
Quantitative Structure–Activity Relationship Models for the Angiotensin-Converting Enzyme Inhibitory Activities of Short-Chain Peptides of Goat Milk Using Quasi-SMILES





Abstract
1. Introduction
2. Materials and Methods
2.1. Data
2.2. Splitting Available Data into Training and Validation Sets
2.3. Optimal Descriptor
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Speck-Planche, A.; Cordeiro, M.N.D.S. Computer-aided drug design methodologies toward the design of anti-hepatitis C agents. Curr. Top. Med. Chem. 2012, 12, 802–813. [Google Scholar] [CrossRef]
- Chen, C.Y.-C. A novel integrated framework and improved methodology of computer-aided drug design. Curr. Top. Med. Chem. 2013, 13, 965–988. [Google Scholar] [CrossRef]
- Raevsky, O.A.; Mukhametov, A.; Grigorev, V.Y.; Ustyugov, A.; Tsay, S.-C.; Hwu, R.J.-R.; Yarla, N.S.; Tarasov, V.V.; Aliev, G.; Bachurin, S.O. Applications of multi-target computer-aided methodologies in molecular design of CNS drugs. Curr. Med. Chem. 2018, 25, 5293–5314. [Google Scholar] [CrossRef]
- Klopman, G.; Ptchelintsev, D. Antifungal triazole alcohols: A comparative analysis of structure-activity, structure-teratogenicity and structure-therapeutic index relationships using the Multiple Computer-Automated Structure Evaluation (Multi-CASE) methodology. J. Comput.-Aided Mol. Des. 1993, 7, 349–362. [Google Scholar] [CrossRef]
- Klopman, G.; Ptchelintsev, D. Application of the Computer Automated Structure Evaluation Methodology to a QSAR Study of Chemoreception. Aromatic Musky Odorants. J. Agric. Food Chem. 1992, 40, 2244–2251. [Google Scholar] [CrossRef]
- Gordeeva, E.V.; Molchanova, M.S.; Zefirov, N.S. General methodology and computer program for the exhaustive restoring of chemical structures by molecular connectivity indexes. Solution of the inverse problem in QSAR/QSPR. Tetrahedron Comput. Methodol. 1990, 3 Pt B, 389–415. [Google Scholar] [CrossRef]
- Speck-Planche, A.; Luan, F.; Cordeiro, M. Abelson tyrosine-protein kinase 1 as principal target for drug discovery against leukemias role of the current computer-aided drug design methodologies. Curr. Top. Med. Chem. 2012, 12, 2745–2762. [Google Scholar] [CrossRef]
- Bordás, B.; Kömíves, T.; Lopata, A. Ligand-based computer-aided pesticide design. A review of applications of the CoMFA and CoMSIA methodologies. Pest Manag. Sci. 2003, 59, 393–400. [Google Scholar] [CrossRef]
- Scotti, L.; Scotti, M.T.; De Oliveira Lima, E.; Da Silva, M.S.; Do Carmo Alves De Lima, M.; Da Rocha Pitta, I.; De Moura, R.O.; De Oliveira, J.G.B.; Da Cruz, R.M.D.; Mendonça, F.J.B., Jr. Experimental methodologies and evaluations of computer-aided drug design methodologies applied to a series of 2-aminothiophene derivatives with antifungal activities. Molecules 2012, 17, 2298–2315. [Google Scholar] [CrossRef]
- de Sousa, N.F.; Scotti, L.; de Moura, É.P.; Dos Santos Maia, M.; Rodrigues, G.C.S.; de Medeiros, H.I.R.; Lopes, S.M.; Scotti, M.T. Computer Aided Drug Design Methodologies with Natural Products in the Drug Research Against Alzheimer’s Disease. Curr. Neuropharmacol. 2022, 20, 857–885. [Google Scholar] [CrossRef]
