Efficient Purification of Polyhistidine-Tagged Recombinant Proteins Using Functionalized Corundum Particles


 and
and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
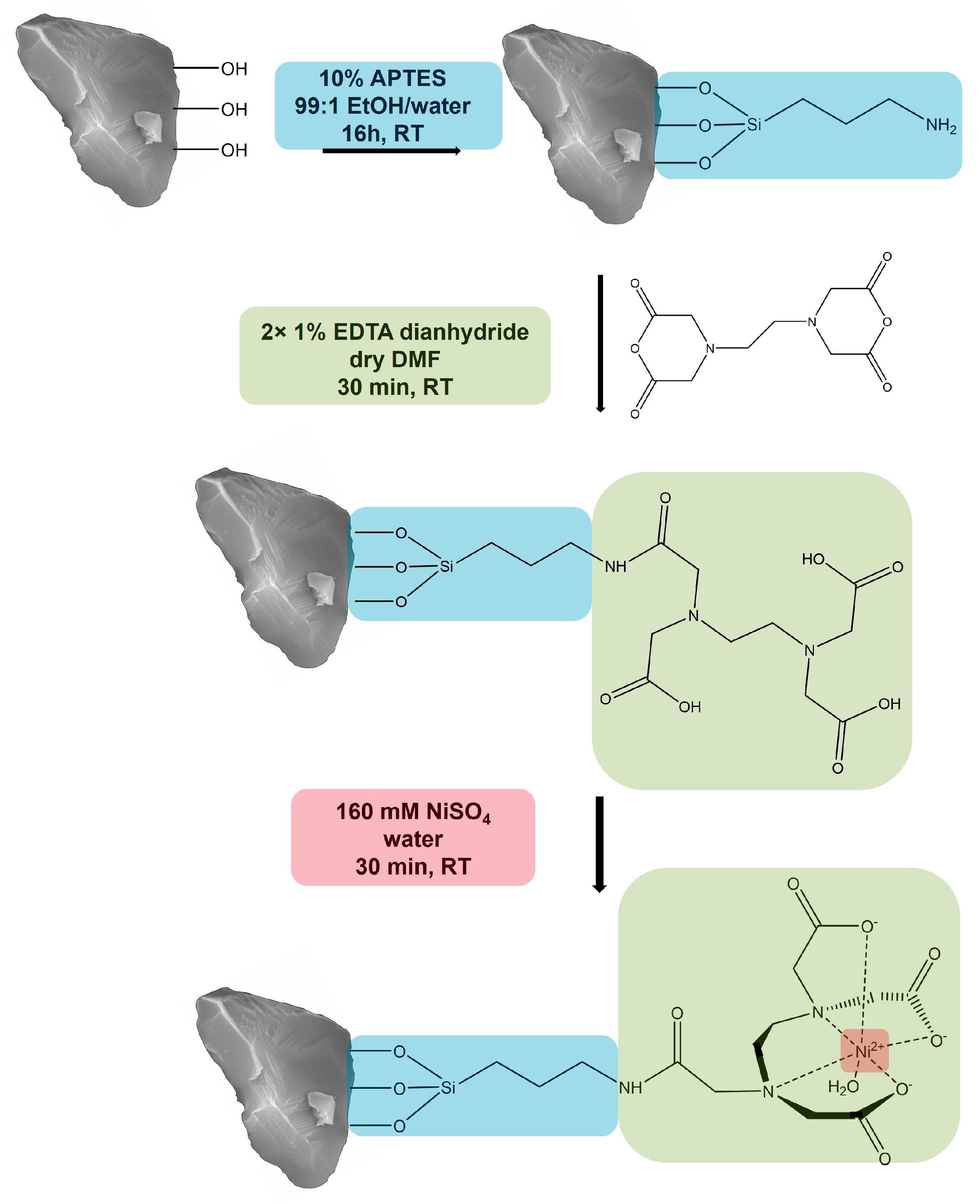
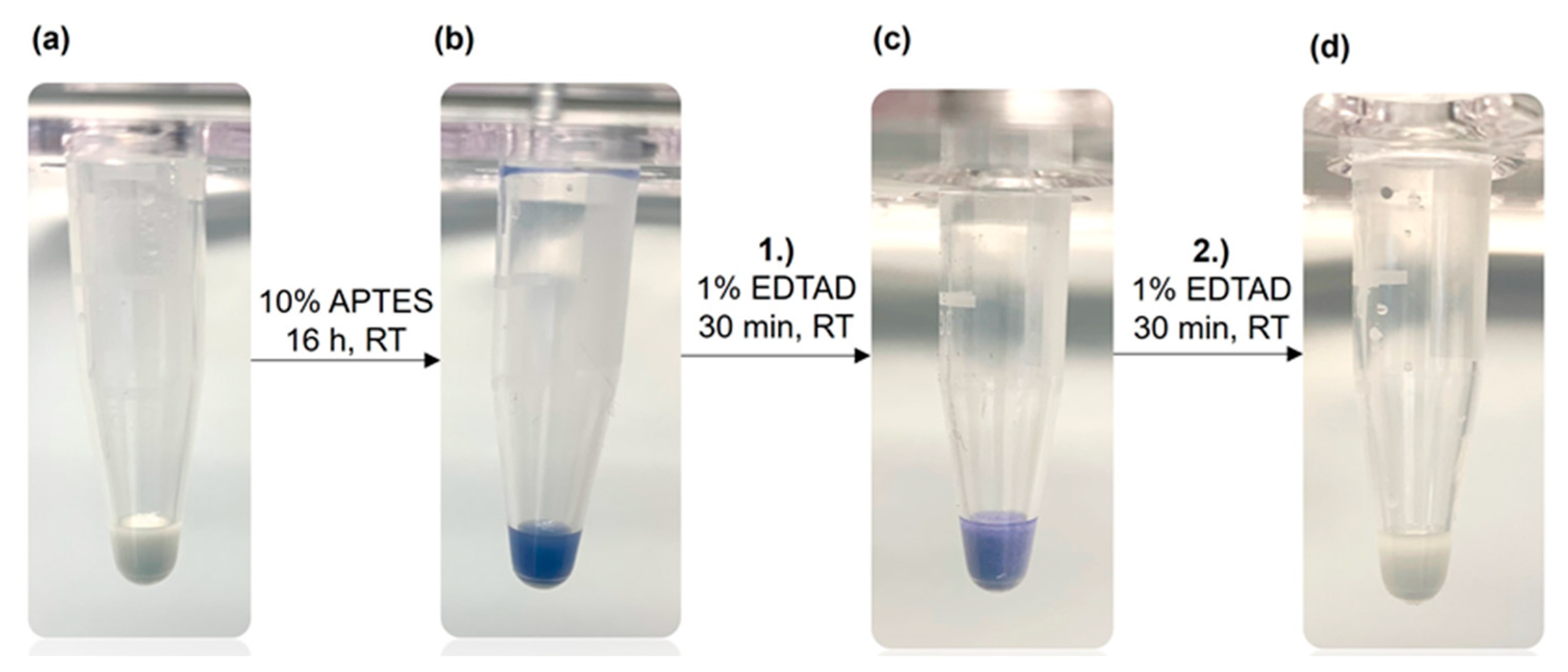
2.2. Functionalization of Corundum
2.3. Kaiser Test
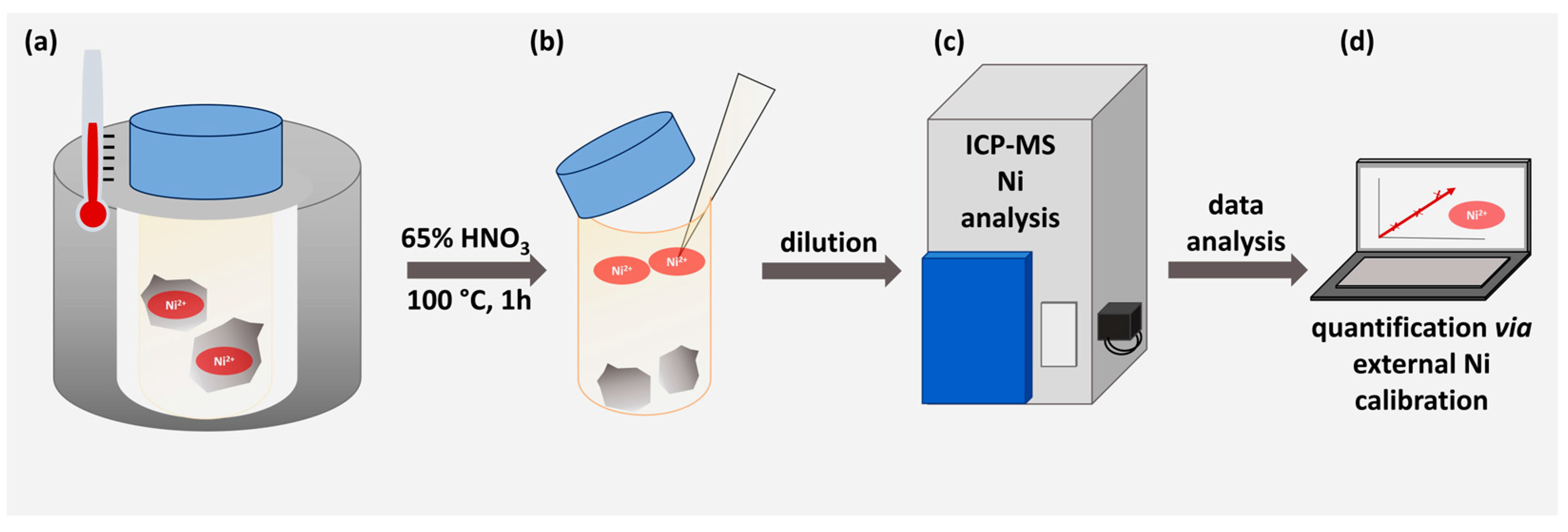
2.4. ICP-MS Determination
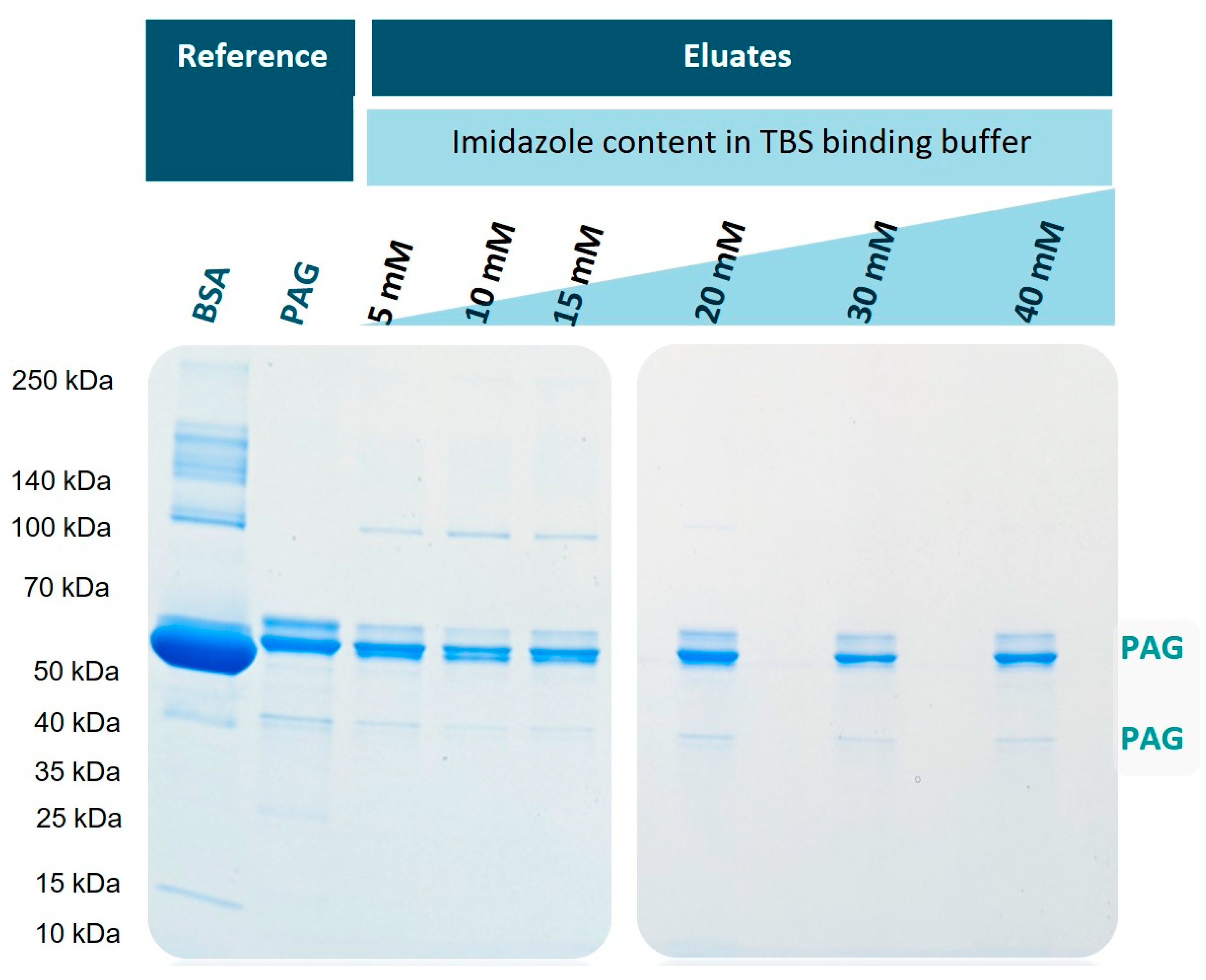
2.5. Optimization of Imidazole Content in Binding Buffer
2.6. SDS-PAGE
2.7. BCA Assay for the Determination of Protein Binding Capacity
2.8. Production of His6-MBP-mSA2 and Cytoplasm Blanks in Different E. coli Strains
2.9. Production of SARS-CoV-2-S-RBD-His8 Domain
3. Results and Discussion
3.1. Functionalization of Purified Corundum
3.2. Determination of Nickel Content via ICP-MS
3.3. Optimization of Imidazole Concentration in Binding Buffer
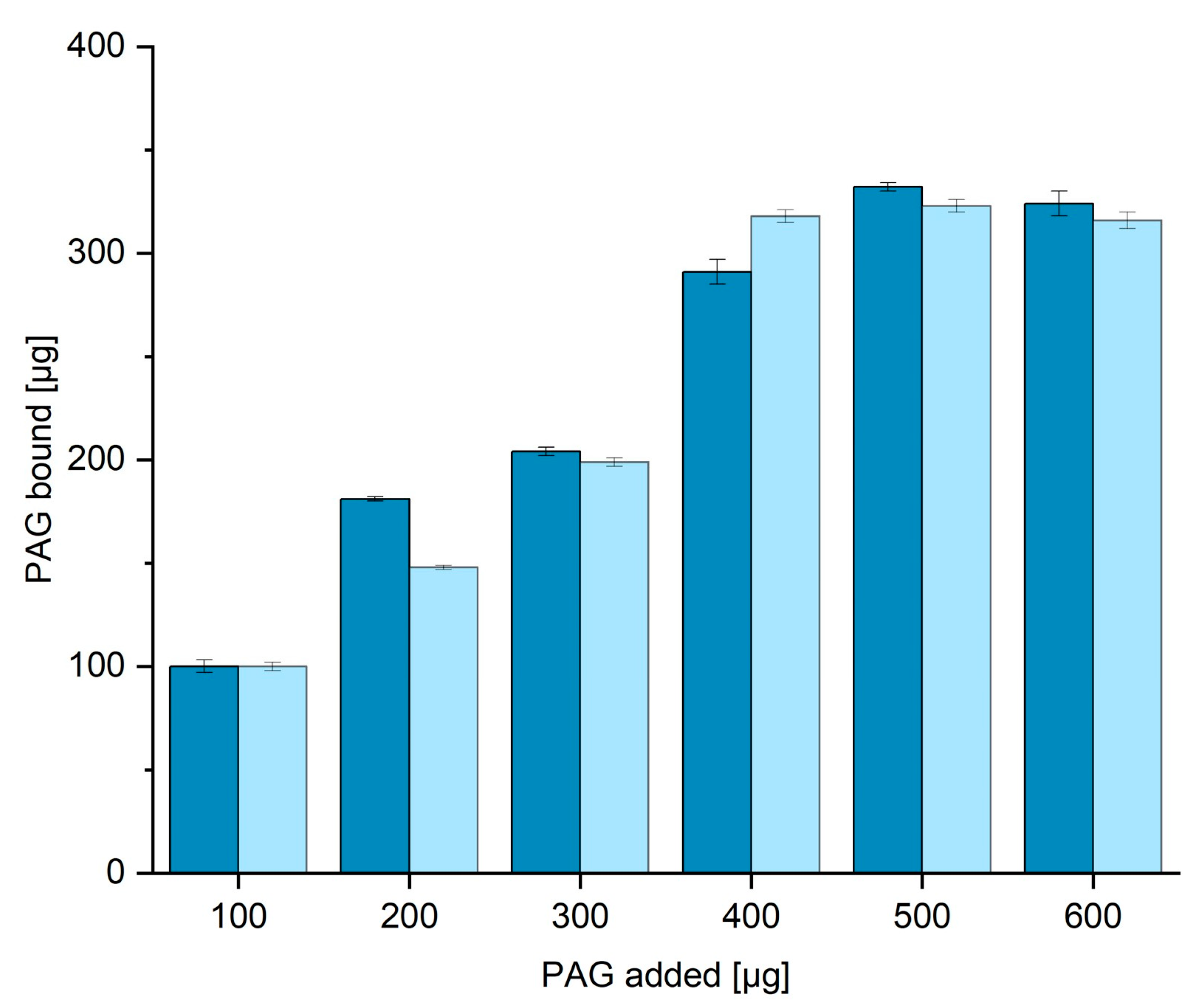
3.4. BCA Assay for the Determination of Protein Binding Capacity in TBS20 and TBS40
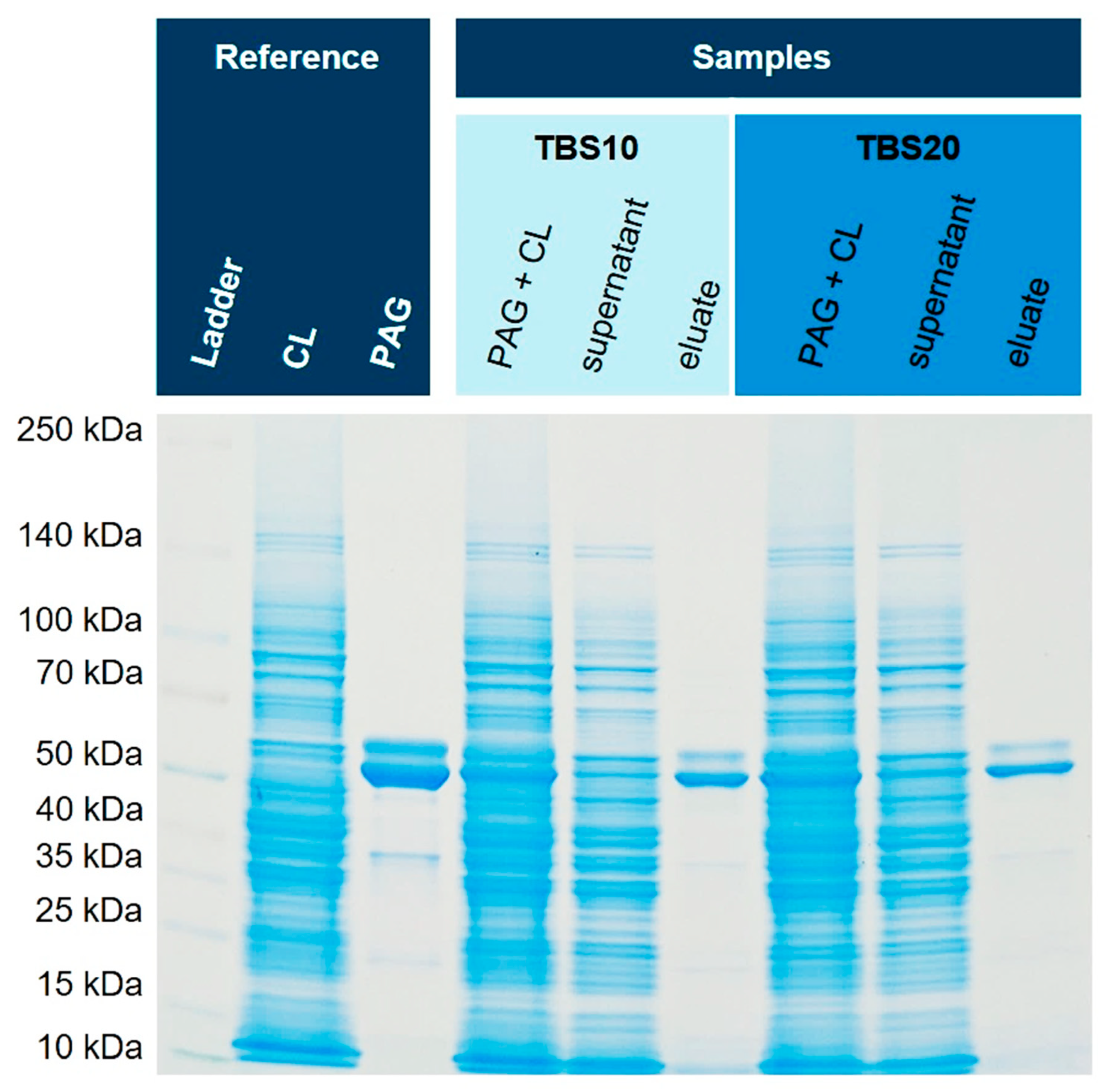
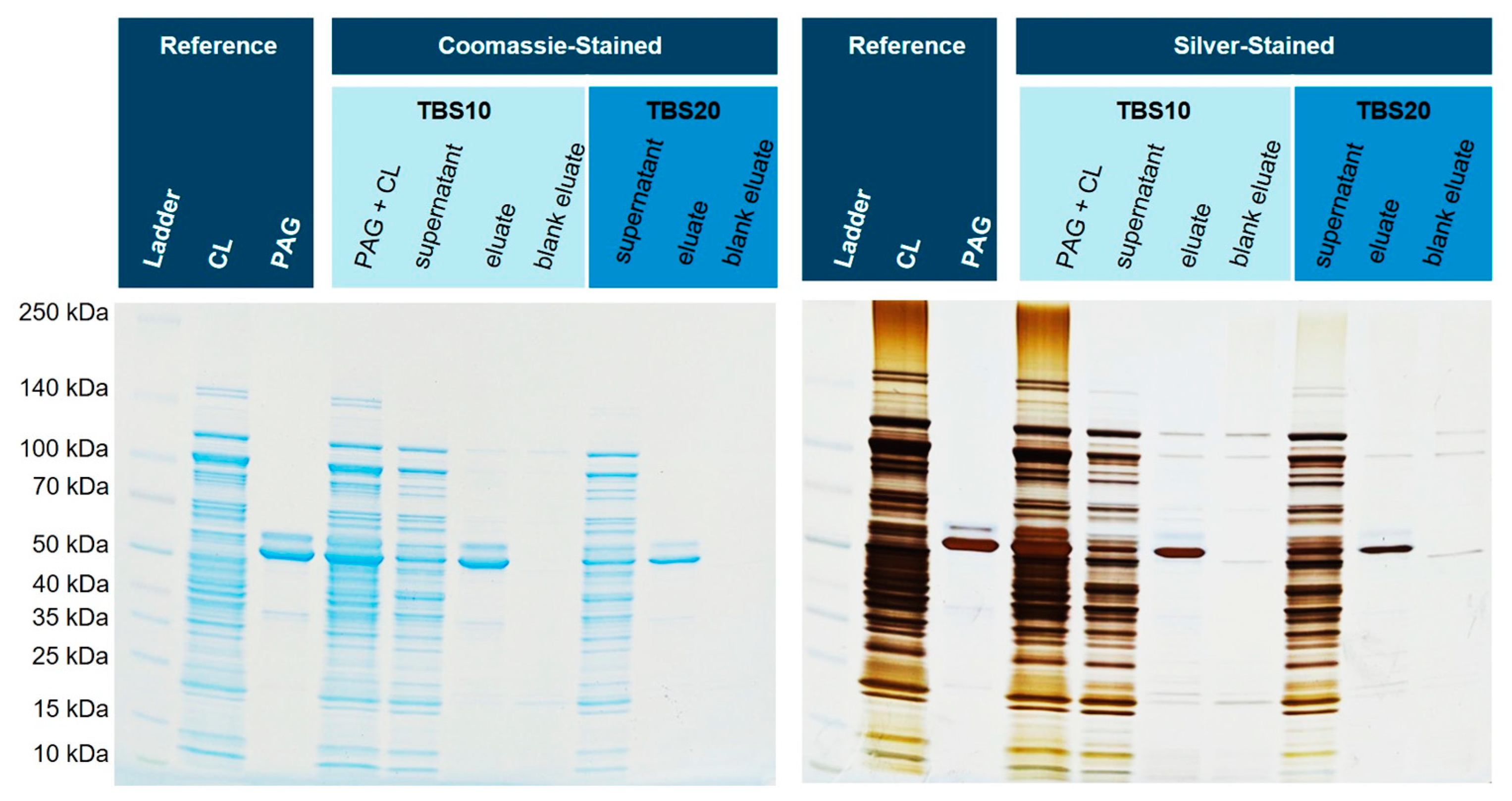
3.5. Purification from E. coli Cell Lysates Spiked with His6-PAG
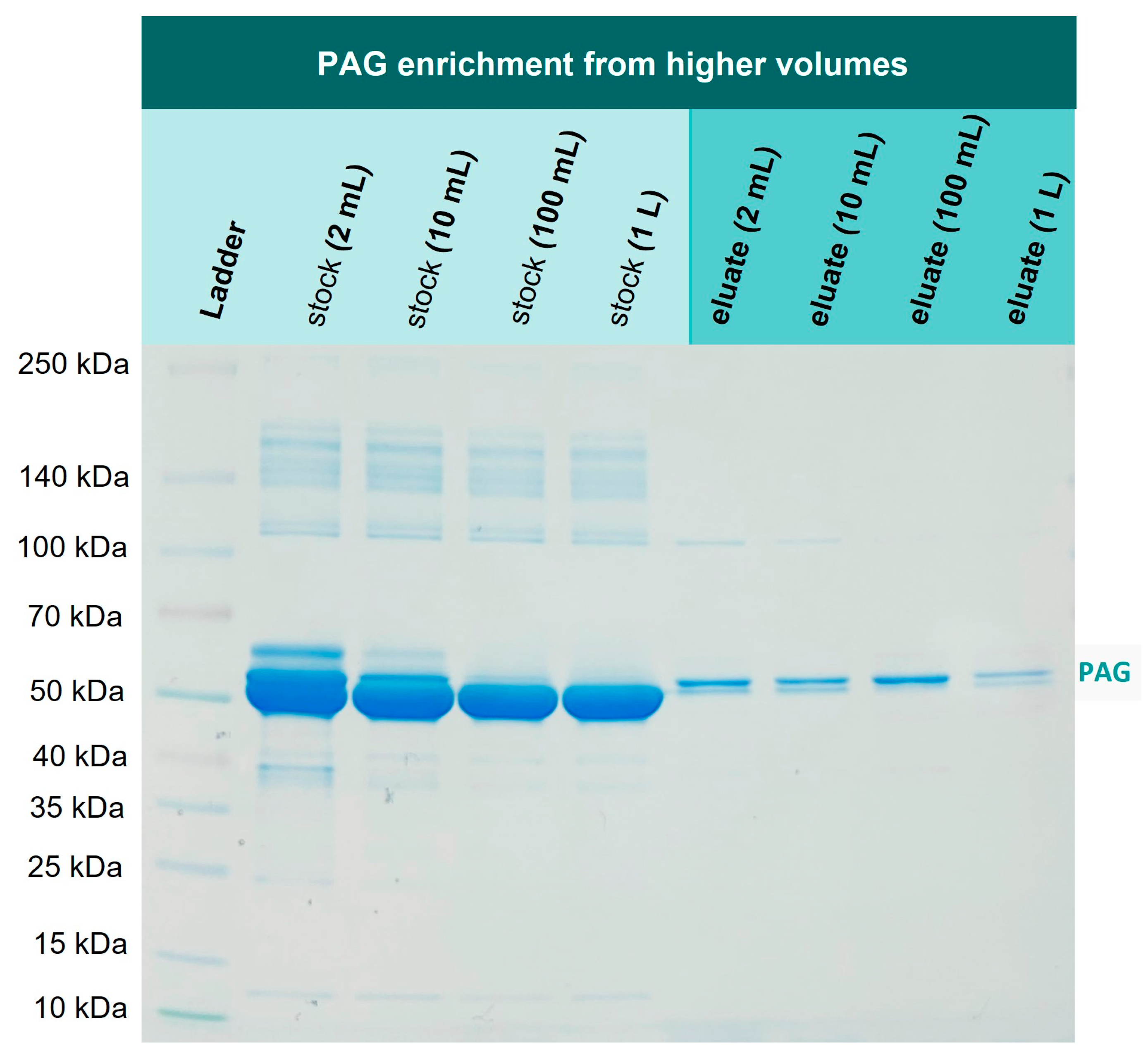
3.6. Enrichment of His6-PAG from Higher Volumes
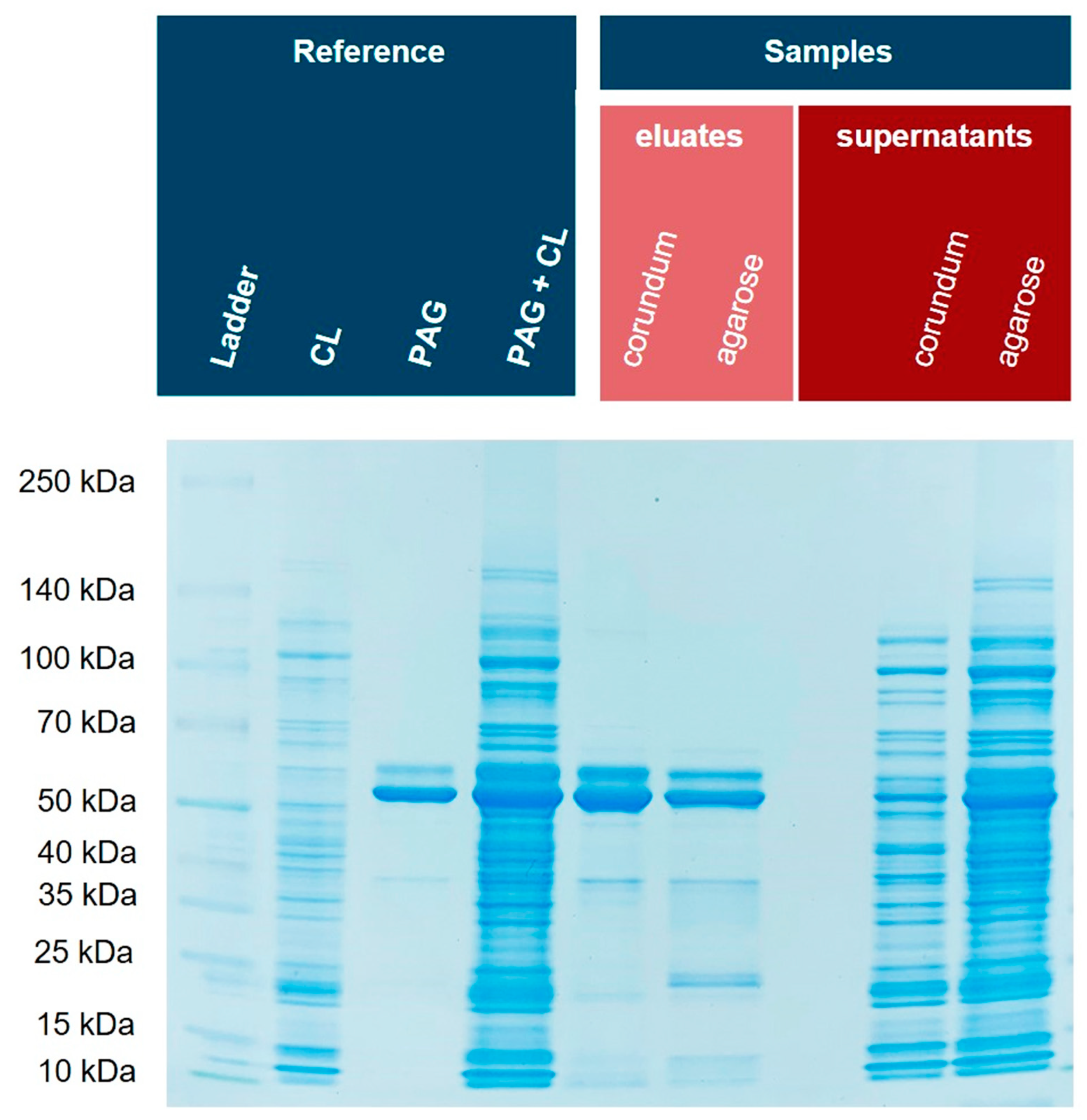
3.7. Comparison of Corundum Platform with Commercial Ni–NTA Agarose Beads
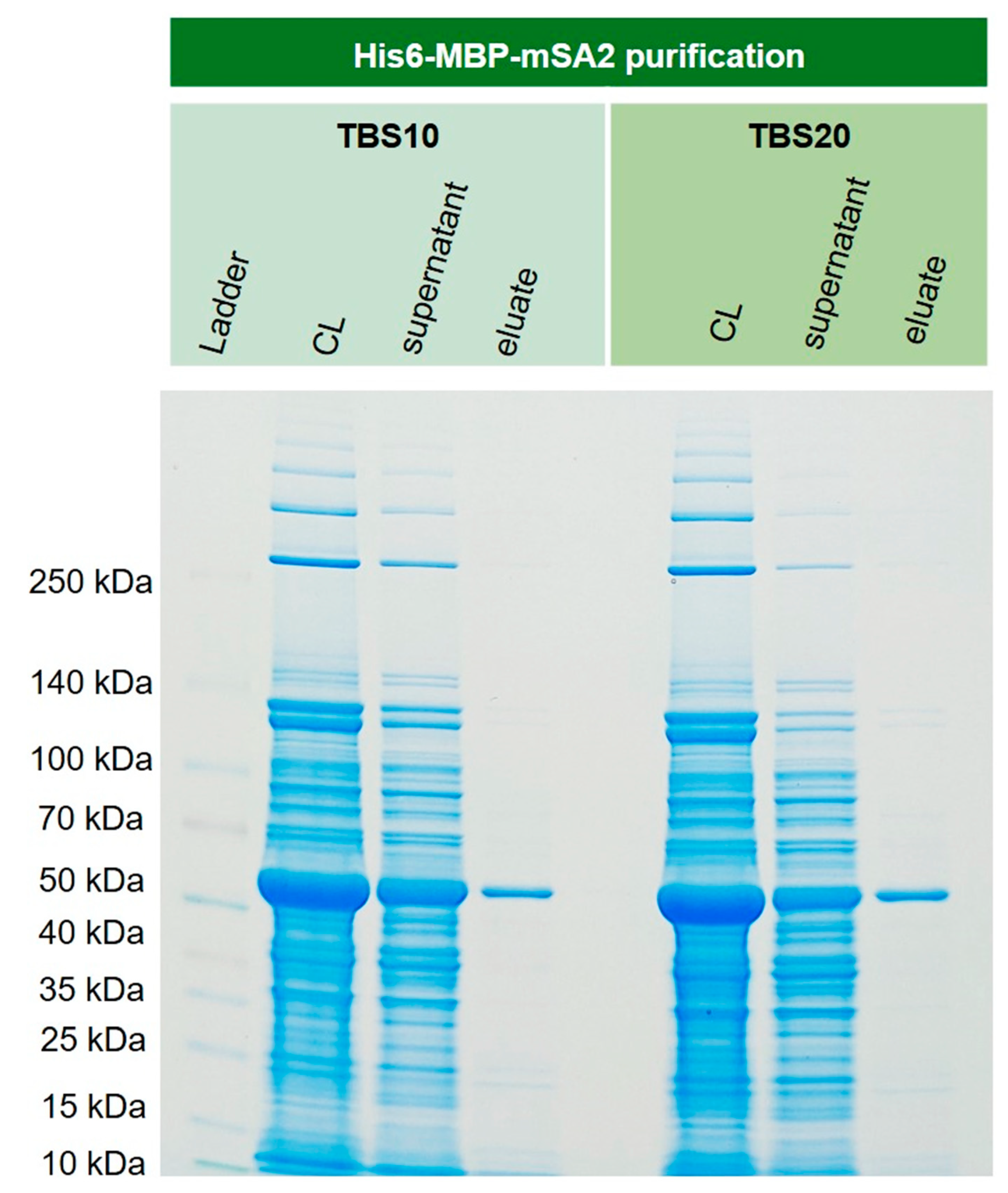
