability were calculated with Molecular Operating Environment (MOE) software, version 2009.10, available at http://www.chemcomp.com (accessed on 15 February 2017). These descriptors include a variety of properties, such as molecular weight, log P, hydrogen-bond donors and acceptors, solubility, total molecular charge, the distribution of the molecular charge, the types and numbers of atoms within the molecule, topological indices, connectivity indices such as Chi indices, and the number of aromatic rings [47]. To evaluate and confirm the model’s accuracy in predicting hERG liability, the dataset containing both active and inactive ligands was divided, allocating two-thirds for training and one-third for testing. The division of these sets was carried out using a random selection module that was developed in-house.
4. Results
To address the increasing complexity of managing vast amounts of data from newly sequenced proteins and DNA, innovative computational methods are essential. These methods are designed to establish links between sequences and functions (commonly known as classification) and to predict 3D structures from sequences. In this paper, we demonstrate our approach by explaining how we determined whether a given protein belonged to the GPCR family and highlight the utility of this approach for identifying and classifying proteins (ICP).
Protein identification and classification and multiple sequence alignment involve significant computational challenges. These tasks are known to be nondeterministic polynomial-time hard (NP-hard), such that the number of potential solutions increases exponentially with the length of the amino acid sequence or the number of residues in a moiety.
Our study analyzed a total of 167 proteins, which included 31 acetylcholine receptors, 44 adrenoreceptors, 38 dopamine receptors, and 54 serotonin receptors. The outcomes were benchmarked against two established methods in this field, the covariant-discrimination algorithm [48] and the support vector machine algorithm, tailored for amine receptor classification [49].
Using the first method, the true positive (TP) classification rates for the acetylcholine, adrenoreceptor, dopamine, and serotonin items were 67.74%, 88.64%, 81.58%, and 88.89%, respectively, leading to an overall accuracy of 83.23%. The second method was more accurate, correctly classifying 93.6% of the TP acetylcholine items, 100.00% of the TP adrenoreceptor items, 92.1% of the TP dopamine items, and 98.2% of the TP serotonin items, resulting in an overall accuracy of 96.4%.
Remarkably, our algorithm, the ILE, achieved perfect accuracy, classifying 100% of the TP items associated with the acetylcholine, adrenoreceptor, dopamine, and serotonin receptors. This translates to an overall accuracy of 100%. Table 1 presents the results of our virtual high-throughput screening, comparing the performance of the established algorithms with the ILE method in identifying GPCR proteins.

Table 1.
Comparison of the performance of established algorithms with the ILE method in the virtual high-throughput screening and identification of GPCR proteins.
Table 2 shows the results from a virtual high-throughput screening, showcasing the efficacy of various established algorithms for categorizing GPCR proteins into their respective superfamilies, A, B, or C. For purposes of comparison, these outcomes are juxtaposed with the performance of the ILE method.
Table 2.
Performance of established algorithms for GPCR protein classification into superfamilies A, B, or C in virtual high-throughput screening, compared to the ILE method.
Table 3 presents the results of a virtual high-throughput screening, depicting the classification of GPCR proteins at their initial subfamily level—amine, peptide, and olfactory—using various well-known algorithms. These results are compared with the performance of the ILE method.
Table 3.
Performance of established algorithms for GPCR protein classification at the first-subfamily level in virtual high-throughput screening, compared to the ILE method.
Table 4 presents the results from a virtual high-throughput screening, demonstrating the classification of GPCR proteins at their secondary subfamily level into adrenergic, dopamine, and histamine categories. The classifications, achieved using various well-recognized algorithms, are compared with the performance of the ILE method.
Table 4.
Performance of established algorithms for GPCR protein classification at the second-subfamily level in virtual high-throughput screening, compared to the ILE method.
Reviewing Table 1, Table 2, Table 3 and Table 4 clearly demonstrates that the application of ILE technology in protein classification offers notable improvements over other methods such as SVMs [43] and HMMs [44], which have faced limitations and required performance enhancements, particularly in specificity, selectivity, and overall accuracy. The advancements offered by ILE, especially in accurately classifying proteins, highlight its potential to replace or complement existing technologies [45,46].
Homology Modeling: Achieving precise multiple sequence alignment (MSA) is essential for improving the accuracy of pairwise sequence alignments, reducing misalignments, and enabling the creation of more reliable 3D models. The ILE method is particularly advantageous when working with protein families that share a common fold and include multiple members, as it allows for the effective interpretation of sequences in the database. This facilitates accurate MSA and the construction of optimal comparative models.
To assess the ILE method’s effectiveness, we analyzed 124 unique proteins from the serine protease family, sourced from the Brookhaven Protein Data Bank (PDB). We calculated the sequence identity score for each pair and employed the ILE method to optimize sequence alignment. This MSA process identified residues in 98 of the proteins, while 28 proteins had incomplete coordinates for at least one residue in their experimentally determined 3D structures. We extracted the alpha carbons (Cα) of residues from selected PDB structures and performed structural superimposition. The quality of the models was evaluated by comparing superimpositions of our homology-based models on X-ray structures of the proteins and by measuring the Cα root-mean-square deviations (RMSDs).
Table 5 presents the results of our homology modeling study, which demonstrate the efficacy of the ILE method across various target–template identity classes within the serine protease family.
Table 5.
Performance of the ILE method for homology modeling for different target–template identity classes within the serine protease family.
The multiple sequence alignment (MSA) matrix, generated using the ILE method on a dataset of serine proteases, was utilized to identify sequence segments suitable for comparative modeling. This process involved a voting approach, where the contribution of each amino acid conservation at a given sequence position was determined based on its frequency at that position, as calculated by Equation (1). These frequency measurements were performed for all the sequences in the dataset.
In this context, Cij represents the conservation factor for residue type i at sequence position j. The term nij denotes the number of sequences containing amino acid i at position j in the multiple sequence alignment, while k refers to the total number of sequences in the dataset. A positional conservation threshold (PCT) was established to indicate whether the conservation factor for residue type i at sequence position j, as per Equation (1), was above a certain point. Demarcating a PCT in refining models is advantageous, as it leads to the development of more accurate homology-based models.
In addition, ILE technology was harnessed to develop an in silico prediction system aimed at identifying chemicals with potential hERG inhibition liability. Our dataset consisted of 300 compounds classified as “active,” each with a half-maximal inhibitory concentration (IC50) of less than 10 µM. Additionally, we included 3000 compounds categorized as “non-active,” randomly selected from the ZINC database. Although hERG activity among some molecules in the non-active class was a possibility, their impact on the overall predictive accuracy of our model was considered minimal.