- Kumar, A.; Sindhu, J.; Kumar, P. In-silico identification of fingerprint of pyrazolyl sulfonamide responsible for inhibition of N-myristoyltransferase using Monte Carlo method with index of ideality of correlation. J. Biomol. Struct. Dyn. 2021, 39, 5014–5025. [Google Scholar] [CrossRef]
- Chen, J.; Intes, X. Comparison of Monte Carlo methods for fluorescence molecular tomography-computational efficiency. Med. Phys. 2011, 38, 5788–5798. [Google Scholar] [CrossRef]
- Harvey, J.-P.; Gheribi, A.E.; Chartrand, P. Accurate determination of the Gibbs energy of Cu-Zr melts using the thermodynamic integration method in Monte Carlo simulations. J. Chem. Phys. 2011, 135, 084502. [Google Scholar] [CrossRef]
- Chen, H.-C.; Lin, L.-C. Computing Mixture Adsorption in Porous Materials through Flat Histogram Monte Carlo Methods. Langmuir 2023, 39, 15380–15390. [Google Scholar] [CrossRef]
- Golubović, M.; Lazarević, M.; Zlatanović, D.; Krtinić, D.; Stoičkov, V.; Mladenović, B.; Milić, D.J.; Sokolović, D.; Veselinović, A.M. The anesthetic action of some polyhalogenated ethers—Monte Carlo method based QSAR study. Comput. Biol. Chem. 2018, 75, 32–38. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. Nucleobase sequence based building up of reliable QSAR models with the index of ideality correlation using Monte Carlo method. J. Biomol. Struct. Dyn. 2020, 38, 3296–3306. [Google Scholar] [CrossRef]
- Geoghegan, T.J.; Nelson, N.P.; Flynn, R.T.; Hill, P.M.; Rana, S.; Hyer, D.E. Design of a focused collimator for proton therapy spot scanning using Monte Carlo methods. Med. Phys. 2020, 47, 2725–2734. [Google Scholar] [CrossRef]
- Chopdar, K.S.; Dash, G.C.; Mohapatra, P.K.; Nayak, B.; Raval, M.K. Monte-Carlo method-based QSAR model to discover phytochemical urease inhibitors using SMILES and GRAPH descriptors. J. Biomol. Struct. Dyn. 2022, 40, 5090–5099. [Google Scholar] [CrossRef]
- Zhang, X.; Chong, K.H.; Zhu, L.; Zheng, J. A Monte Carlo method for in silico modeling and visualization of Waddington’s epigenetic landscape with intermediate details. BioSystems 2020, 198, 104275. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A. In silico enhancement of azo dye adsorption affinity for cellulose fibre through mechanistic interpretation under guidance of QSPR models using Monte Carlo method with index of ideality correlation. SAR QSAR Environ. Res. 2020, 31, 697–715. [Google Scholar] [CrossRef]
- Estrada, E.; Guevara, N.; Gutman, I. Extension of edge connectivity index. Relationships to line graph indices and QSPR applications. J. Chem. Inf. Comput. Sci. 1998, 38, 428–431. [Google Scholar] [CrossRef]
- Estrada, E.; González, H. What are the limits of applicability for graph theoretic descriptors in QSPR/QSAR? Modeling dipole moments of aromatic compounds with TOPS-MODE descriptors. J. Chem. Inf. Comput. Sci. 2003, 43, 75–84. [Google Scholar] [CrossRef]