3.8. Purification of Recombinant His6-MBP-mSA2 Fusion Protein from E. coli Cytoplasm
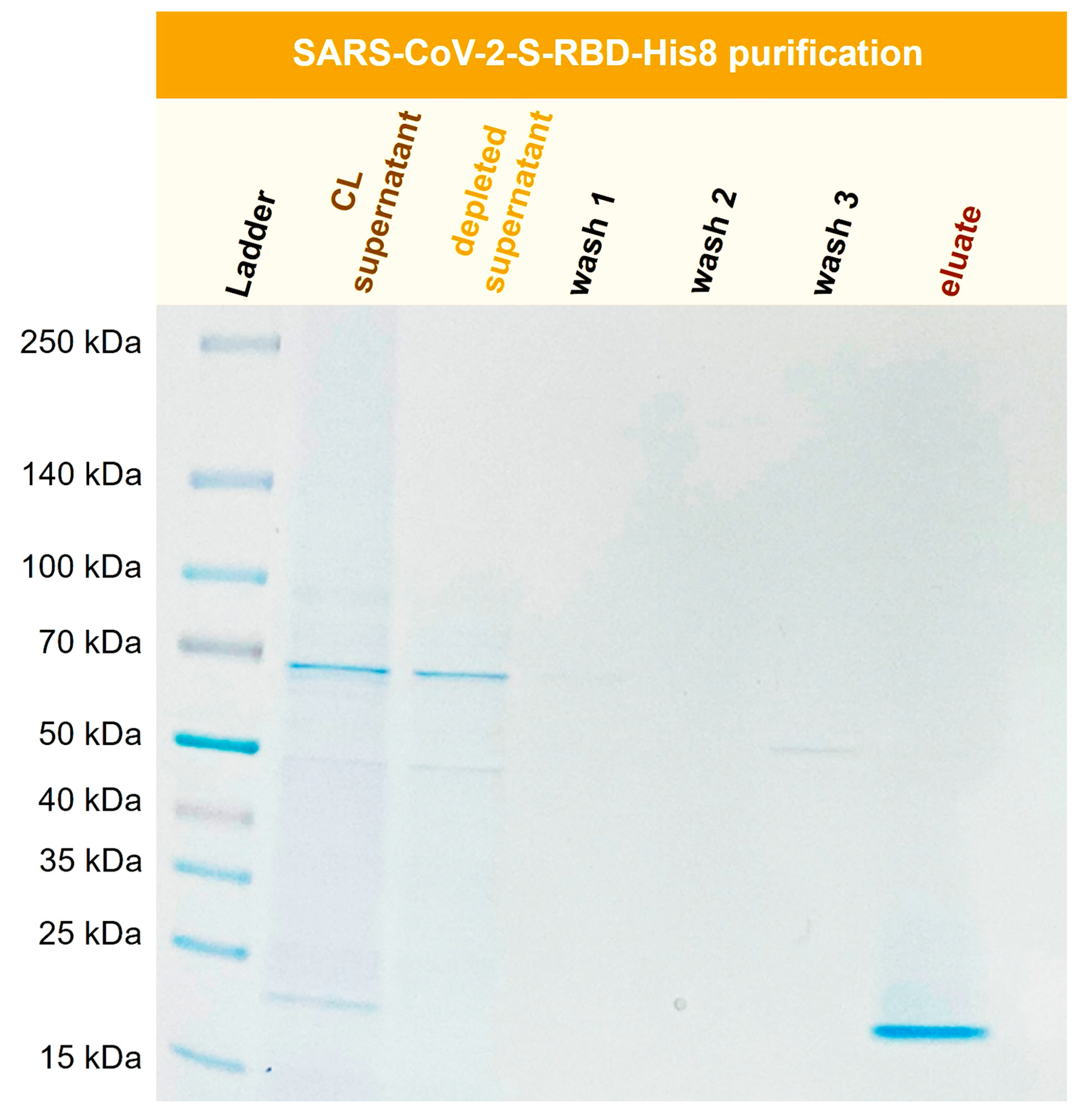
3.9. Purification of Recombinant SARS-CoV-2-S-RBD-His8 from Expi293F Cells
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- De Jesus, M.; Wurm, F.M. Manufacturing recombinant proteins in kg-ton quantities using animal cells in bioreactors. Eur. J. Pharm. Biopharm. 2011, 78, 184–188. [Google Scholar] [CrossRef]
- Tripathi, N.K.; Shrivastava, A. Recent Developments in Bioprocessing of Recombinant Proteins: Expression Hosts and Process Development. Front. Bioeng. Biotechnol. 2019, 7, 420. [Google Scholar] [CrossRef]
- Buerger, C.; Groner, B. Bifunctional recombinant proteins in cancer therapy: Cell penetrating peptide aptamers as inhibitors of growth factor signaling. J. Cancer Res. Clin. Oncol. 2003, 129, 669–675. [Google Scholar] [CrossRef] [PubMed]
- Kalim, M.; Liang, K.; Khan, M.S.I.; Zhan, J. Efficient development and expression of scFv recombinant proteins against PD-L1 surface domain and potency in cancer therapy. Cytotechnology 2019, 71, 705–722. [Google Scholar] [CrossRef] [PubMed]
- Burnett, M.J.B.; Burnett, A.C. Therapeutic recombinant protein production in plants: Challenges and opportunities. Plants People Planet 2020, 2, 121–132. [Google Scholar] [CrossRef]
- Pollet, J.; Chen, W.-H.; Strych, U. Recombinant protein vaccines, a proven approach against coronavirus pandemics. Adv. Drug Deliv. Rev. 2021, 170, 71–82. [Google Scholar] [CrossRef]
