Integrating Multi–Omics Data for Gene-Environment Interactions



Abstract
1. Introduction
2. Method
2.1. Analysis Framework
2.2. Stage 1: The Linear Regulatory Model (LRM)
2.3. Stage 2: The Penalized G×E Interaction Model
2.4. Computation
| Algorithm 1 The Integrative analysis for G×E Interaction |
|
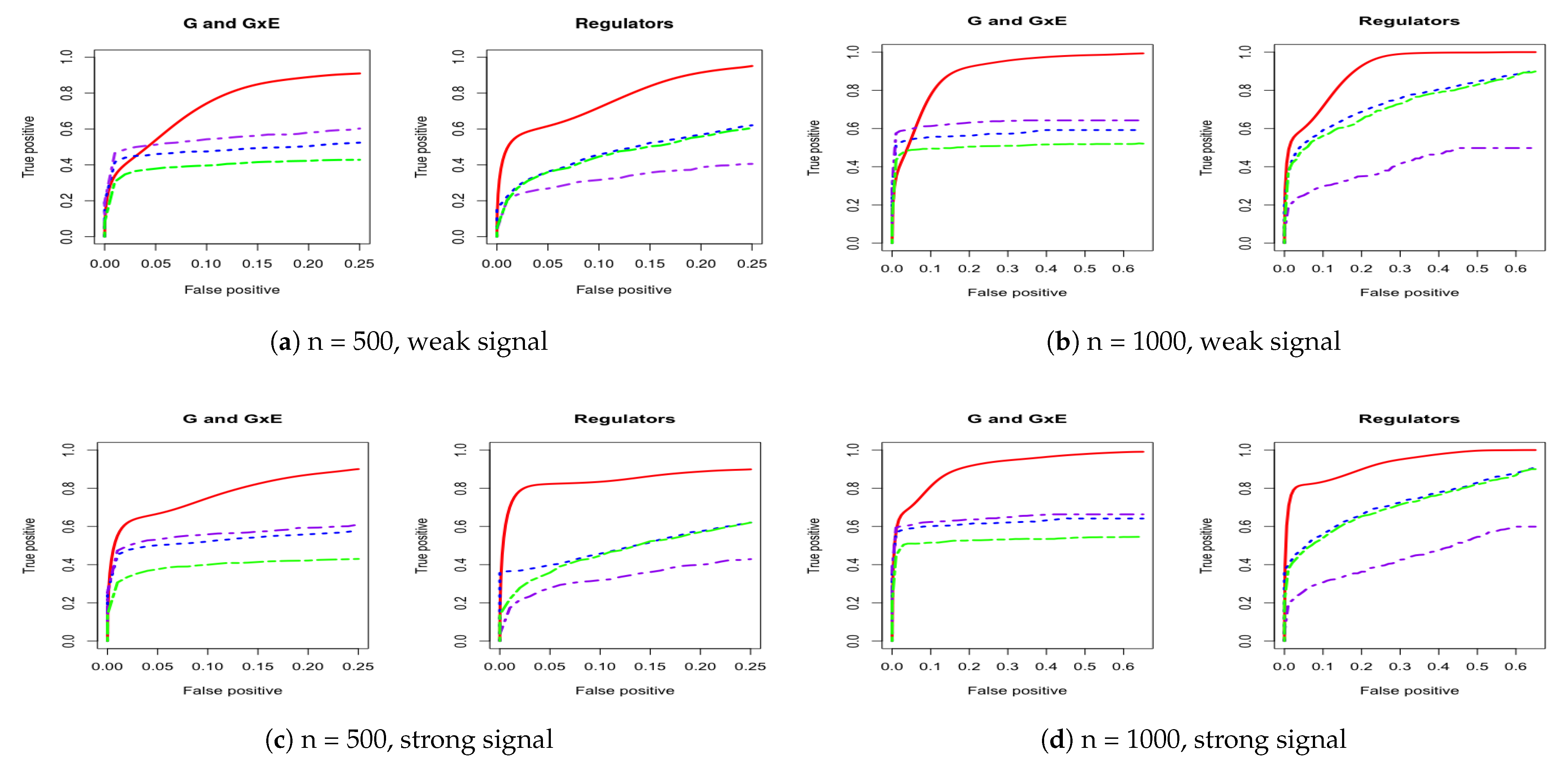
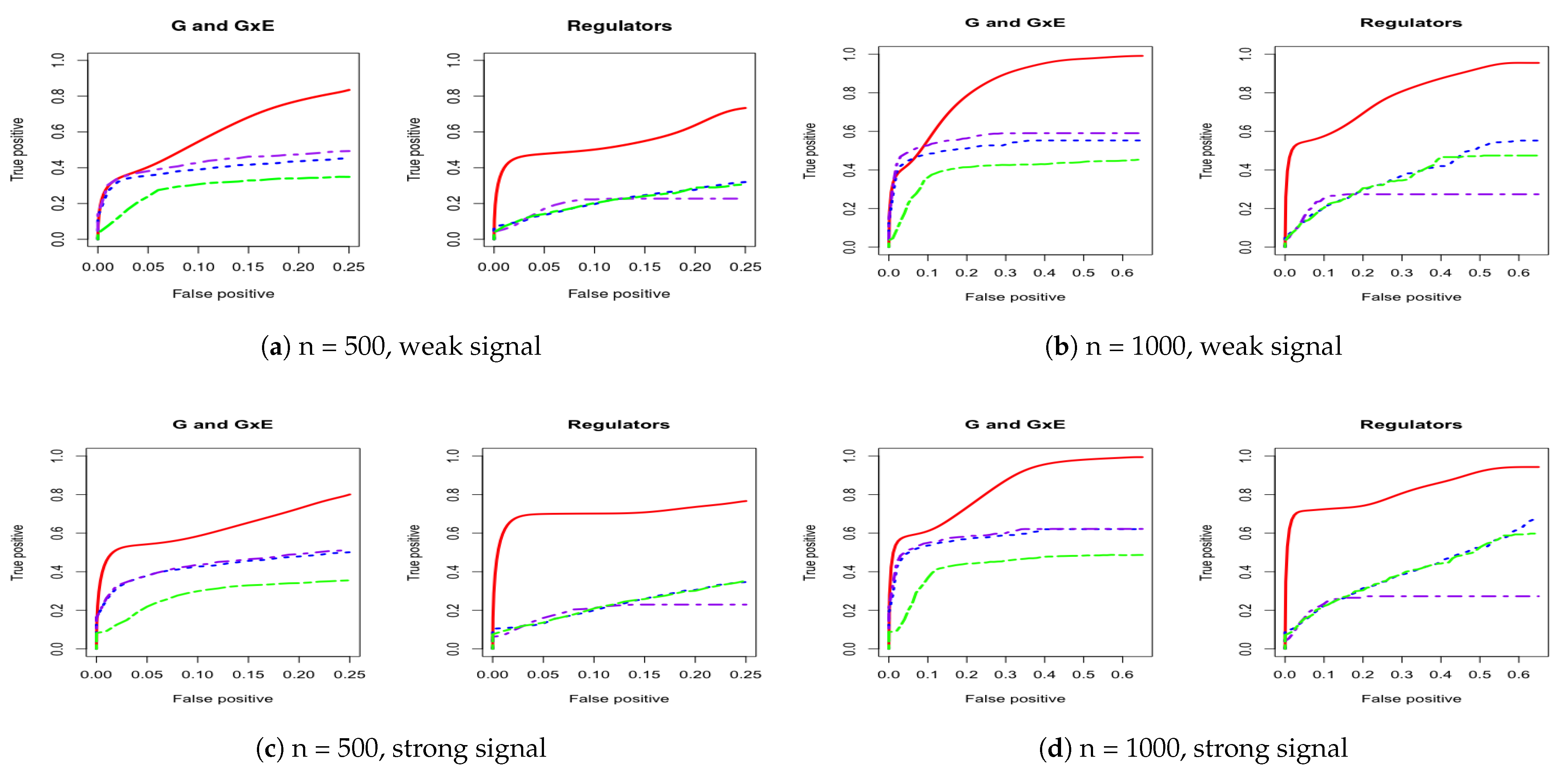
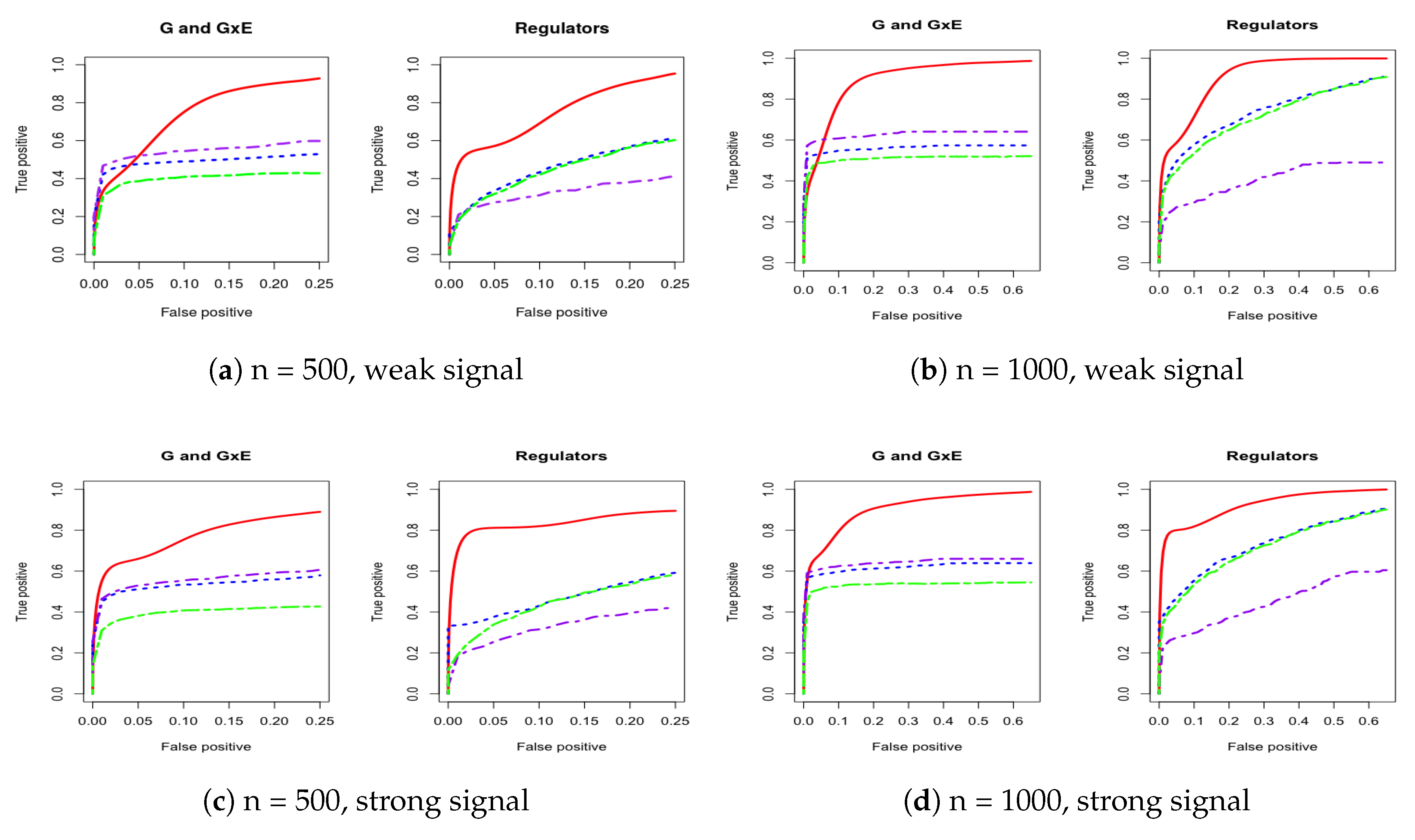
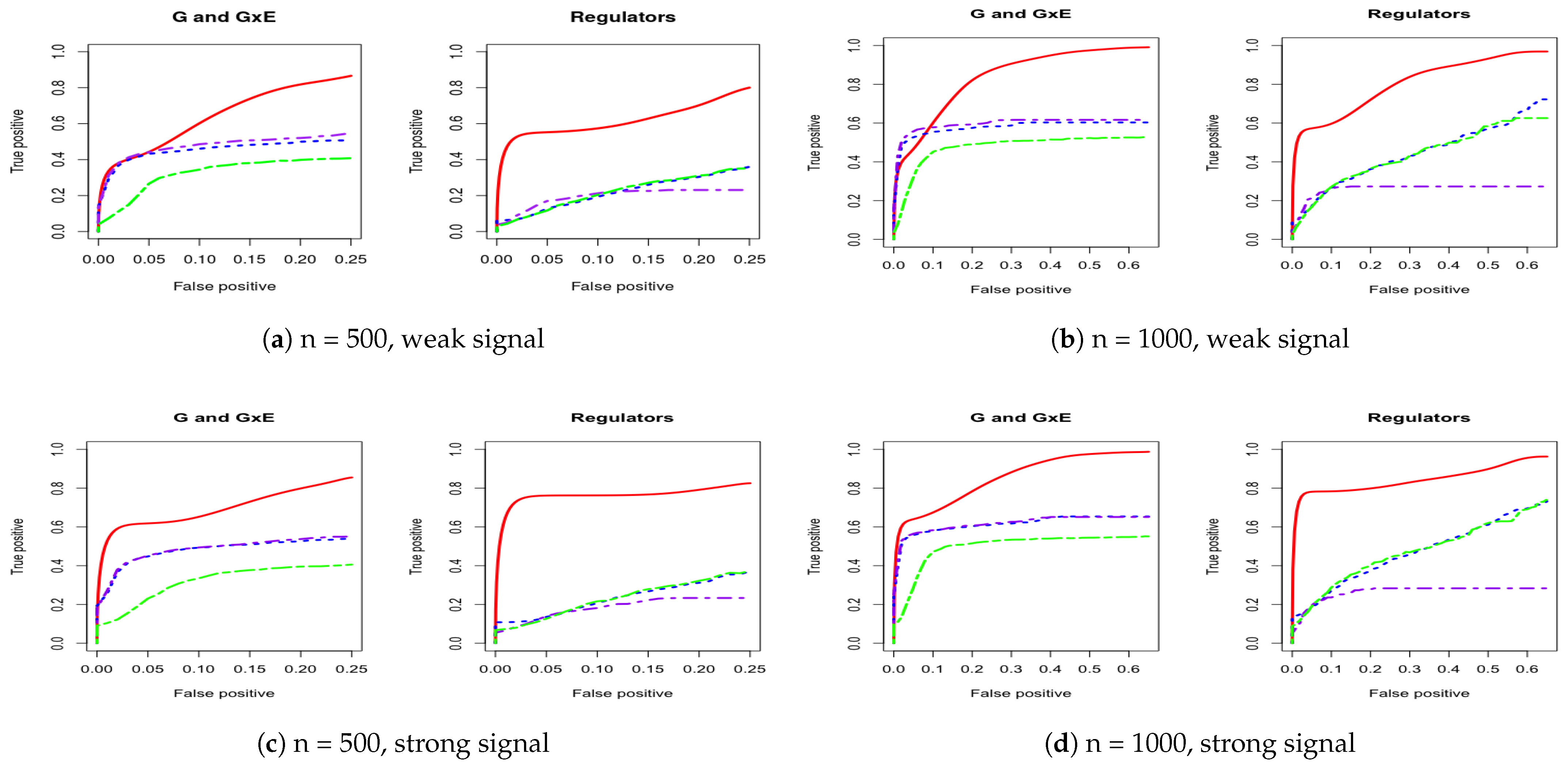
3. Simulation
4. Analysis of TCGA Data
4.1. Lung Adenocarcinoma (LUAD) Data
4.2. Lung Squamous Cell Carcinoma (LUSC) Data
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Other Simulation Scenarios


Appendix B. Accelerated Failure Time (AFT) Model
References
- Simonds, N.I.; Ghazarian, A.A.; Pimentel, C.B.; Schully, S.D.; Ellison, G.L.; Gillanders, E.M.; Mechanic, L.E. Review of the gene-environment interaction literature in cancer: What do we know? Genet. Epidemiol. 2016, 40, 356–365. [Google Scholar] [CrossRef]
- Dempfle, A.; Scherag, A.; Hein, R.; Beckmann, L.; Chang-Claude, J.; Schäfer, H. Gene-environment interactions for complex traits: Definitions, methodological requirements and challenges. Eur. J. Hum. Genet. 2008, 16, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Hirschhorn, J.N.; Lohmueller, K.; Byrne, E.; Hirschhorn, K. A comprehensive review of genetic association studies. Genet. Med. 2002, 4, 45–61. [Google Scholar] [PubMed]
- Wu, C.; Li, S.; Cui, Y. Genetic association studies: An information content perspective. Curr. Genom. 2012, 13, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhou, F.; Ren, J.; Li, X.; Jiang, Y.; Ma, S. A Selective Review of Multi-Level Omics Data Integration Using Variable Selection. High-throughput 2019, 8, 4. [Google Scholar]
- Zhu, R.; Zhao, Q.; Zhao, H.; Ma, S. Integrating multidimensional omics data for cancer outcome. Biostatistics 2016, 17, 605–618. [Google Scholar] [CrossRef]
- Zhou, F.; Ren, J.; Lu, X.; Ma, S.; Wu, C. Gene-Environment Interaction: A Variable Selection Perspective. Epistasis Methods Mol. Biol. 2021, in press. [Google Scholar]
- Wang, W.; Baladandayuthapani, V.; Morris, J.S.; Broom, B.M.; Manyam, G.; Do, K.A. iBAG: Integrative Bayesian analysis of high-dimensional multiplatform genomics data. Bioinformatics 2013, 29, 149–159. [Google Scholar] [CrossRef]
- Ciriello, G.; Cerami, E.; Sander, C.; Schultz, N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012, 22, 398–406. [Google Scholar] [CrossRef]