- Ahmadi, S.; Mehrabi, M.; Rezaei, S.; Mardafkan, N. Structure-activity relationship of the radical scavenging activities of some natural antioxidants based on the graph of atomic orbitals. J. Mol. Struct. 2019, 1191, 165–174. [Google Scholar] [CrossRef]
- Toropova, A.P.; Toropov, A.A.; Rasulev, B.F.; Benfenati, E.; Gini, G.; Leszczynska, D.; Leszczynski, J. QSAR models for ACE-inhibitor activity of tripeptides based on representation of the molecular structure by graph of atomic orbitals and SMILES. Struct. Chem. 2012, 23, 1873–1878. [Google Scholar] [CrossRef]
- Toropov, A.A.; Toropova, A.P. QSPR modeling of alkanes properties based on graph of atomic orbitals. J. Mol. Struct. THEOCHEM 2003, 637, 1–10. [Google Scholar] [CrossRef]
- Toropov, A.A.; Toropova, A.P.; Raska, I., Jr.; Benfenati, E.; Gini, G. QSAR modeling of endpoints for peptides which is based on representation of the molecular structure by a sequence of amino acids. Struct. Chem. 2012, 23, 1891–1904. [Google Scholar] [CrossRef]
- Toropova, A.P.; Toropov, A.A.; Kumar, P.; Kumar, A.; Achary, P.G.R. Fragments of local symmetry in a sequence of amino acids: Does one can use for QSPR/QSAR of peptides? J. Mol. Struct. 2023, 1293, 136300. [Google Scholar] [CrossRef]
- Weininger, D. SMILES, a Chemical Language and Information System: 1: Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Toropova, A.P.; Toropov, A.A.; Rallo, R.; Leszczynska, D.; Leszczynski, J. Optimal descriptor as a translator of eclectic data into prediction of cytotoxicity for metal oxide nanoparticles under different conditions. Ecotoxicol. Environ. Saf. 2015, 112, 39–45. [Google Scholar] [CrossRef]
- Trinh, T.X.; Choi, J.-S.; Jeon, H.; Byun, H.-G.; Yoon, T.-H.; Kim, J. Quasi-SMILES-based Nano-Quantitative Structure-Activity Relationship model to predict the cytotoxicity of multiwalled carbon nanotubes to human lung cells. Chem. Res. Toxicol. 2018, 31, 183–190. [Google Scholar] [CrossRef]
- Toropov, A.A.; Toropova, A.P.; Leszczynska, D.; Leszczynski, J. “Ideal correlations” for biological activity of peptides. BioSystems 2019, 181, 51–57. [Google Scholar] [CrossRef]
- Toropova, A.P.; Toropov, A.A.; Benfenati, E.; Leszczynska, D.; Leszczynski, J. Prediction of antimicrobial activity of large pool of peptides using quasi-SMILES. BioSystems 2018, 169–170, 5–12. [Google Scholar] [CrossRef]
- Moinul, M.; Khatun, S.; Abdul Amin, S.; Jha, T.; Gayen, S. Quasi-SMILES as a tool for peptide QSAR modelling. In QSPR/QSAR Analysis Using SMILES and Quasi-SMILES; Toropova, A.P., Toropov, A.A., Eds.; Challenges and Advances in Computational Chemistry and Physics; Springer: Cham, Switzerland, 2023; Volume 33, pp. 269–294. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A.; Sindhu, J.; Lal, S. Quasi-SMILES as a basis for the development of QSPR models to predict the CO2 capture capacity of deep eutectic solvents using correlation intensity index and consensus modelling. Fuel 2023, 345, 128237. [Google Scholar] [CrossRef]