- Wingfield, P.T. Overview of the Purification of Recombinant Proteins Produced in Escherichia coli. Curr. Protoc. Protein Sci. 2002, 30, 6.1.1–6.1.37. [Google Scholar] [CrossRef]
- Wood, D.W. New trends and affinity tag designs for recombinant protein purification. Curr. Opin. Struct. Biol. 2014, 26, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Magnusdottir, A.; Johansson, I.; Dahlgren, L.-G.; Nordlund, P.; Berglund, H. Enabling IMAC purification of low abundance recombinant proteins from E. coli lysates. Nat. Methods 2009, 6, 477–478. [Google Scholar] [CrossRef]
- Dybczyński, R.S.; Samczyński, Z.; Chajduk, E. Comparison of usefulness of four chelating agents (EDTA, NTA, ODA and IDA) for the chromatographic separation of micro and macro amounts of rare earth elements. Crit. Rev. Anal. Chem. 2021, 1–15. [Google Scholar]
- Völzke, J.L.; Shamami, P.H.; Gawlitza, K.; Feldmann, I.; Zimathies, A.; Meyer, K.; Weller, M.G. High-Purity Corundum as Support for Affinity Extractions from Complex Samples. Separations 2022, 9, 252. [Google Scholar] [CrossRef]
- Hsiao, V.K.S.; Waldeisen, J.R.; Zheng, Y.; Lloyd, P.F.; Bunning, T.J.; Huang, T.J. Aminopropyltriethoxysilane (APTES)-functionalized nanoporous polymeric gratings: Fabrication and application in biosensing. J. Mater. Chem. 2007, 17, 4896–4901. [Google Scholar] [CrossRef]