- Kristensen, V.N.; Lingjærde, O.C.; Russnes, H.G.; Vollan, H.K.M.; Frigessi, A.; Børresen-Dale, A.L. Principles and methods of integrative genomic analyses in cancer. Nat. Rev. Cancer 2014, 14, 299. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B (Methodol.) 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Lee, M.; Shen, H.; Huang, J.Z.; Marron, J. Biclustering via sparse singular value decomposition. Biometrics 2010, 66, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Zou, H. The adaptive lasso and its oracle properties. J. Am. Stat. Assoc. 2006, 101, 1418–1429. [Google Scholar] [CrossRef]
- Fan, J.; Li, R. Variable selection via nonconcave penalized likelihood and its oracle properties. J. Am. Stat. Assoc. 2001, 96, 1348–1360. [Google Scholar]
- Zhang, C.H. Nearly unbiased variable selection under minimax concave penalty. Ann. Stat. 2010, 38, 894–942. [Google Scholar] [CrossRef]
- Gross, S.M.; Tibshirani, R. Collaborative regression. Biostatistics 2014, 16, 326–338. [Google Scholar]
- Subramanian, J.; Govindan, R. Lung cancer in never smokers: A review. J. Clin. Oncol. 2007, 25, 561–570. [Google Scholar] [CrossRef]
- Couraud, S.; Zalcman, G.; Milleron, B.; Morin, F.; Souquet, P.J. Lung cancer in never smokers—A review. Eur. J. Cancer 2012, 48, 1299–1311. [Google Scholar]
- Kenfield, S.A.; Wei, E.K.; Stampfer, M.J.; Rosner, B.A.; Colditz, G.A. Comparison of aspects of smoking among the four histological types of lung cancer. Tob. Control 2008, 17, 198–204. [Google Scholar] [CrossRef]
- Kumar, V.; Abbas, A.K.; Aster, J.C. Robbins Basic Pathology e-book; Elsevier Health Sciences: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Chen, Y.; Tang, J.; Lu, T.; Liu, F. CAPN1 promotes malignant behavior and erlotinib resistance mediated by phosphorylation of c-Met and PIK3R2 via degrading PTPN1 in lung adenocarcinoma. Thorac. Cancer 2020, 11, 1848–1860. [Google Scholar] [CrossRef]
- Huang, N.; Lin, W.; Shi, X.; Tao, T. STK24 expression is modulated by DNA copy number/methylation in lung adenocarcinoma and predicts poor survival. Future Oncol. 2018, 14, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- Pombo, C.M.; Force, T.; Kyriakis, J.; Nogueira, E.; Fidalgo, M.; Zalvide, J. The GCK II and III subfamilies of the STE20 group kinases. Front Biosci 2007, 12, 850–859. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hameed, Y.; Ejaz, S. Up-regulation of FN1, Activation of Maturation Promoting Factor and Associated Signaling Pathway Facilitates Epithelial-Mesenchymal Transition, Inhibits Apoptosis and Elevates Proliferation Rate of Breast Cancer Cells. Silico Anal. Microarray Datasets 2020. [Google Scholar] [CrossRef]
- Guo, W.; Sun, S.; Guo, L.; Song, P.; Xue, X.; Zhang, H.; Zhang, G.; Li, R.; Gao, Y.; Qiu, B.; et al. Elevated SLC2A1 Expression Correlates with Poor Prognosis in Patients with Surgically Resected Lung Adenocarcinoma: A Study Based on Immunohistochemical Analysis and Bioinformatics. DNA Cell Biol. 2020, 39, 631–644. [Google Scholar]