5. hERG Liability Indexing Model
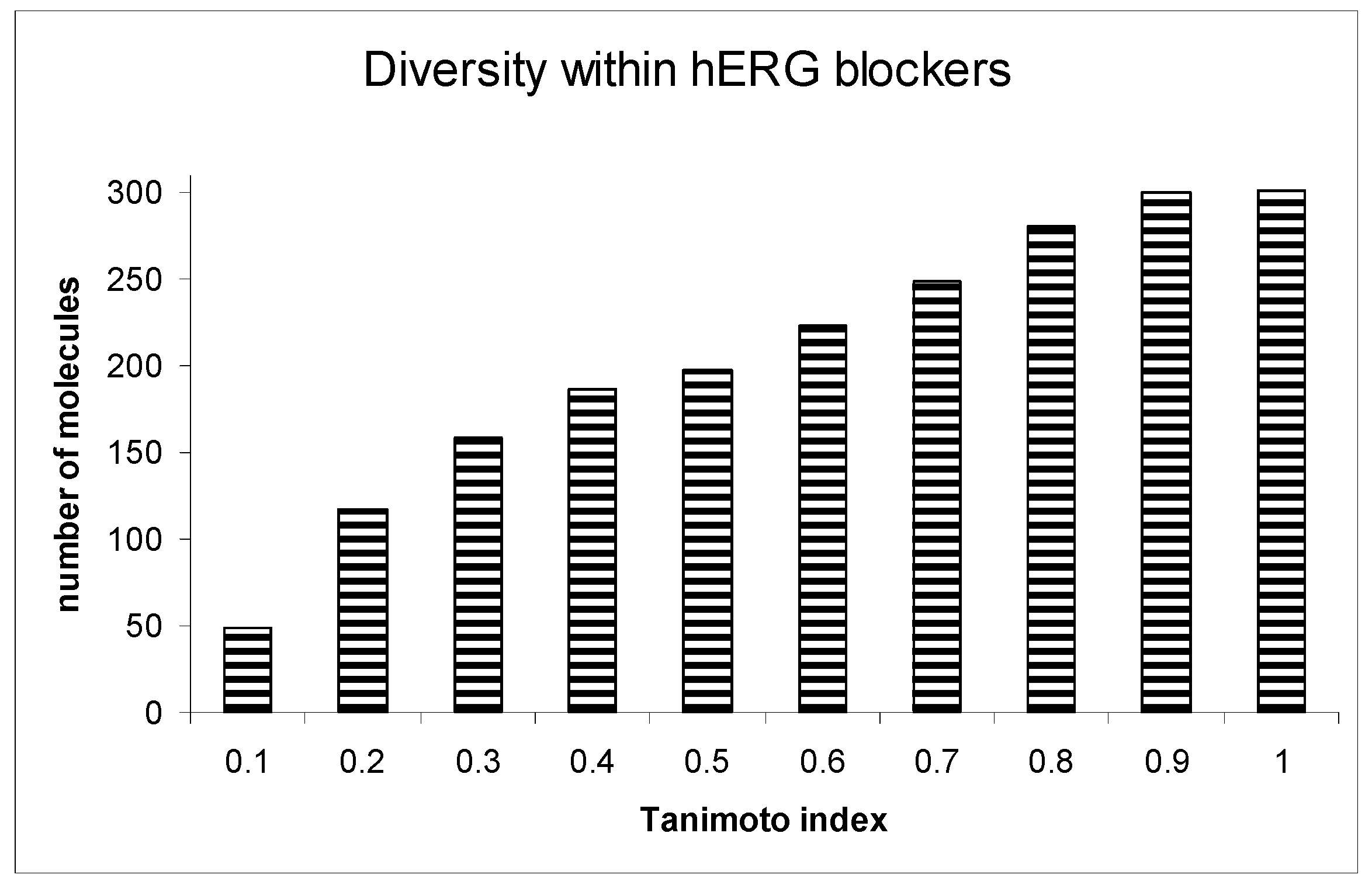
A crucial aspect of our analysis was examining the diversity within the hERG blockers. Among the 300 compounds identified as hERG blockers, a significant majority (249 compounds) displayed a Tanimoto index of similarity below 0.7, indicating a high degree of chemical diversity. This diversity was critical for ensuring the robustness and generalizability of the prediction model.
Further extending this analysis to the proposed non-blockers from the ZINC database, we observed even greater diversity. An overwhelming 99.4% of these molecules had a Tanimoto similarity index below 0.7. This high level of diversity among non-blockers is particularly notable, as it underscores the wide-ranging applicability of our prediction system across various chemical entities.