- Sypabekova, M.; Hagemann, A.; Rho, D.; Kim, S. Review: 3-Aminopropyltriethoxysilane (APTES) Deposition Methods on Oxide Surfaces in Solution and Vapor Phases for Biosensing Applications. Biosensors 2022, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Sánta-Bell, E.; Kovács, N.K.; Alács, B.; Molnár, Z.; Hornyánszky, G. Immobilization of Phenylalanine Ammonia-lyase via EDTA Based Metal Chelate Complexes—Optimization and Prospects. Period. Polytech. Chem. Eng. 2021, 65, 308–319. [Google Scholar] [CrossRef]
- Patchornik, G. Purification of His-Tagged Proteins with [Desthiobiotin−BSA−EDTA] Conjugates Exhibiting Resistance to EDTA. Bioconjug. Chem. 2008, 19, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F.V.; Gurgel, L.V.A.; Gil, L.F. Removal of Zn2+ from aqueous single metal solutions and electroplating wastewater with wood sawdust and sugarcane bagasse modified with EDTA dianhydride (EDTAD). J. Hazard. Mater. 2010, 176, 856–863. [Google Scholar] [CrossRef]
- Yu, J.; Tong, M.; Sun, X.; Li, B. Enhanced and selective adsorption of Pb2+ and Cu2+ by EDTAD-modified biomass of baker’s yeast. Bioresour. Technol. 2008, 99, 2588–2593. [Google Scholar] [CrossRef]
- Wilke, M.; Röder, B.; Paul, M.; Weller, M.G. Sintered Glass Monoliths as Supports for Affinity Columns. Separations 2021, 8, 56. [Google Scholar] [CrossRef]