- Silva, V.M.; Gomes, J.A.; Tenório, L.P.G.; de Omena Neta, G.C.; da Costa Paixão, K.; Duarte, A.K.F.; da Silva, G.C.B.; Ferreira, R.J.S.; Koike, B.D.V.; de Sales Marques, C.; et al. Schwann cell reprogramming and lung cancer progression: A meta-analysis of transcriptome data. Oncotarget 2019, 10, 7288. [Google Scholar] [CrossRef]
- Misono, S.; Seki, N.; Mizuno, K.; Yamada, Y.; Uchida, A.; Sanada, H.; Moriya, S.; Kikkawa, N.; Kumamoto, T.; Suetsugu, T.; et al. Molecular pathogenesis of gene regulation by the miR-150 duplex: miR-150-3p regulates TNS4 in lung adenocarcinoma. Cancers 2019, 11, 601. [Google Scholar] [CrossRef]
- Yang, J.; Liu, Y.; Mai, X.; Lu, S.; Jin, L.; Tai, X. STAT1-induced upregulation of LINC00467 promotes the proliferation migration of lung adenocarcinoma cells by epigenetically silencing DKK1 to activate Wnt/β-catenin signaling pathway. Biochem. Biophys. Res. Commun. 2019, 514, 118–126. [Google Scholar] [CrossRef]
- Zhang, S.; Lu, Y.; Liu, Z.; Li, X.; Wang, Z.; Cai, Z. Identification Six Metabolic Genes as Potential Biomarkers for Lung Adenocarcinoma. J. Comput. Biol. 2020, 27, 1532–1543. [Google Scholar] [CrossRef]
- Lussier, M.P.; Lepage, P.K.; Bousquet, S.M.; Boulay, G. RNF24, a new TRPC interacting protein, causes the intracellular retention of TRPC. Cell Calcium 2008, 43, 432–443. [Google Scholar] [CrossRef]
- Lin, T.; Gu, J.; Qu, K.; Zhang, X.; Ma, X.; Miao, R.; Xiang, X.; Fu, Y.; Niu, W.; She, J.; et al. A new risk score based on twelve hepatocellular carcinoma-specific gene expression can predict the patients’ prognosis. Aging (Albany N. Y.) 2018, 10, 2480. [Google Scholar] [CrossRef]
- Wang, X.W.; Wei, W.; Wang, W.Q.; Zhao, X.Y.; Guo, H.; Fang, D.C. RING finger proteins are involved in the progression of barrett esophagus to esophageal adenocarcinoma: A preliminary study. Gut Liver 2014, 8, 487. [Google Scholar] [CrossRef] [PubMed]
- Anand, S.; Khan, M.A.; Khushman, M.; Dasgupta, S.; Singh, S.; Singh, A.P. Comprehensive Analysis of Expression, Clinicopathological Association and Potential Prognostic Significance of RABs in Pancreatic Cancer. Int. J. Mol. Sci. 2020, 21, 5580. [Google Scholar]
- Zahra, A.; Rubab, I.; Malik, S.; Khan, A.; Khan, M.J.; Fatmi, M.Q. Meta-Analysis of miRNAs and their involvement as biomarkers in oral cancers. BioMed Res. Int. 2018, 2018, 8439820. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Yu, J.; Huang, T.; Jia, H.; Dong, Q.; He, F.; Yuan, W.; Qin, L.; Li, Y.; Xie, L. Differential combinatorial regulatory network analysis related to venous metastasis of hepatocellular carcinoma. BMC Genom. 2012, 13, S14. [Google Scholar] [CrossRef]