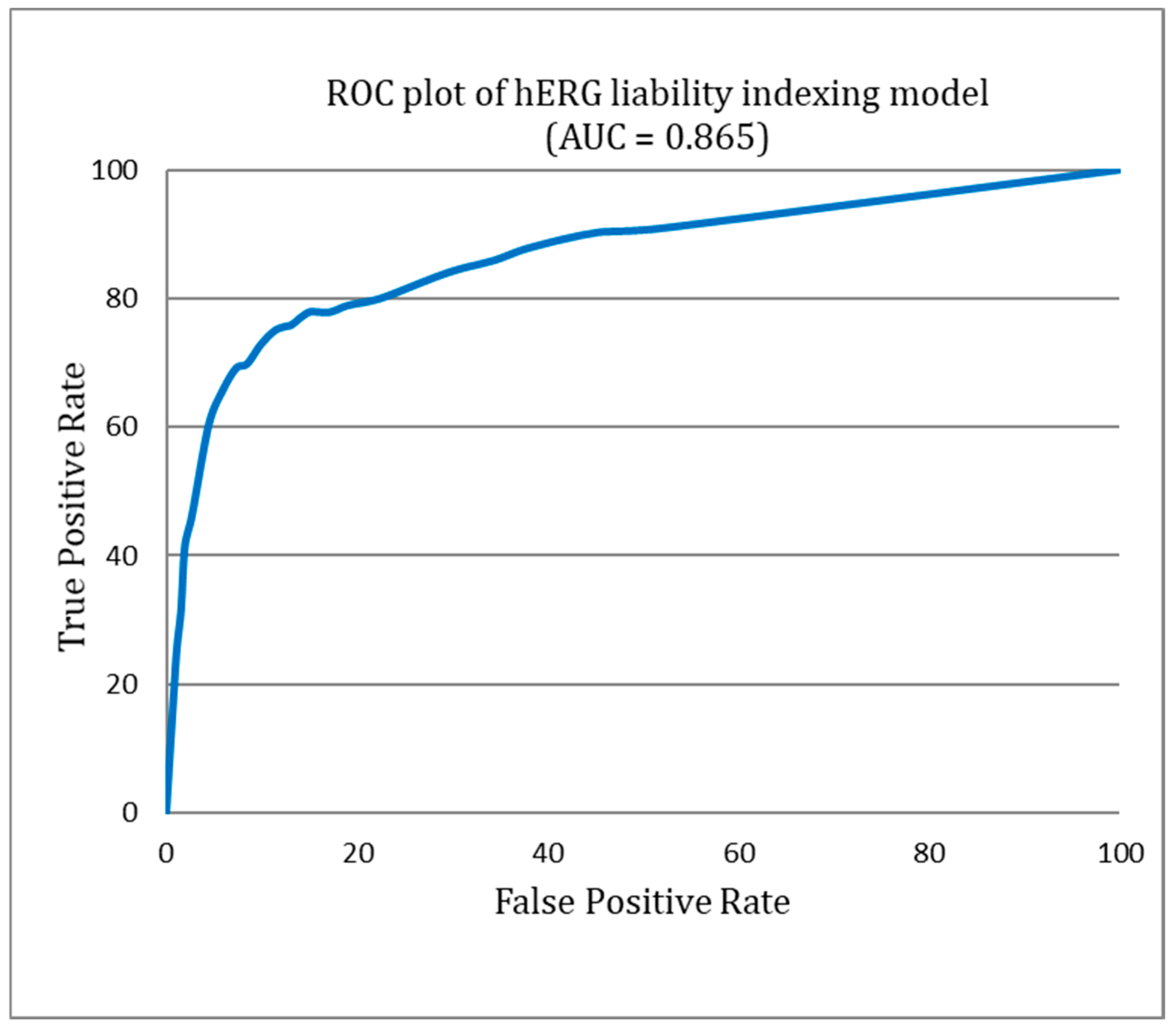
Figure 4 provides a detailed illustration of the diversity within both the hERG blockers and non-blockers. This visual representation illuminates the significant chemical variability encompassed by our study, a factor that plays a pivotal role in the robustness and efficacy of the ILE-based hERG inhibition liability prediction system. Figure 5 presents the receiver operating characteristic (ROC) curve for the indexing model of hERG blockers, with an area under the curve (AUC) of 0.865. This indicates that the model is highly accurate in distinguishing between hERG blockers and non-blockers.
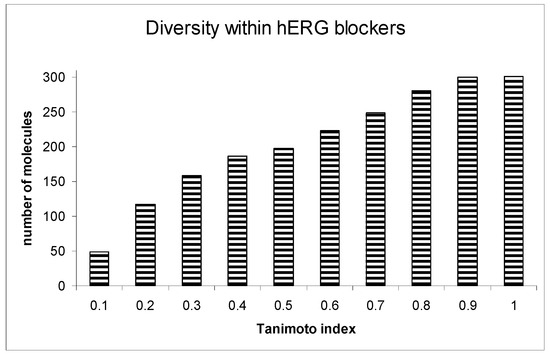
Figure 4.
Diversity within hERG blockers.
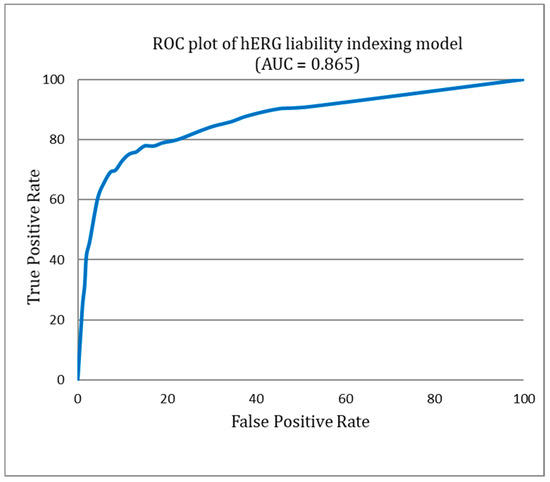
Figure 5.
Receiver operating characteristic (ROC) curve of the hERG liability indexing model.
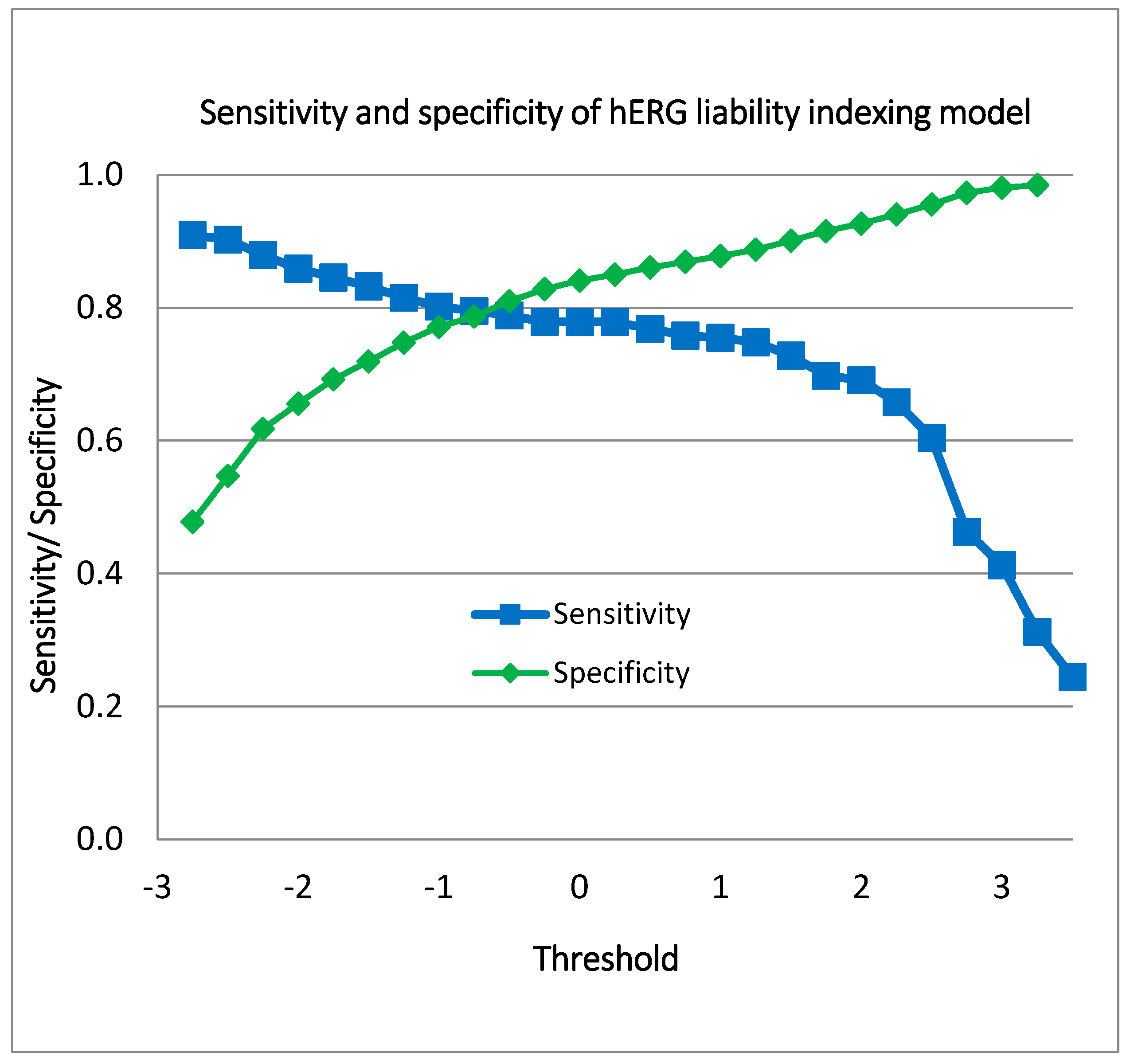
Figure 6 illustrates the performance of the indexing model for hERG blockers, showcasing the range of sensitivity and specificity values achieved at various hERG liability thresholds. The sensitivity values span from 0.245 to 0.909, indicating that the model’s ability to correctly identify hERG blockers varies significantly with the threshold. Similarly, the specificity values range from 0.478 to 0.99, reflecting the model’s capability to accurately exclude non-blockers across different threshold settings.
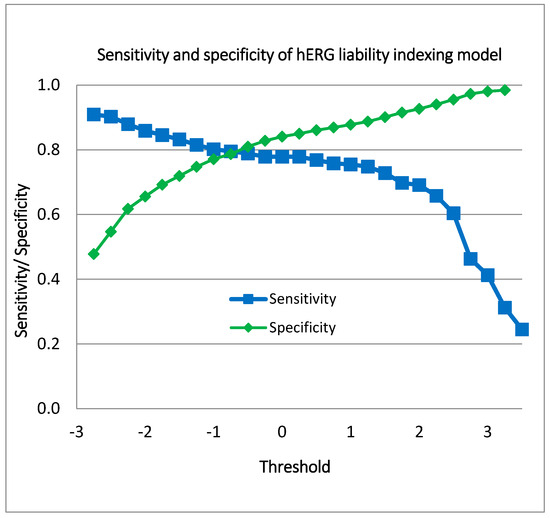