- Toropova, A.P.; Toropov, A.A.; Roncaglioni, A.; Benfenati, E.; Leszczynska, D.; Leszczynski, J. CORAL: Model of Ecological Impact of Heavy Metals on Soils via the Study of Modification of Concentration of Biomolecules in Earthworms (Eisenia fetida). Arch. Environ. Contam. Toxicol. 2023, 84, 504–515. [Google Scholar] [CrossRef]
- Manganelli, S.; Benfenati, E. Nano-QSAR model for predicting cell viability of human embryonic kidney cells. Methods Mol. Biol. 2017, 1601, 275–290. [Google Scholar] [CrossRef]
- Toropova, A.P.; Toropov, A.A. Nanomaterials: Quasi-SMILES as a flexible basis for regulation and environmental risk assessment. Sci. Total Environ. 2022, 823, 153747. [Google Scholar] [CrossRef]
- Toropova, A.P.; Toropov, A.A. Quasi-SMILES as a basis to build up models of endpoints for nanomaterials. Environ. Technol. 2023, 44, 4460–4467. [Google Scholar] [CrossRef]
- Toropov, A.A.; Kjeldsen, F.; Toropova, A.P. Use of quasi-SMILES to build models based on quantitative results from experiments with nanomaterials. Chemosphere 2022, 303, 135086. [Google Scholar] [CrossRef]
- Toropova, A.P.; Meneses, J.; Alfaro-Moreno, E.; Toropov, A.A. The system of self-consistent models based on quasi-SMILES as a tool to predict the potential of nano-inhibitors of human lung carcinoma cell line A549 for different experimental conditions. Drug Chem. Toxicol. 2023, 47, 306–313. [Google Scholar] [CrossRef]
- Muhammad, F.; Awais, M.M.; Akhtar, M.; Anwar, M.I. Quantitative structure activity relationship and risk analysis of some pesticides in the goat milk. Iran. J. Environ. Health Sci. Eng. 2013, 10, 4. [Google Scholar] [CrossRef]
- Du, A.; Jia, W. Bioaccessibility of novel antihypertensive short-chain peptides in goat milk using the INFOGEST static digestion model by effect-directed assays. Food Chem. 2023, 427, 136735. [Google Scholar] [CrossRef]
- Toropov, A.A.; Di Nicola, M.R.; Toropova, A.P.; Roncaglioni, A.; Dorne, J.L.C.M.; Benfenati, E. Quasi-SMILES: Self-consistent models for toxicity of organic chemicals to tadpoles. Chemosphere 2023, 312, 137224. [Google Scholar] [CrossRef] [PubMed]
- Toropova, A.P.; Toropov, A.A.; Roncaglioni, A.; Benfenati, E. Monte Carlo technique to study the adsorption affinity of azo dyes by applying new statistical criteria of the predictive potential. SAR QSAR Environ. Res. 2022, 33, 621–630. [Google Scholar] [CrossRef]
- Toropov, A.A.; Toropova, A.P.; Roncaglioni, A.; Benfenati, E. In silico prediction of the mutagenicity of nitroaromatic compounds using correlation weights of fragments of local symmetry. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2023, 891, 503684. [Google Scholar] [CrossRef] [PubMed]
- Sigala-Robles, R.; Santiago-López, L.; Hernández-Mendoza, A.; Vallejo-Cordoba, B.; Mata-Haro, V.; Wall-Medrano, A.; González-Córdova, A.F. Peptides, exopolysaccharides, and short-chain fatty acids from fermented milk and perspectives on inflammatory bowel diseases. Dig. Dis. Sci. 2022, 67, 4654–4665. [Google Scholar] [CrossRef] [PubMed]