- Ke, D.; Guo, Q.; Fan, T.Y.; Xiao, X. Analysis of the Role and Regulation Mechanism of hsa-miR-147b in Lung Squamous Cell Carcinoma Based on The Cancer Genome Atlas Database. Cancer Biother. Radiopharm. 2020. [Google Scholar] [CrossRef]
- Relli, V.; Trerotola, M.; Guerra, E.; Alberti, S. Abandoning the notion of non-small cell lung cancer. Trends Mol. Med. 2019, 25, 585–594. [Google Scholar] [CrossRef]
- Zhang, Q.; Huang, R.; Hu, H.; Yu, L.; Tang, Q.; Tao, Y.; Liu, Z.; Li, J.; Wang, G. Integrative analysis of hypoxia-associated signature in pan-cancer. iScience 2020, 23, 101460. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.; Xiao, X.; Liu, H.; Wang, F.; Li, S.; Wen, Y.; Wei, Y.; Su, J.; Zhang, Y.; et al. The identification of age-associated cancer markers by an integrative analysis of dynamic DNA methylation changes. Sci. Rep. 2016, 6, 22722. [Google Scholar]
- Bae, J.M.; Wen, X.; Kim, T.S.; Kwak, Y.; Cho, N.Y.; Lee, H.S.; Kang, G.H. Fibroblast growth factor receptor 1 (FGFR1) amplification detected by droplet digital polymerase chain reaction (ddPCR) is a prognostic factor in colorectal cancers. Cancer Res. Treat. Off. J. Korean Cancer Assoc. 2020, 52, 74. [Google Scholar] [CrossRef]
- Hu, J.; Xu, L.; Shou, T.; Chen, Q. Systematic analysis identifies three-lncRNA signature as a potentially prognostic biomarker for lung squamous cell carcinoma using bioinformatics strategy. Transl. Lung Cancer Res. 2019, 8, 614. [Google Scholar] [CrossRef]
- Wang, L.; Jia, P.; Wolfinger, R.D.; Chen, X.; Zhao, Z. Gene set analysis of genome-wide association studies: Methodological issues and perspectives. Genomics 2011, 98, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Cui, Y. Boosting signals in gene-based association studies via efficient SNP selection. Briefings Bioinform. 2014, 15, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zuo, X.Y.; Su, W.Y.; Zhao, X.L.; Yuan, M.Q.; Han, L.Z.; Zhao, X.; Chen, Y.D.; Rao, S.Q. Pathway-based analysis tools for complex diseases: A review. Genom. Proteom. Bioinform. 2014, 12, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Huang, Y.; Du, Y.; Zhao, Y.; Ren, J.; Ma, S.; Wu, C. Identification of prognostic genes and pathways in lung adenocarcinoma using a Bayesian approach. Cancer Inform. 2017, 16, 1176935116684825. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, H. Network-constrained regularization and variable selection for analysis of genomic data. Bioinformatics 2008, 24, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, S. Penalized logistic regression for high-dimensional DNA methylation data with case-control studies. Bioinformatics 2012, 28, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; He, T.; Li, Y.; Liu, S.; Du, Y.; Jiang, Y.; Wu, C. Network-based regularization for high dimensional SNP data in the case–control study of Type 2 diabetes. BMC Genet. 2017, 18, 44. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, Q.; Jiang, Y.; Ma, S. Robust network-based analysis of the associations between (epi) genetic measurements. J. Multivar. Anal. 2018, 168, 119–130. [Google Scholar] [CrossRef]
- Ren, J.; Du, Y.; Li, S.; Ma, S.; Jiang, Y.; Wu, C. Robust network-based regularization and variable selection for high-dimensional genomic data in cancer prognosis. Genet. Epidemiol. 2019, 43, 276–291. [Google Scholar]
- Wu, C.; Jiang, Y.; Ren, J.; Cui, Y.; Ma, S. Dissecting gene-environment interactions: A penalized robust approach accounting for hierarchical structures. Stat. Med. 2018, 37, 437–456. [Google Scholar] [CrossRef]
- Li, J.; Wang, Z.; Li, R.; Wu, R. Bayesian group lasso for nonparametric varying-coefficient models with application to functional genome-wide association studies. Ann. Appl. Stat. 2015, 9, 640. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Cui, Y. A novel method for identifying nonlinear gene–environment interactions in case–control association studies. Hum. Genet. 2013, 132, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhong, P.S.; Cui, Y. Additive varying-coefficient model for nonlinear gene-environment interactions. Stat. Appl. Genet. Mol. Biol. 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Shi, X.; Cui, Y.; Ma, S. A penalized robust semiparametric approach for gene–environment interactions. Stat. Med. 2015, 34, 4016–4030. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Xu, S. Semiparametric nonlinear regression for detecting gene and environment interactions. J. Stat. Plan. Inference 2015, 156, 31–47. [Google Scholar] [CrossRef]
- Ren, J.; Zhou, F.; Li, X.; Chen, Q.; Zhang, H.; Ma, S.; Jiang, Y.; Wu, C. Semiparametric Bayesian variable selection for gene-environment interactions. Stat. Med. 2020, 39, 617–638. [Google Scholar] [CrossRef]
- Li, J.; Lu, Q.; Wen, Y. Multi-kernel linear mixed model with adaptive lasso for prediction analysis on high-dimensional multi-omics data. Bioinformatics 2020, 36, 1785–1794. [Google Scholar] [CrossRef]
- Zhou, F.; Ren, J.; Li, G.; Jiang, Y.; Li, X.; Wang, W.; Wu, C. Penalized Variable Selection for Lipid–Environment interactions in a longitudinal lipidomics study. Genes 2019, 10, 1002. [Google Scholar] [CrossRef]