Figure 6.
Sensitivity and specificity values of the indexing model for hERG liability.
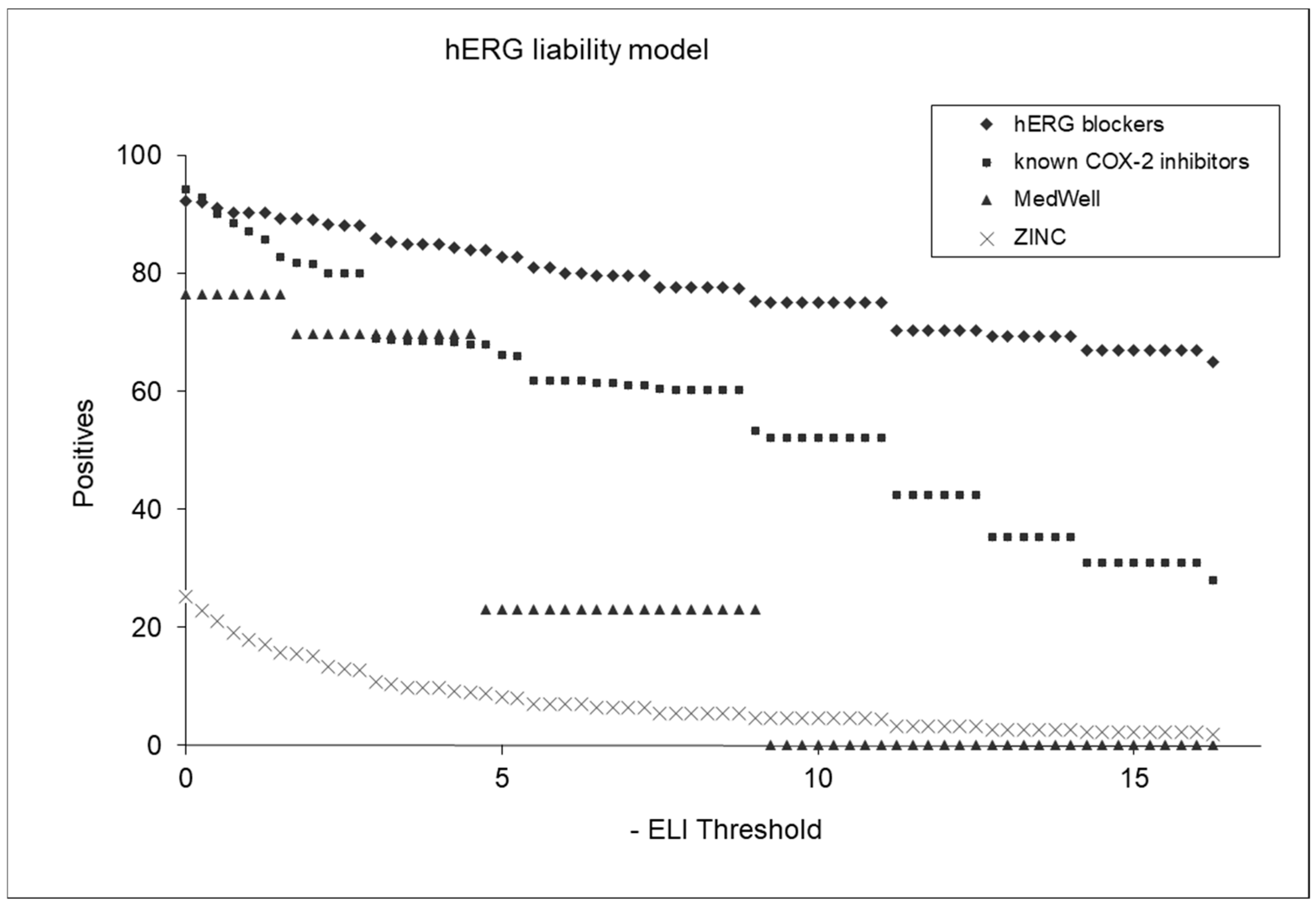
Figure 7 displays plots of positive factors that underlie the hERG liability model’s predictions for various compound categories. The hERG liability index (ELI) is an index that indicates the likelihood of a molecule’s being hERG-liable. The higher the ELI value, the greater the chance that the molecule will bind to hERG and act on it. The plot features four distinct groups: hERG blockers, known COX-2 inhibitors, MedWell agents, and a selection of molecules randomly chosen from the ZINC database (referred to as “chemicals on the shelf”). Each group is represented by a different color or marker in the plot, allowing for a clear visual comparison of their hERG liability profiles. The hERG blockers are expected to show higher hERG liability scores, which serve as a benchmark for comparison. The known COX-2 inhibitors and MedWell agents are plotted to assess their relative hERG liabilities as predicted by the model, providing insights into their cardiac safety. The chemicals from the ZINC database offer a broader perspective, representing a diverse range of compounds. This comparative plot is useful for evaluating the effectiveness and discriminative power of the hERG liability model, illustrating its capability to differentiate between compounds with varying degrees of hERG inhibition.