- Spriestersbach, A.; Kubicek, J.; Schäfer, F.; Block, H.; Maertens, B. Purification of his-tagged proteins. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2015; Volume 559, pp. 1–15. [Google Scholar]
- Bornhorst, J.A.; Falke, J.J. Purification of proteins using polyhistidine affinity tags. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2000; Volume 326, pp. 245–254. [Google Scholar]
- Vançan, S.; Miranda, E.A.; Bueno, S.M.A. IMAC of human IgG: Studies with IDA-immobilized copper, nickel, zinc, and cobalt ions and different buffer systems. Process. Biochem. 2002, 37, 573–579. [Google Scholar] [CrossRef]
- Lee, J.J.; Bruley, D.F.; Kang, K.A. Effect of pH And Imidazole on Protein C Purification from Cohn Fraction Iv-1 By IMAC. In Oxygen Transport to Tissue XXVIII; Springer: Berlin/Heidelberg, Germany, 2008; pp. 61–66. [Google Scholar]
- Kay, C.; Lorthioir, O.E.; Parr, N.J.; Congreve, M.; McKeown, S.C.; Scicinski, J.J.; Ley, S.V. Solid-phase reaction monitoring--chemical derivatization and off-bead analysis. Biotechnol. Bioeng. 2000, 71, 110–118. [Google Scholar] [CrossRef]
- Palomo, J.M. Solid-phase peptide synthesis: An overview focused on the preparation of biologically relevant peptides. RSC Adv. 2014, 4, 32658–32672. [Google Scholar] [CrossRef]