- Stute, W.; Wang, J.L. The strong law under random censorship. Ann. Stat. 1993, 21, 1591–1607. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Covariance | Signal | Approaches | G and G×E | Regulators |
|---|---|---|---|---|
| AR-1 | weak | IGE | 0.73 (0.07) | 0.76 (0.10) |
| S-LASSO | 0.47 (0.04) | 0.46 (0.13) | ||
| J-LASSO | 0.54 (0.04) | 0.32 (0.05) | ||
| ColReg | 0.39 (0.03) | 0.45 (0.15) | ||
| strong | IGE | 0.77 (0.07) | 0.85 (0.06) | |
| S-LASSO | 0.52 (0.05) | 0.48 (0.14) | ||
| J-LASSO | 0.55 (0.04) | 0.33 (0.05) | ||
| ColReg | 0.39 (0.03) | 0.46 (0.15) | ||
| Banded | weak | IGE | 0.74 (0.06) | 0.74 (0.10) |
| S-LASSO | 0.48 (0.03) | 0.44 (0.11) | ||
| J-LASSO | 0.54 (0.05) | 0.32 (0.04) | ||
| ColReg | 0.39 (0.03) | 0.43 (0.12) | ||
| strong | IGE | 0.77 (0.08) | 0.84 (0.06) | |
| S-LASSO | 0.52 (0.04) | 0.46 (0.11) | ||
| J-LASSO | 0.55 (0.05) | 0.32 (0.04) | ||
| ColReg | 0.39 (0.03) | 0.43 (0.12) | ||
| LUSC | weak | IGE | 0.59 (0.09) | 0.55 (0.15) |
| S-LASSO | 0.39 (0.04) | 0.21 (0.06) | ||
| J-LASSO | 0.42 (0.05) | 0.19 (0.06) | ||
| ColReg | 0.28 (0.04) | 0.21 (0.07) | ||
| strong | IGE | 0.63 (0.10) | 0.71 (0.13) | |
| S-LASSO | 0.42 (0.05) | 0.22 (0.07) | ||
| J-LASSO | 0.43 (0.05) | 0.19 (0.06) | ||
| ColReg | 0.28(0.05) | 0.22 (0.07) | ||
| LUAD | weak | IGE | 0.64 (0.09) | 0.62 (0.15) |
| S-LASSO | 0.45 (0.04) | 0.21 (0.06) | ||
| J-LASSO | 0.47 (0.05) | 0.19 (0.05) | ||
| ColReg | 0.32 (0.03) | 0.22 (0.07) | ||
| strong | IGE | 0.70 (0.08) | 0.77 (0.11) | |
| S-LASSO | 0.47 (0.05) | 0.23 (0.08) | ||
| J-LASSO | 0.48 (0.05) | 0.18 (0.05) | ||
| ColReg | 0.31 (0.04) | 0.23 (0.08) |
| Covariance | Signal | Approaches | G and G×E | Regulators |
|---|---|---|---|---|
| AR-1 | weak | IGE | 0.89 (0.02) | 0.91 (0.02) |
| S-LASSO | 0.57 (0.04) | 0.73 (0.09) | ||
| J-LASSO | 0.62 (0.04) | 0.40 (0.04) | ||
| ColReg | 0.50 (0.03) | 0.71 (0.09) | ||
| strong | IGE | 0.91 (0.02) | 0.93 (0.02) | |
| S-LASSO | 0.61 (0.04) | 0.71 (0.08) | ||
| J-LASSO | 0.64 (0.05) | 0.43 (0.04) | ||
| ColReg | 0.52 (0.03) | 0.70 (0.09) | ||
| Banded | weak | IGE | 0.89 (0.03) | 0.91 (0.03) |
| S-LASSO | 0.55 (0.04) | 0.73 (0.07) | ||
| J-LASSO | 0.62 (0.04) | 0.40 (0.05) | ||
| ColReg | 0.50 (0.03) | 0.71 (0.08) | ||
| strong | IGE | 0.90 (0.04) | 0.92 (0.02) | |
| S-LASSO | 0.61 (0.04) | 0.72 (0.08) | ||
| J-LASSO | 0.64 (0.04) | 0.44 (0.06) | ||
| ColReg | 0.53 (0.04) | 0.70 (0.08) | ||
| LUSC | weak | IGE | 0.82 (0.04) | 0.78 (0.06) |
| S-LASSO | 0.51 (0.05) | 0.36 (0.07) | ||
| J-LASSO | 0.56 (0.05) | 0.25 (0.07) | ||
| ColReg | 0.39 (0.04) | 0.35 (0.08) | ||
| strong | IGE | 0.83 (0.04) | 0.82 (0.06) | |
| S-LASSO | 0.57 (0.05) | 0.39 (0.07) | ||