- Lewandowski, B.; Wennemers, H. Asymmetric catalysis with short-chain peptides. Curr. Opin. Chem. Biol. 2014, 22, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Sun, Z.; Yang, Z.; Qiao, X. Microbiota-derived short-chain fatty acids and modulation of host-derived peptides formation: Focused on host defense peptides. Biomed. Pharmacother. 2023, 162, 114586. [Google Scholar] [CrossRef] [PubMed]
- Lenstra, R. The graph, geometry and symmetries of the genetic code with hamming metric. Symmetry 2015, 7, 1211–1260. [Google Scholar] [CrossRef]
- Rehm, L.; Morshed, M.G.; Misra, S.; Shukla, A.; Rakheja, S.; Pinarbasi, M.; Ghosh, A.W.; Kent, A.D. Temperature-resilient random number generation with stochastic actuated magnetic tunnel junction devices. Appl. Phys. Lett. 2024, 124, 052401. [Google Scholar] [CrossRef]
- Liman, W.; Oubahmane, M.; Hdoufane, I.; Bjij, I.; Villemin, D.; Daoud, R.; Cherqaoui, D.; Allali, A.E. Monte Carlo method and GA-MLR-based QSAR modeling of NS5A inhibitors against the hepatitis C virus. Molecules 2022, 27, 2729. [Google Scholar] [CrossRef]
- Gálvez-Llompart, M.; Sastre, G. Machine Learning Search for Suitable Structure Directing Agents for the Synthesis of Beta (BEA) Zeolite Using Molecular Topology and Monte Carlo Techniques. In AI-Guided Design and Property Prediction for Zeolites and Nanoporous Materials; Sastre, G., Daeyaert, F., Eds.; Wiley: Hoboken, NJ, USA, 2023; pp. 61–80. [Google Scholar] [CrossRef]
- Ahmadi, S.; Lotfi, S.; Afshari, S.; Kumar, P.; Ghasemi, E. CORAL: Monte Carlo based global QSAR modelling of Bruton tyrosine kinase inhibitors using hybrid descriptors. SAR QSAR Environ. Res. 2021, 32, 1013–1031. [Google Scholar] [CrossRef]
- Antović, A.R.; Karadžić, R.; Veselinović, A.M. Monte Carlo optimization method based QSAR modeling of postmortem redistribution of structurally diverse drugs. New J. Chem. 2022, 46, 14731–14737. [Google Scholar] [CrossRef]
- Ouabane, M.; Tabti, K.; Hajji, H.; Elbouhi, M.; Khaldan, A.; Elkamel, K.; Sbai, A.; Ajana, M.A.; Sekkate, C.; Bouachrine, M.; et al. Structure-odor relationship in pyrazines and derivatives: A physicochemical study using 3D-QSPR, HQSPR, Monte Carlo, molecular docking, ADME-Tox and molecular dynamics. Arab. J. Chem. 2023, 16, 105207. [Google Scholar] [CrossRef]
- Antović, A.; Karadžić, R.; Živković, J.V.; Veselinović, A.M. Development of QSAR Model Based on Monte Carlo optimization for predicting GABAA receptor binding of newly emerging benzodiazepines. Acta Chim. Slov. 2023, 70, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Tabti, K.; Abdessadak, O.; Sbai, A.; Maghat, H.; Bouachrine, M.; Lakhlifi, T. Design and development of novel spiro-oxindoles as potent antiproliferative agents using quantitative structure activity based Monte Carlo method, docking molecular, molecular dynamics, free energy calculations, and pharmacokinetics/toxicity studies. J. Mol. Struct. 2023, 1284, 135404. [Google Scholar] [CrossRef]