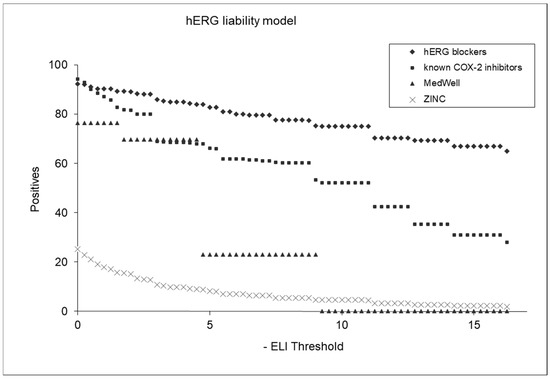
Figure 7.
Comparative analysis of hERG liability across different compound classes.
Model Validation and Performance Assessment
To rigorously assess the predictive performance of our models, we conducted a series of leave-1/3-out validation runs. In each run, a subset comprising 100 active and 1000 non-active compounds was randomly selected to serve as the holdout test set. This approach ensured a comprehensive evaluation across various subsets of the data.
Our results were encouraging; the models achieved a Matthews correlation coefficient (MCC) of approximately 0.8, based on the average of three independent test runs. This high MCC value indicates a strong correlation between the predicted and actual classifications and underscores the models’ robustness.
In terms of specific predictions, the models successfully identified nearly 89% of the hERG blockers (true positives), demonstrating their efficacy at detecting compounds with hERG liability. In addition, only about 10% of the molecules from the ZINC database were incorrectly classified as hERG blockers (false positives), showcasing the models’ precision.
Further analysis was performed by applying an ELI (hERG liability index) threshold of 5.0. This yielded a variety of identification percentages across different compound classes: 83% for hERG agents, 66% for COX-2 inhibitors, 23% for MedWell molecules, and 8.5% for ZINC chemicals potentially liable to hERG. These results highlight the models’ discriminative capability, effectively distinguishing between compounds with varying levels of hERG inhibition potential.
Overall, the validation and performance metrics demonstrate the high accuracy and reliability of our models for predicting hERG liability, affirming their potential utility in the drug discovery and development process.
6. Discussion
Accurate multiple sequence alignment (MSA) is essential for refining pairwise sequence alignments, reducing misalignments, and improving the construction of 3D models. Our method is particularly effective when analyzing protein families with a shared fold and multiple members, as it adeptly interprets the extensive data within sequence databases. It facilitates the precise alignment of multiple sequences and the development of superior comparative models.
To assess the performance of our method, we examined 124 unique proteins from the serine protease family, obtained from the Brookhaven Protein Data Bank (PDB). We calculated sequence identity scores for each sequence pair and employed the ILE method for optimal alignment. From the MSA, residues were identified in 98 proteins, while 28 proteins lacked coordinates for at least one residue in their 3D experimental structures. We extracted the alpha carbons (Cα) of these residues and performed structural superimpositions.
To evaluate the quality of our homology-based models, we compared the results with the proteins’ X-ray structures by measuring the Cα root-mean-square deviations (RMSDs). Table 5 presents a detailed overview of our method’s performance in homology modeling; it categorizes the results based on sequence identity ranges between the target and template sequences and reports the percentage of models that had RMSD values below the specified thresholds.
Furthermore, we processed the MSA matrix of the serine protease dataset, using our method to select sequence segments for comparative modeling. A voting approach was used, which determined each amino acid’s contribution to conservation at a sequence position, based on its frequency. This led to the definition of positional conservation thresholds (PCTs) to refine our models.
In conclusion, our method performed with exceptional accuracy on the MSA, resulting in highly reliable homology-based models. The integration of a voting-based approach and PCTs further enhanced the models’ accuracy. These findings highlight the effectiveness of the ILE method for addressing complex tasks in protein identification, classification, and structural modeling and extend its applicability beyond bioinformatics.
Model for hERG-Toxic Molecules
In the realm of drug discovery, the ILE-based model emerges as a crucial tool for assessing the hERG liability of drug candidates. Its significance lies in its ability to inform decision-making processes, potentially leading to a notable reduction in attrition rates during drug development. By accurately predicting hERG toxicity, the model helps to identify risks early in the drug discovery pipeline, thereby streamlining the development process and reducing costly late-stage failures.
One of the key strengths of the ILE-based model is its utilization of 1D-2D descriptors. These descriptors not only effectively represent the necessary chemical properties for hERG liability prediction but are also advantageous in respect to the ease and speed of calculation. Using them enhances the model’s applicability in fast-paced research environments, where timely and efficient decision making is critical.
Furthermore, the simplicity and computational efficiency of the 1D-2D descriptors make the ILE model highly practical for widespread use in pharmaceutical research. They enable rapid screening of large libraries of compounds and early identification in the development cycle of those with hERG toxicity issues. This capability is particularly valuable in an industry where time and resource optimization are paramount.
The ILE model’s predictive power and practicality underscore its potential to become an integral part of the drug development toolkit. By providing researchers with a swift and reliable method for assessing hERG liability, the model can contribute significantly to the development of safer pharmaceuticals. Moreover, its integration into drug discovery pipelines will lead to a more streamlined, cost-effective, and ultimately successful drug development process.