| J-LASSO | 0.58 (0.05) | 0.25 (0.08) | ||
| ColReg | 0.42 (0.04) | 0.38 (0.07) | ||
| LUAD | weak | IGE | 0.83 (0.04) | 0.80 (0.06) |
| S-LASSO | 0.57 (0.04) | 0.43 (0.06) | ||
| J-LASSO | 0.59 (0.04) | 0.25 (0.06) | ||
| ColReg | 0.47 (0.03) | 0.43 (0.06) | ||
| strong | IGE | 0.85 (0.03) | 0.84 (0.04) | |
| S-LASSO | 0.61 (0.04) | 0.46 (0.07) | ||
| J-LASSO | 0.61 (0.04) | 0.26 (0.06) | ||
| ColReg | 0.49 (0.03) | 0.46 (0.07) |
| LRMs | ||||
|---|---|---|---|---|
| #1 (0.07) | #2 (−0.01) | #3 (−0.02) | #4 (−0.03) | |
| mRNA | PIK3R2 (0.35) | PIK3R2 (0.98) | ECT2 (−0.98) | INTS7 (−0.77) |
| STK3 (−0.74) | STK3 (0.11) | PSMD2 (−0.17) | PIK3R2 (−0.62) | |
| NCKAP5L (0.74) | NCKAP5L (−0.08) | |||
| CUL9 (0.14) | ||||
| CNA | NEK2(−0.22) | CECR1 (0.65) | KPNA4 (−0.44) | INTS7 (−0.70) |
| LPGAT1 (0.22) | C1QTNF6 (−0.75) | B3GALNT1 (0.43) | DTL (0.70) | |
| INTS7 (0.65) | PSMD2 (−0.55) | |||
| DTL (−0.65) | LIPH (0.55) | |||
| CECR1 (−0.19) | ||||
| #5 (−0.05) | #6 (0.08) | #7 (−0.06) | #8 (0.06) | |
| mRNA | PIK3R2 (0.12) | INTS7 (0.73) | PIK3R2 (−0.10) | PSMD2 (0.31) |
| STK3 (−0.78) | PIK3R2 (0.63) | STK3 (−0.24) | TMOD 3(0.61) | |
| NCKAP5L (0.57) | STK3 (0.18) | CUL9 (−0.96) | DIAPH3 (0.72) | |
| CUL9 (0.16) | NCKAP5L (−0.14) | |||
| CNA | INTS7 (−0.16) | NEK2 (−0.69) | INTS7 (−0.34) | MAPRE3 (0.70) |
| DTL (0.16) | LPGAT1 (0.71) | DTL (0.36) | IFT172 (−0.67) | |
| CECR1 (−0.78) | CECR1 (0.61) | PSMD2 (0.09) | ||
| C1QTNF6 (−0.57) | C1QTNF6 (−0.61) | ITGB1 (0.09) | ||
| ADAM10 (0.14) | ||||
| Residual effects | ||||
| mRNA | MAST3 (0.01) | |||
| DM | ADSS (0.01) | SLC2A1 (0.01) | PTCH2 (0.01) | ECT2 (0.09) |
| TNS4 (0.02) | MUSTN1 (0.05) | DKK1 (0.02) | FSCN1 (0.05) | |
| GNPNAT1 (0.04) | HPS1 (−0.04) | MAPRE3 (−0.02) | ||
| CNA | LAMC2 (−0.01) | CD5 (−0.03) | E2F7 (−0.01) | |
| LRMs | AGE | GENDER | SMOKING |
|---|---|---|---|
| #1 | 0.08 | −0.25 | |
| #2 | 0.02 | ||
| #3 | 0.01 | ||
| #4 | 0.01 | 0.01 | |
| #5 | 0.01 | ||
| mRNA Residual | AGE | GENDER | SMOKING |
| MAST3 | 0.27 | ||
| HPS1 | 0.01 | ||
| BBS5 | −0.04 | −0.03 | |
| TLE1 | −0.01 | ||
| ADAM10 | 0.02 | 0.03 | |
| SLC16A3 | 0.07 | ||
| BTN2A2 | −0.02 | −0.06 | |
| FAM71E1 | 0.02 |
| LRMs | ||||
|---|---|---|---|---|
| #1 (−0.01) | #2 (0.01) | #3 (0.01) | #4 (−0.02) | |
| mRNA | RNF24 (−0.17) | SEC23B (0.23) | REEP3 (−0.76) | AP2A2 (−0.59) |
| ESM1 (−0.53) | RNF24 (−0.97) | FUT11 (−0.64) | PNPLA6 (−0.37) | |
| RASAL2 (−0.39) | RFX1 (−0.55) | |||
| LAMC1 (−0.34) | XRN2 (0.45) | |||
| DLGAP4 (−0.63) | ||||
| DM | DCBLD1 (0.09) | TCF7L2 (0.22) | RGP1 (−0.52) | |
| CHI3L1 (0.18) | NCOR2 (0.27) | |||
| CNA | CD163L1 (−0.16) | ENTPD6 (0.68) | RERE (−0.89) | CD163L1 (0.70) |