- Sarin, V.K.; Kent, S.B.; Tam, J.P.; Merrifield, R.B. Quantitative monitoring of solid-phase peptide synthesis by the ninhydrin reaction. Anal. Biochem. 1981, 117, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Tchipilov, T.; Raysyan, A.; Weller, M.G. Methods for the quantification of particle-bound protein–application to reagents for lateral-flow immunoassays (LFIA). Preprints.org 2022, 2022030332. [Google Scholar] [CrossRef]
- Pina, A.S.; Batalha, Í.L.; Roque, A.C.A. Affinity Tags in Protein Purification and Peptide Enrichment: An Overview. In Protein Downstream Processing; 2014; Volume 1129, pp. 147–168. Humana Press: Totowa, NJ, USA. [Google Scholar] [CrossRef]
- Lata, S.; Gavutis, M.; Tampé, R.; Piehler, J. Specific and Stable Fluorescence Labeling of Histidine-Tagged Proteins for Dissecting Multi-Protein Complex Formation. J. Am. Chem. Soc. 2006, 128, 2365–2372. [Google Scholar] [CrossRef]
- Rezwan, K.; Meier, L.P.; Rezwan, M.; Vörös, J.; Textor, M.; Gauckler, L.J. Bovine serum albumin adsorption onto colloidal Al2O3 particles: A new model based on zeta potential and UV-Vis measurements. Langmuir 2004, 20, 10055–10061. [Google Scholar] [CrossRef]
- Bujacz, A. Structures of bovine, equine and leporine serum albumin. Acta Crystallogr. Sect. D Biol. Crystallogr. 2012, 68, 1278–1289. [Google Scholar] [CrossRef]
- Reinmuth-Selzle, K.; Tchipilov, T.; Backes, A.T.; Tscheuschner, G.; Tang, K.; Ziegler, K.; Lucas, K.; Pöschl, U.; Fröhlich-Nowoisky, J.; Weller, M.G. Determination of the protein content of complex samples by aromatic amino acid analysis, liquid chromatography-UV absorbance, and colorimetry. Anal. Bioanal. Chem. 2022, 414, 4457–4470. [Google Scholar] [CrossRef] [PubMed]