Advantages and Limitations of ILE Technology: The ILE technology offers several significant advantages in the context of bioinformatics and drug discovery. One of its primary strengths is its exceptional accuracy in protein classification, as evidenced by its outstanding performance in classifying GPCRs in our study. The technology also excels in virtual high-throughput screening, efficiently identifying candidate molecules with desirable properties, and in homology-based modeling, where it has shown remarkable precision in creating 3D models. Furthermore, its straightforward design and processing efficiency make it a practical tool for rapid screening of chemicals for their bioactivity, significantly reducing the time and resources required to discover new hits in the drug discovery process.
However, despite these strengths, ILE technology does have certain limitations. A primary challenge is that, while the ILE model is highly effective within the specific contexts tested, its generalizability to other domains or different types of proteins may require further validation. Additionally, the reliance on predefined descriptors means the model’s performance can be constrained by the quality and relevance of the input data, potentially affecting its predictive accuracy in cases involving small or noisy datasets.
In conclusion, the ILE-based model for hERG-toxic molecules represents a significant advancement in drug safety assessment. Its ability to combine accuracy with practicality makes it a valuable asset to the pursuit of effective and safe therapeutic agents.
Future Perspectives: ILE technology presents promising potential for expanding its application beyond its current use. Future developments may include its integration into additional areas in the fields of bioinformatics, cheminformatics, and drug discovery. Specifically, the adaptability of the ILE algorithm could be harnessed to optimize processes in these fields, thereby enhancing the precision and efficiency of data analysis. We also plan to further refine the algorithmic processes of ILE, particularly to optimize the indexing chemicals for their bioactivity, potentially leading to even greater performance improvements. These advancements could establish ILE as a vital tool in various scientific endeavors, providing robust solutions to complex challenges in computational biology and drug discovery.
7. Conclusions
This paper introduces intelligent learning engine (ILE) optimization technology, a significant leap forward in the realm of bioinformatics and protein science. The ILE method, with its proficiency in multiple sequence alignment (MSA) and protein identification, classification, and structural modeling, effectively overcomes some of the most complex challenges in protein analysis. The empirical evaluation and results of this study, based on a dataset of 124 unique proteins from the serine protease family, validate the remarkable accuracy of the ILE method for MSA. Notably, its capability to identify residues in 98 proteins, even in the absence of experimental 3D structures, underscores its efficacy. Additionally, the method’s success in creating highly precise 3D models, as evidenced by Cα root-mean-square deviations (Cα RMSDs) compared with those observed in X-ray images, marks its significant potential for homology-based modeling. The incorporation of positional consensus templates (PCTs), developed through an innovative voting-based approach, considerably elevates the quality of these models.
The impact of the ILE method reaches beyond protein science, making a notable mark in the field of drug discovery. The technology’s advanced and discriminative model, utilizing 1D-2D descriptors for assessing hERG blockage by chemical entities, provides critical insights for developers of cardio-safe drug candidates. Particularly, the hERG toxicity indexing model is a key innovation, offering valuable data that can suggest a preference for certain types of molecules, for example, a preference for MedWell agents over COX-2 inhibitors in relation to cardio-safety.
The implications of this study are far-reaching, extending well beyond theoretical applications to practical uses across diverse scientific and technological sectors. By enhancing our understanding and manipulation of biological data and structures, ILE optimization technology lays the groundwork for significant advancements in areas such as drug discovery, healthcare, finance, and telecommunications. Its ability to improve screening accuracy across various domains stands as a testament to its transformative potential.
In conclusion, the ILE method adeptly addresses the intricate challenges of protein analysis and paves the way for novel applications in a plethora of fields. Its wide-ranging applicability and demonstrated effectiveness in both bioinformatics and other areas establish it as an indispensable tool in the continual pursuit of scientific and technological advancement.
In future work, we aim to explore the application of the simulated annealing algorithm, as reported by M. A. Rufal et al. [50], to improve the efficiency and accuracy of our ILE optimization technology. By integrating the principles of simulated annealing, we anticipate enhancing our algorithm’s ability to navigate complex optimization landscapes, potentially leading to even more significant breakthroughs in protein analysis, drug discovery, and beyond.
Author Contributions
J.R. and A.R. (Anwar Rayan) were instrumental in conceiving and designing this study, securing funding, and refining the final manuscript. J.R. and S.A.-L. significantly contributed to interpreting the data, developing the study’s conclusions, and drafting the initial manuscript. Furthermore, J.R. worked in collaboration with A.R. (Adam Rayan) on the methodological design, validation processes, enhancement of visual representations, and comprehensive formal analysis. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated or analyzed during this study are included within the manuscript.
Acknowledgments
Anwar Rayan acknowledges the Al-Qasemi Research Foundation for covering the costs of language editing.
Conflicts of Interest
Adam Rayan was employed by NGS Ac-Tech—Next Generation Scholars Ltd. The remaining authors declare that the research was conducted without any commercial or financial relationships that could be interpreted as a potential conflict of interest.