- Nikolić, N.; Kostić, T.; Golubović, M.; Nikolić, T.; Marinković, M.; Perić, V.; Mladenović, S.; Veselinović, A.M. Monte Carlo optimization based QSAR modeling of angiotensin II receptor antagonists. Acta Chim. Slov. 2023, 70, 318–326. [Google Scholar] [CrossRef]
- Lotfi, S.; Ahmadi, S.; Kumar, P. Ecotoxicological prediction of organic chemicals toward Pseudokirchneriella subcapitata by Monte Carlo approach. RSC Adv. 2022, 12, 24988–24997. [Google Scholar] [CrossRef] [PubMed]
- Ahmadi, S.; Ghanbari, H.; Lotfi, S.; Azimi, N. Predictive QSAR modeling for the antioxidant activity of natural compounds derivatives based on Monte Carlo method. Mol. Divers. 2021, 25, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Drefahl, A. CurlySMILES: A chemical language to customize and annotate encodings of molecular and nanodevice structures. J. Cheminf. 2011, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Toropova, A.P.; Toropov, A.A. The coefficient of conformism of a correlative prediction (CCCP): Building up reliable nano-QSPRs/QSARs for endpoints of nanoparticles in different experimental conditions encoded via quasi-SMILES. Sci. Total Environ. 2024, 927, 172119. [Google Scholar] [CrossRef]
- Toropova, A.P.; Toropov, A.A.; Manganelli, S.; Leone, C.; Baderna, D.; Benfenati, E.; Fanelli, R. Quasi-SMILES as a tool to utilize eclectic data for predicting the behavior of nanomaterials. NanoImpact 2016, 1, 60–64. [Google Scholar] [CrossRef]
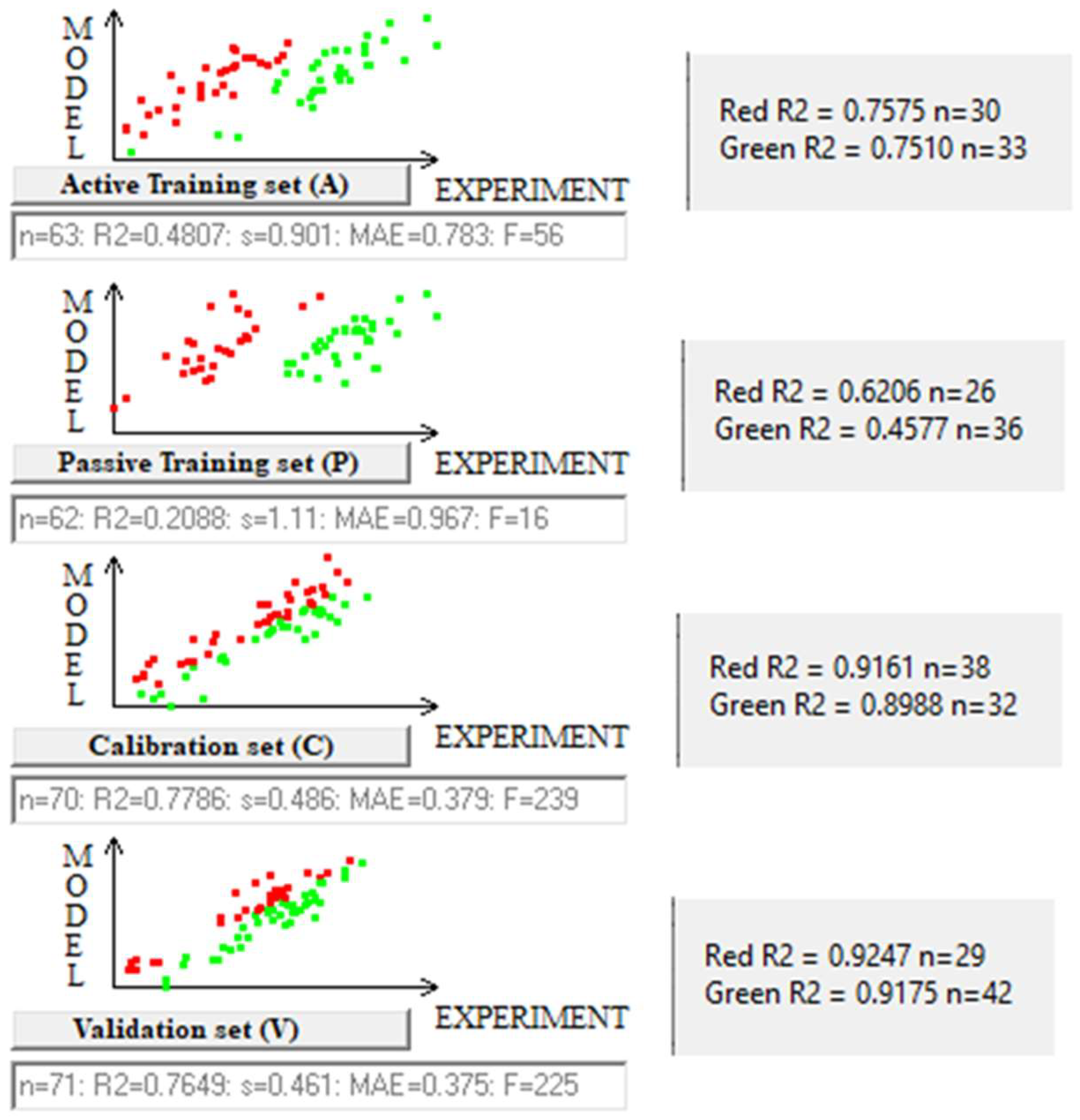
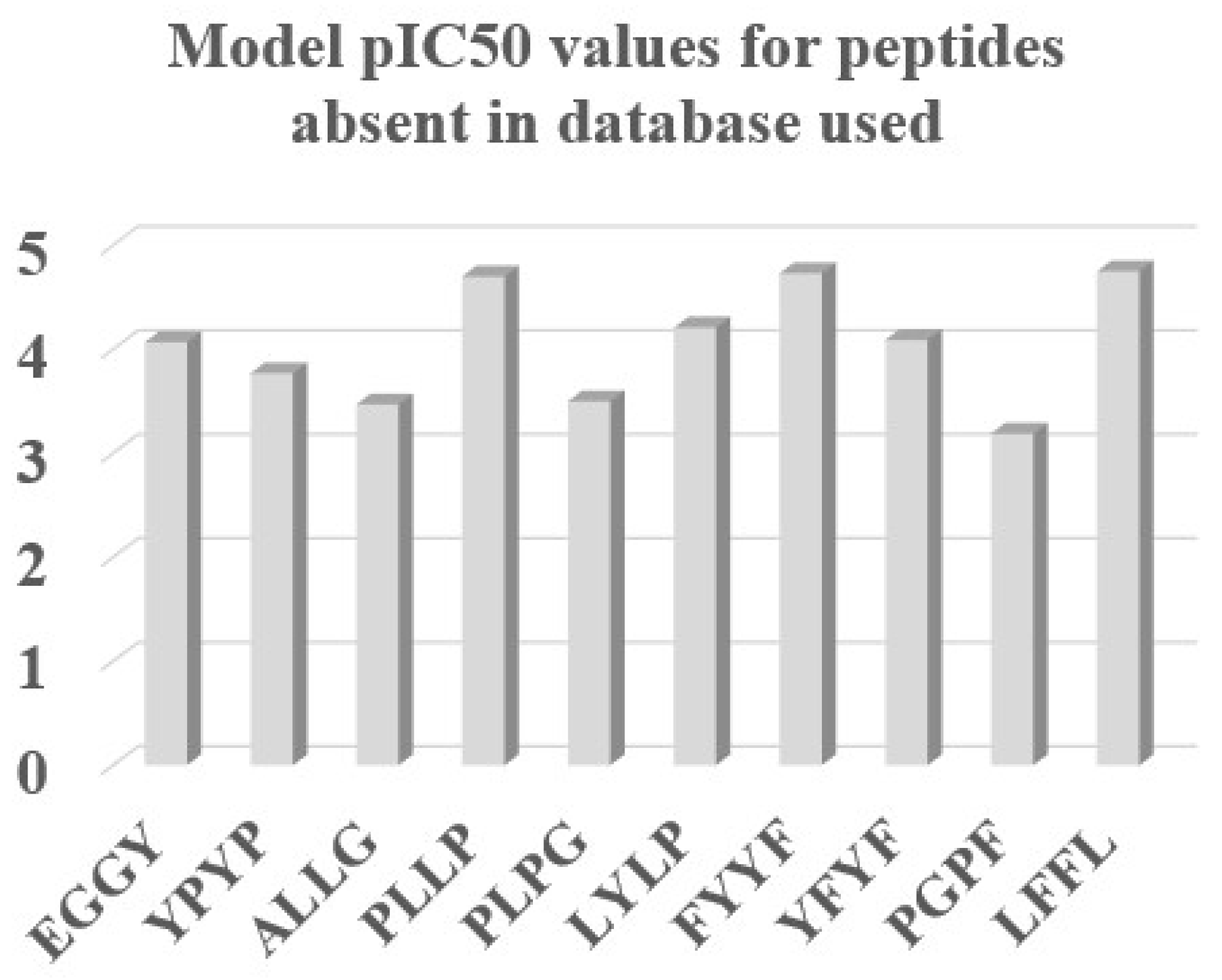

{kind=link}
{kind=link}
| Split/Eq. | n * | R2 | IIC | CII | Q2 | Q2F1 | Q2F2 | Q2F3 | <Rm2> | MAE | F | Nact | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/3 | A | 63 | 0.4807 | 0.6303 | 0.7322 | 0.4501 | 0.783 | 56 | |||||
| P | 62 | 0.2088 | 0.4045 | 0.7011 | 0.1430 | 0.967 | 16 | ||||||
| C | 70 | 0.7786 | 0.8822 | 0.8910 | 0.7638 | 0.7844 | 0.7583 | 0.8475 | 0.6895 | 0.379 | 239 | ||
| V | 71 | 0.7649 | - | - | - | - | - | - | - | 0.38 | - | 28 | |
| 2/4 | A | 69 | 0.3732 | 0.5600 | 0.7187 | 0.3399 | 0.892 | 40 | |||||
| P | 66 | 0.3837 | 0.6073 | 0.7125 | 0.3486 | 0.840 | 40 | ||||||
| C | 65 | 0.7876 | 0.8869 | 0.8767 | 0.7745 | 0.8024 | 0.7875 | 0.8681 | 0.6886 | 0.375 | 234 | ||
| V | 66 | 0.7867 | - | - | - | - | - | - | - | 0.33 | - | 25 | |
| 3/5 | A | 71 | 0.2836 | 0.5178 | 0.6979 | 0.2391 | 0.939 | 27 | |||||
| P | 64 | 0.2507 | 0.3968 | 0.7318 | 0.2000 | 0.945 | 21 | ||||||
| C | 65 | 0.7654 | 0.8748 | 0.8793 | 0.7493 | 0.7869 | 0.7580 | 0.8763 | 0.5959 | 0.342 | 206 | ||
| V | 66 | 0.8339 | - | - | - | - | - | - | - | 0.30 | - | 25 | |
| 4/6 | A | 71 | 0.2832 | 0.4622 | 0.6927 | 0.2418 | 0.938 | 27 | |||||
| P | 69 | 0.4217 | 0.5024 | 0.7158 | 0.3913 | 0.904 | 49 | ||||||
| C | 62 | 0.8326 | 0.9120 | 0.8942 | 0.8219 | 0.8106 | 0.8051 | 0.8913 | 0.5924 | 0.341 | 299 | ||
| V | 64 | 0.8083 | - | - | - | - | - | - | - | 0.32 | - | 22 | |