- Elowitz, M.B.; Surette, M.G.; Wolf, P.-E.; Stock, J.B.; Leibler, S. Protein Mobility in the Cytoplasm of Escherichia coli. J. Bacteriol. 1999, 181, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Robichon, C.; Luo, J.; Causey, T.B.; Benner, J.S.; Samuelson, J.C. Engineering Escherichia coli BL21(DE3) Derivative Strains To Minimize E. coli Protein Contamination after Purification by Immobilized Metal Affinity Chromatography. Appl. Environ. Microbiol. 2011, 77, 4634–4646. [Google Scholar] [CrossRef]
- Hochuli, E. Large-scale chromatography of recombinant proteins. J. Chromatogr. A 1988, 444, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; He, L.; Zhang, X.; Pu, J.; Voronin, D.; Jiang, S.; Zhou, Y.; Du, L. Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: Implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol. Immunol. 2020, 17, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-H.; Hotez, P.J.; Bottazzi, M.E. Potential for developing a SARS-CoV receptor-binding domain (RBD) recombinant protein as a heterologous human vaccine against coronavirus infectious disease (COVID)-19. Hum. Vaccines Immunother. 2020, 16, 1239–1242. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Ni µmol/g Corundum | Ni2+ µg/g Corundum |
|---|---|---|
| Nickel-loaded corundum | 5.2 ± 0.1 | 304 ± 6 |
| Blank material | 0.0015 ± 0.0001 | 0.09 ± 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Völzke, J.L.; Smatty, S.; Döring, S.; Ewald, S.; Oelze, M.; Fratzke, F.; Flemig, S.; Konthur, Z.; Weller, M.G. Efficient Purification of Polyhistidine-Tagged Recombinant Proteins Using Functionalized Corundum Particles. BioTech 2023, 12, 31. https://doi.org/10.3390/biotech12020031
Völzke JL, Smatty S, Döring S, Ewald S, Oelze M, Fratzke F, Flemig S, Konthur Z, Weller MG. Efficient Purification of Polyhistidine-Tagged Recombinant Proteins Using Functionalized Corundum Particles. BioTech. 2023; 12(2):31. https://doi.org/10.3390/biotech12020031
Chicago/Turabian StyleVölzke, Jule L., Sarah Smatty, Sarah Döring, Shireen Ewald, Marcus Oelze, Franziska Fratzke, Sabine Flemig, Zoltán Konthur, and Michael G. Weller. 2023. "Efficient Purification of Polyhistidine-Tagged Recombinant Proteins Using Functionalized Corundum Particles" BioTech 12, no. 2: 31. https://doi.org/10.3390/biotech12020031
APA StyleVölzke, J. L., Smatty, S., Döring, S., Ewald, S., Oelze, M., Fratzke, F., Flemig, S., Konthur, Z., & Weller, M. G. (2023). Efficient Purification of Polyhistidine-Tagged Recombinant Proteins Using Functionalized Corundum Particles. BioTech, 12(2), 31. https://doi.org/10.3390/biotech12020031