References
- Da’adoosh, B.; Marcus, D.; Rayan, A.; King, F.; Che, J.; Goldblum, A. Discovering highly selective and diverse PPAR-delta agonists by ligand based machine learning and structural modeling. Sci. Rep. 2019, 9, 1106. [Google Scholar] [CrossRef] [PubMed]
- Rayan, A.; Raiyn, J.; Falah, M. Nature is the best source of anticancer drugs: Indexing natural products for their anticancer bioactivity. PLoS ONE 2017, 12, e0187925. [Google Scholar] [CrossRef] [PubMed]
- Rayan, A. New vistas in GPCR 3D structure prediction. J. Mol. Model. 2010, 16, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, M.; Rayan, M.; Abu-Lafi, S.; Leonardi, M.E.; Milardi, D.; Guccione, S.; Rayan, A. Homology-based Modeling of Rhodopsin-like Family Members in the Inactive State: Structural Analysis and Deduction of Tips for Modeling and Optimization. Mol. Inform. 2017, 36, 1700014. [Google Scholar] [CrossRef] [PubMed]
- Aswad, M.; Rayan, M.; Abu-Lafi, S.; Falah, M.; Raiyn, J.; Abdallah, Z.; Rayan, A. Nature is the best source of anti-inflammatory drugs: Indexing natural products for their anti-inflammatory bioactivity. Inflamm. Res. 2018, 67, 67–75. [Google Scholar] [CrossRef]
- Awhangbo, L.; Schmitt, V.; Marcilhac, C.; Charnier, C.; Latrille, E.; Steyer, J.P. Determination of the optimal feed recipe of anaerobic digesters using a mathematical model and a genetic algorithm. Bioresour. Technol. 2023, 393, 130091. [Google Scholar] [CrossRef]
- Ghaleb, F.A.; Al-Rimy, B.A.S.; Boulila, W.; Saeed, F.; Kamat, M.; Foad Rohani, M.; Razak, S.A. Fairness-Oriented Semichaotic Genetic Algorithm-Based Channel Assignment Technique for Node Starvation Problem in Wireless Mesh Networks. Comput. Intell. Neurosci. 2021, 2021, 2977954. [Google Scholar] [CrossRef]
- Shi, K.; Huang, L.; Jiang, D.; Sun, Y.; Tong, X.; Xie, Y.; Fang, Z. Path Planning Optimization of Intelligent Vehicle Based on Improved Genetic and Ant Colony Hybrid Algorithm. Front. Bioeng. Biotechnol. 2022, 10, 905983. [Google Scholar] [CrossRef] [PubMed]
- Onizawa, N.; Katsuki, K.; Shin, D.; Gross, W.J.; Hanyu, T. Fast-Converging Simulated Annealing for Ising Models Based on Integral Stochastic Computing. IEEE Trans. Neural Netw. Learn. Syst. 2023, 34, 10999–11005. [Google Scholar] [CrossRef]
- Zhou, X.; Ling, M.; Lin, Q.; Tang, S.; Wu, J.; Hu, H. Effectiveness Analysis of Multiple Initial States Simulated Annealing Algorithm, A Case Study on the Molecular Docking Tool AutoDock Vina. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 3830–3841. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.B.; Jowett, T.; Yu, D.; Pahl, E.; Garden, A.L. Comparison of Taboo Search Methods for Atomic Cluster Global Optimization with a Basin-Hopping Algorithm. J. Chem. Inf. Model. 2023, 63, 5784–5793. [Google Scholar] [CrossRef]
- Rayan, A.; Falah, M.; Raiyn, J.; Da’adoosh, B.; Kadan, S.; Zaid, H.; Goldblum, A. Indexing molecules for their hERG liability. Eur. J. Med. Chem. 2013, 65, 304–314. [Google Scholar] [CrossRef] [PubMed]
- Rayan, A.; Marcus, D.; Goldblum, A. Predicting oral druglikeness by iterative stochastic elimination. J. Chem. Inf. Model. 2010, 50, 437–445. [Google Scholar] [CrossRef]
- Rayan, M.; Abdallah, Z.; Abu-Lafi, S.; Masalha, M.; Rayan, A. Indexing Natural Products for their Antifungal Activity by Filters-based Approach: Disclosure of Discriminative Properties. Curr. Comput. Aided Drug Des. 2019, 15, 235–242. [Google Scholar] [CrossRef]
- Jensen, S.T.; Liu, J.S. BioOptimizer: A Bayesian scoring function approach to motif discovery. Bioinformatics 2004, 20, 1557–1564. [Google Scholar] [CrossRef]
- Carstens, S.; Nilges, M.; Habeck, M. Bayesian inference of chromatin structure ensembles from population-averaged contact data. Proc. Natl. Acad. Sci. USA 2020, 117, 7824–7830. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Yao, H.; Lin, K. An overview of neural networks for drug discovery and the inputs used. Expert Opin. Drug Discov. 2018, 13, 1091–1102. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ghose, U.; Buckley, N.J.; Engelborghs, S.; Sleegers, K.; Frisoni, G.B.; Wallin, A.; Lleó, A.; Popp, J.; Martinez-Lage, P.; et al. Predicting AT(N) pathologies in Alzheimer’s disease from blood-based proteomic data using neural networks. Front. Aging Neurosci. 2022, 14, 1040001. [Google Scholar] [CrossRef] [PubMed]
- Hasanzadeh, A.; Hamblin, M.R.; Kiani, J.; Noori, H.; Hardie, J.M.; Karimi, M.; Shafiee, H. Could artificial intelligence revolutionize the development of nanovectors for gene therapy and mRNA vaccines? Nano Today 2022, 47, 101665. [Google Scholar] [CrossRef]
- Barbero-Aparicio, J.A.; Olivares-Gil, A.; Díez-Pastor, J.F.; García-Osorio, C. Deep learning and support vector machines for transcription start site identification. PeerJ Comput. Sci. 2023, 9, e1340. [Google Scholar] [CrossRef]
- Machicao, J.; Craighero, F.; Maspero, D.; Angaroni, F.; Damiani, C.; Graudenzi, A.; Antoniotti, M.; Bruno, O.M. On the Use of Topological Features of Metabolic Networks for the Classification of Cancer Samples. Curr. Genom. 2021, 22, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Siddique, A.; Shirzaei, S.; Smith, A.E.; Valenta, J.; Garner, L.J.; Morey, A. Acceptability of Artificial Intelligence in Poultry Processing and Classification Efficiencies of Different Classification Models in the Categorisation of Breast Fillet Myopathies. Front. Physiol. 2021, 12, 712649. [Google Scholar] [CrossRef]
- Lepage, T.; Junier, I. Modeling Bacterial DNA: Simulation of Self-Avoiding Supercoiled Worm-Like Chains Including Structural Transitions of the Helix. Methods Mol. Biol. 2017, 1624, 323–337. [Google Scholar] [PubMed]
- Pan, X.; Chen, L.; Feng, K.Y.; Hu, X.H.; Zhang, Y.H.; Kong, X.Y.; Huang, T.; Cai, Y.D. Analysis of Expression Pattern of snoRNAs in Different Cancer Types with Machine Learning Algorithms. Int. J. Mol. Sci. 2019, 20, 2185. [Google Scholar] [CrossRef]