| DLGAP4 (−0.96) | ABHD12 (−0.69) | DLGAP4 (−0.43) | PARD6G (−0.39) | |
| #5 (0.16) | #6 (0.05) | #7 (−0.05) | #8 (0.01) | |
| mRNA | COL5A3 (0.45) | MGST3 (0.33) | TPM4 (0.68) | TCTN2 (−0.45) |
| DCBLD1 (0.57) | OSBPL5 (0.31) | UBB (0.59) | ANGPT2 (−0.40) | |
| PDGFA (0.31) | SNX9 (0.56) | NCOR2 (−0.42) | UBE4B (−0.37) | |
| CHST15 (0.45) | MYO1C (0.46) | MBTPS1 (−0.47) | ||
| LGALS1 (0.39) | CCDC68 (0.49) | FAM178B (−0.50) | ||
| DM | DCBLD1 (−0.86) | CHST15 (−0.97) | RGP1 (−0.55) | NCOR2 (0.16) |
| FAM178B (−0.37) | RGP1 (0.13) | |||
| CHST15 (−0.17) | NCOR2 (−0.10) | |||
| LGALS1 (−0.15) | ||||
| CNA | DLGAP4 (0.27) | STK40 (−0.26) | CD163L1 (−0.35) | |
| TCTN2 (−0.78) | DLGAP4 (−0.92) | |||
| Residual effects | ||||
| mRNA | LRAT (−0.02) | PLEKHA6 (−0.02) | ||
| DM | BAMBI (0.01) | PYGB (0.02) | FUT11 (−0.18) | ZNF394 (0.03) |
| CCIN (−0.01) | DEAF1 (−0.10) | ACOT7 (0.04) | KLK6 (−0.12) | |
| LHX8 (−0.01) | PLEKHB1 (0.09) | |||
| CNA | FGFRL1 (−0.05) | DCBLD1 (−0.04) | NEFL (−0.04) | CHST1 (0.02) |
| ULK1 (−0.03) | FPR2 (0.02) | PYGB (−0.10) | ||
| LRMs | AGE | GENDER | SMOKING |
|---|---|---|---|
| #1 | 0.02 | 0.03 | |
| #2 | 0.03 | ||
| #4 | −0.02 | ||
| #5 | 0.01 | 0.05 | −0.02 |
| #6 | 0.01 | −0.01 | |
| #7 | −0.36 | ||
| #8 | 0.02 | ||
| mRNA Residual | AGE | GENDER | SMOKING |
| LRAT | −0.17 | ||
| PLEKHA6 | −0.30 | ||
| AP2A2 | 0.02 | ||
| SLC12A7 | −0.10 | 0.07 | |
| TCTN2 | −0.15 | −0.09 | |
| CLEC5A | 0.01 | ||
| RNF24 | −0.06 | 0.04 | |
| PRRX2 | 0.04 | −0.04 | |
| CCDC74A | 0.14 | −0.13 | |
| FGF9 | 0.03 | −0.06 | |
| IGF2R | 0.05 | −0.02 | |
| CHMP4C | 0.24 | 0.13 | −0.01 |
| SLC45A4 | −0.11 | ||
| SULF2 | −0.05 | −0.03 | |
| UBB | −0.11 | ||
| DVL1 | −0.07 | ||
| NID1 | 0.08 | 0.20 | |
| KLK8 | 0.01 | ||
| DOCK6 | 0.26 | −0.10 | |
| FHDC1 | 0.01 | −0.16 | |
| OPLAH | −0.12 | ||
| VSTM1 | −0.02 | ||
| SLC28A1 | −0.07 | ||
| TCF7L2 | 0.12 | ||
| DLGAP4 | −0.04 | ||
| CRNKL1 | −0.25 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Du, Y.; Fan, K.; Lu, X.; Wu, C. Integrating Multi–Omics Data for Gene-Environment Interactions. BioTech 2021, 10, 3. https://doi.org/10.3390/biotech10010003
Du Y, Fan K, Lu X, Wu C. Integrating Multi–Omics Data for Gene-Environment Interactions. BioTech. 2021; 10(1):3. https://doi.org/10.3390/biotech10010003
Chicago/Turabian StyleDu, Yinhao, Kun Fan, Xi Lu, and Cen Wu. 2021. "Integrating Multi–Omics Data for Gene-Environment Interactions" BioTech 10, no. 1: 3. https://doi.org/10.3390/biotech10010003
APA StyleDu, Y., Fan, K., Lu, X., & Wu, C. (2021). Integrating Multi–Omics Data for Gene-Environment Interactions. BioTech, 10(1), 3. https://doi.org/10.3390/biotech10010003