| 5/7 | A | 65 | 0.3926 | 0.6076 | 0.7115 | 0.3582 | 0.859 | 41 | |||||
| P | 67 | 0.5001 | 0.6846 | 0.7267 | 0.4753 | 0.908 | 65 | ||||||
| C | 66 | 0.7889 | 0.8880 | 0.8561 | 0.7760 | 0.7930 | 0.7827 | 0.9122 | 0.7011 | 0.299 | 239 | ||
| V | 68 | 0.7748 | - | - | - | - | - | - | - | 0.34 | - | 26 |
| Quasi-SMILES | DCW(3, 15) | pIC50(Expr) | pIC50(Calc) | Defect of Quasi-SMILES | Applicability Domain * |
|---|---|---|---|---|---|
| ADDA | 4.9121 | 3.8300 | 4.1611 | 5.0143 | YES |
| AEEL | 5.5666 | 4.2400 | 4.5168 | 5.0078 | YES |
| AFFL | 4.6788 | 4.2000 | 4.0344 | 3.0151 | YES |
| AGAG | 0.5240 | 2.6000 | 1.7763 | 3.0179 | YES |
| AKKK | 7.6277 | 5.4900 | 5.6370 | 2.0211 | YES |
| AYAY | 4.2358 | 4.0600 | 3.7936 | 3.0133 | YES |
| DGDG | 1.5879 | 2.1500 | 2.3545 | 2.0099 | YES |
| FAAL | 5.1709 | 4.5800 | 4.3018 | 2.0226 | YES |
| FFFP | 4.5058 | 4.9200 | 3.9403 | 3.0170 | YES |
| FGGK | 3.8671 | 3.8000 | 3.5932 | 2.0221 | YES |
| GSGS | 2.0150 | 2.4200 | 2.5866 | 5.0099 | YES |
| GYGY | 2.6355 | 3.6300 | 2.9238 | 3.0110 | YES |
| IAAE | 5.5930 | 4.4600 | 4.5312 | 3.0191 | YES |
| IAAQ | 5.5433 | 4.4600 | 4.5042 | 3.0191 | YES |
| IGIG | 2.2514 | 2.9200 | 2.7151 | 3.0142 | YES |
| IKKP | 7.4584 | 5.6800 | 5.5450 | 3.0184 | YES |
| IPIP | 3.3166 | 3.8900 | 3.2940 | 3.0171 | YES |
| IRRA | 5.2918 | 5.0100 | 4.3675 | 2.0370 | YES |
| ITTF | 5.9388 | 4.3100 | 4.7191 | 4.0076 | YES |
| LWLW | 5.6253 | 4.4500 | 4.5488 | 3.0262 | YES |
| LYLY | 4.5575 | 4.4100 | 3.9684 | 3.0093 | YES |
| MYMY | 5.8750 | 3.7100 | 4.6845 | 5.0054 | YES |
| PLPL | 4.4238 | 3.4700 | 3.8957 | 0.0292 | YES |
| RARA | 2.1150 | 3.3400 | 2.6409 | 0.0413 | YES |
| RFRF | 2.8395 | 3.7900 | 3.0347 | 3.0406 | YES |
| RGGP | 4.2545 | 4.2700 | 3.8037 | 1.0406 | YES |
| YEEY | 6.6675 | 5.4000 | 5.1152 | 3.0074 | YES |
| YGGY | 5.2962 | 4.8300 | 4.3699 | 2.0189 | YES |
| YLYL | 4.5575 | 3.9900 | 3.9684 | 3.0093 | YES |
| YNYN | 4.9150 | 4.2900 | 4.1627 | 5.0054 | YES |
| YPPR | 5.7054 | 4.7800 | 4.5923 | 2.0278 | YES |
| YPYY | 4.8573 | 4.0500 | 4.1314 | 2.0182 | YES |
| FPFP | 2.7609 | 3.5000 | 2.9920 | 3.0201 | YES |
| FPPF | 4.9269 | 4.6800 | 4.1692 | 2.0245 | YES |
| FQQP | 5.4828 | 4.9200 | 4.4713 | 4.0098 | YES |
| FVAP | 6.6277 | 5.0000 | 5.0935 | 1.0501 | YES |
| FYFY | 4.7808 | 4.6300 | 4.0898 | 3.0126 | YES |
| GDGD | 1.5879 | 2.0400 | 2.3545 | 2.0099 | YES |
| GEEG | 4.4691 | 3.7200 | 3.9204 | 5.0120 | YES |
| GLGL | 1.0321 | 2.6000 | 2.0524 | 0.0631 | YES |
| GNGN | 1.7778 | 2.8900 | 2.4577 | 2.0099 | YES |
| GQGQ | 2.1003 | 2.1500 | 2.6330 | 5.0099 | YES |
| GRRP | 4.8240 | 4.7000 | 4.1133 | 3.0380 | YES |
| KAKA | 2.1131 | 3.4200 | 2.6399 | 0.0224 | YES |
| KGKG | 1.9467 | 2.4900 | 2.5495 | 3.0201 | YES |
| KPPF | 5.2113 | 4.4900 | 4.3238 | 2.0214 | YES |
| LDDP | 5.5989 | 4.3700 | 4.5344 | 4.0081 | YES |
| LEEE | 6.4487 | 4.0000 | 4.9963 | 6.0039 | No |
| LEEL | 6.9531 | 4.8100 | 5.2704 | 5.0103 | YES |
| LFLF | 4.1444 | 3.4600 | 3.7439 | 3.0155 | YES |