- Aryanti, C.; Uwuratuw, J.A.; Labeda, I.; Raharjo, W.; Lusikooy, R.E.; Rauf, M.A.; Mappincara, A.; Sampetoding, S.; Kusuma, M.I.; Syarifuddin, E. The Mutation Portraits of Oncogenes and Tumor Supressor Genes in Predicting the Overall Survival in Pancreatic Cancer: A Bayesian Network Meta-Analysis. Asian Pac. J. Cancer Prev. 2023, 24, 2895–2902. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Marchant, R.; Morris, R.W.; Baur, L.A.; Simpson, S.J.; Cripps, S. Bayesian network modelling to identify on-ramps to childhood obesity. BMC Med. 2023, 21, 105. [Google Scholar] [CrossRef] [PubMed]
- Emani, P.S.; Geradi, M.N.; Gürsoy, G.; Grasty, M.R.; Miranker, A.; Gerstein, M.B. Assessing and mitigating privacy risks of sparse, noisy genotypes by local alignment to haplotype databases. Genome Res. 2023, 33, 2156–2173. [Google Scholar] [CrossRef]
- Ringbauer, H.; Huang, Y.; Akbari, A.; Mallick, S.; Olalde, I.; Patterson, N.; Reich, D. Accurate detection of identity-by-descent segments in human ancient DNA. Nat. Genet. 2023, 56, 143–151. [Google Scholar] [CrossRef]
- Kwak, K.; Kostic, E.; Kim, D. Gait variability-based classification of the stages of the cognitive decline using partial least squares-discriminant analysis. Sci. Prog. 2023, 106, 368504231218604. [Google Scholar] [CrossRef]
- Teunissen, J.W.; Faber, I.R.; De Bock, J.; Slembrouck, M.; Verstockt, S.; Lenoir, M.; Pion, J. A machine learning approach for the classification of sports based on a coaches’ perspective of environmental, individual and task requirements: A sports profile analysis. J. Sports Sci. 2023, 1–10. [Google Scholar] [CrossRef]
- Zeidan, M.; Rayan, M.; Zeidan, N.; Falah, M.; Rayan, A. Indexing Natural Products for Their Potential Anti-Diabetic Activity: Filtering and Mapping Discriminative Physicochemical Properties. Molecules 2017, 22, 1563. [Google Scholar] [CrossRef]
- SoRelle, R. Warnings strengthened on tranquilizer inapsine (Droperidol). Circulation 2001, 104, E9061–E9062. [Google Scholar] [PubMed]
- Oyekan, P.J.; Gorton, H.C.; Copeland, C.S. Antihistamine-related deaths in England: Are the high safety profiles of antihistamines leading to their unsafe use? Br. J. Clin. Pharmacol. 2021, 87, 3978–3987. [Google Scholar] [CrossRef] [PubMed]
- Paterson, T.; Azizoglu, S.; Gokhale, M.; Chambers, M.; Suphioglu, C. Preserved Ophthalmic Anti-Allergy Medication in Cumulatively Increasing Risk Factors of Corneal Ectasia. Biology 2023, 12, 1036. [Google Scholar] [CrossRef] [PubMed]
- Madeira, L.; Queiroz, G.; Henriques, R. Prepandemic psychotropic drug status in Portugal: A nationwide pharmacoepidemiological profile. Sci. Rep. 2023, 13, 6912. [Google Scholar] [CrossRef]
- Abbas, S.; Ihle, P.; Adler, J.B.; Engel, S.; Günster, C.; Linder, R.; Lehmkuhl, G.; Schübert, I. Psychopharmacological Prescriptions in Children and Adolescents in Germany. Dtsch. Arztebl. Int. 2016, 113, 396–403. [Google Scholar] [CrossRef]
- Kobayashi, K.; Omuro, N.; Takahara, A. The conventional antihistamine drug cyproheptadine lacks QT-interval-prolonging action in halothane-anesthetized guinea pigs: Comparison with hydroxyzine. J. Pharmacol. Sci. 2014, 124, 92–98. [Google Scholar] [CrossRef]
- Binggeli, C.; Candinas, R.; Brunckhorst, C. Psychopharmaceuticals and arrhythmias. Ther. Umsch. 2004, 61, 279–283. [Google Scholar] [CrossRef]
- Haddad, S.; Oktay, L.; Erol, I.; Şahin, K.; Durdagi, S. Utilizing Heteroatom Types and Numbers from Extensive Ligand Libraries to Develop Novel hERG Blocker QSAR Models Using Machine Learning-Based Classifiers. ACS Omega 2023, 8, 40864–40877. [Google Scholar] [CrossRef]
- Venkateshappa, R.; Hunter, D.V.; Muralidharan, P.; Nagalingam, R.S.; Huen, G.; Faizi, S.; Luthra, S.; Lin, E.; Cheng, Y.M.; Hughes, J.; et al. Targeted activation of human ether-à-go-go-related gene channels rescues electrical instability induced by the R56Q+/- long QT syndrome variant. Cardiovasc. Res. 2023, 119, 2522–2535. [Google Scholar] [CrossRef]
- Falah, M.; Rayan, M.; Rayan, A. A Novel Paclitaxel Conjugate with Higher Efficiency and Lower Toxicity: A New Drug Candidate for Cancer Treatment. Int. J. Mol. Sci. 2019, 20, 4965. [Google Scholar] [CrossRef] [PubMed]
- Rayan, M.; Shadafny, S.; Falah, A.; Falah, M.; Abu-Lafi, S.; Asli, S.; Rayan, A. A Novel Docetaxel-Biotin Chemical Conjugate for Prostate Cancer Treatment. Molecules 2022, 27, 961. [Google Scholar] [CrossRef]
- Cai, Y.D.; Liu, X.J.; Xu, X.; Zhou, G.P. Support vector machines for predicting protein structural class. BMC Bioinform. 2001, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Yoon, B.J. Hidden Markov Models and their Applications in Biological Sequence Analysis. Curr. Genom. 2009, 10, 402–415. [Google Scholar] [CrossRef] [PubMed]
- Ferran, E.A.; Ferrara, P.; Pflugfelder, B. Protein classification using neural networks. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1993, 1, 127–135. [Google Scholar]
- Gupta, K.; Sehgal, V.; Levchenko, A. A method for probabilistic mapping between protein structure and function taxonomies through cross training. BMC Struct. Biol. 2008, 8, 40. [Google Scholar] [CrossRef]
- Chemical-Computing-Group MOE. Molecular Operating Environment, version 2011.10; Chemical Computing Group: Montreal, QC, Canada, 2011. [Google Scholar]
- Chou, K.C.; Elrod, D.W. Protein subcellular location prediction. Protein Eng. 1999, 12, 107–118. [Google Scholar] [CrossRef]
- Li, Z.; Zhou, X.; Dai, Z.; Zou, X. Classification of G-protein coupled receptors based on support vector machine with maximum relevance minimum redundancy and genetic algorithm. BMC Bioinform. 2010, 11, 325. [Google Scholar] [CrossRef] [PubMed]
- Rufai, M.; Alabison, R.; Abidemi, A.; Dansu, E. Solution to The Travelling Salesperson Problem Using Simulated Annealing Algorithm. Electron. J. Math. Anal. Appl. 2017, 5, 135–142. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).