| LGGI | 4.1162 | 4.5400 | 3.7286 | 1.0338 | YES |
| LGGL | 4.0173 | 4.4800 | 3.6748 | 0.0546 | YES |
| LIYP | 6.4530 | 5.0000 | 4.9986 | 3.0108 | YES |
| LKKA | 6.3239 | 5.0700 | 4.9284 | 2.0180 | YES |
| LLLF | 5.0405 | 4.1000 | 4.2309 | 1.0239 | YES |
| LLLP | 5.2143 | 4.8000 | 4.3254 | 0.0287 | YES |
| LNNP | 5.7607 | 4.2400 | 4.6223 | 5.0081 | YES |
| LQQW | 7.0176 | 5.4200 | 5.3054 | 4.0128 | YES |
| RPPP | 5.3047 | 4.2200 | 4.3745 | 1.0338 | YES |
| RPRP | 3.3487 | 3.7400 | 3.3115 | 3.0419 | YES |
| RRRR | 6.8579 | 4.2300 | 5.2187 | 3.0600 | YES |
| RWRW | 5.2161 | 4.8000 | 4.3264 | 0.0513 | YES |
| SGSG | 2.0150 | 2.0700 | 2.5866 | 5.0099 | YES |
| SYSY | 4.9354 | 4.1800 | 4.1738 | 5.0054 | YES |
| VAAA | 5.8424 | 4.8900 | 4.6668 | 0.0573 | YES |
| VAAF | 5.2781 | 4.4500 | 4.3601 | 1.0498 | YES |
| VGGP | 4.7551 | 4.5800 | 4.0758 | 1.0298 | YES |
| VIIY | 6.9351 | 5.1200 | 5.2606 | 3.0104 | YES |
| VLLY | 6.1322 | 4.5100 | 4.8242 | 2.0164 | YES |
| VVVF | 5.9261 | 4.4500 | 4.7122 | 1.0386 | YES |
| VYVY | 5.5250 | 4.9200 | 4.4943 | 3.0128 | YES |
| Method | Determination Coefficient for Training or Calibration Set | Determination Coefficient for the Validation Set | Reference |
|---|---|---|---|
| Partial least-squares | 0.61 for training set | 0.40 | [42] |
| Support vector machine | 0.93 for training set | 0.65 | [42] |
| Monte Carlo method | |||
| Split #1 | 0.78 * for calibration set | 0.76 | This work |
| Split #2 | 0.79 for calibration set | 0.79 | - |
| Split #3 | 0.76 for calibration set | 0.83 | - |
| Split #4 | 0.83 for calibration set | 0.81 | - |
| Split #5 | 0.79 for calibration set | 0.77 | - |
| A or FLS | CWs Probe 1 | CWs Probe 2 | CWs Probe 3 | CWs Probe 4 | CWs Probe 5 | NA | NP | NC | Statistical Defect |
|---|---|---|---|---|---|---|---|---|---|
| Increase | |||||||||
| [xyyx0]..... | 0.5362 | 0.7174 | 0.0494 | 0.1663 | 0.2945 | 59 | 58 | 63 | 0.0004 |
| [xyx0]...... | 2.0153 | 1.9219 | 2.4935 | 2.1093 | 1.3410 | 44 | 49 | 48 | 0.0015 |
| P........... | 0.2332 | 0.2868 | 0.5592 | 0.3310 | 0.2543 | 23 | 27 | 20 | 0.0043 |
| L........... | 0.5570 | 0.3970 | 0.7452 | 0.3197 | 0.6662 | 15 | 18 | 20 | 0.0020 |
| Y........... | 0.8356 | 0.7732 | 1.0343 | 0.9938 | 0.7029 | 15 | 14 | 16 | 0.0005 |
| V........... | 0.7098 | 1.0258 | 1.2453 | 0.6531 | 0.2719 | 13 | 9 | 15 | 0.0037 |
| I........... | 0.8481 | 0.5378 | 0.9922 | 0.6733 | 0.5455 | 11 | 9 | 10 | 0.0021 |
| F........... | 0.1322 | 0.2310 | 0.2963 | 0.2405 | 0.1434 | 10 | 14 | 13 | 0.0036 |
| K........... | 0.3180 | 0.7672 | 1.0451 | 0.8752 | 0.1288 | 6 | 5 | 4 | 0.0051 |
| G...G....... | 0.6542 | 1.2557 | 0.7237 | 0.7341 | 0.6380 | 5 | 5 | 3 | 0.0058 |
| W........... | 0.9591 | 1.4843 | 1.7561 | 1.5675 | 1.1600 | 5 | 10 | 5 | 0.0090 |
| A...A....... | 1.1490 | 0.7535 | 0.6467 | 0.9423 | 0.8890 | 4 | 4 | 2 | 0.0072 |
| P...A....... | 1.0247 | 1.4088 | 1.6942 | 1.8574 | 1.1433 | 4 | 4 | 2 | 0.0072 |
| R........... | 0.6049 | 0.5535 | 0.6594 | 0.5883 | 0.2712 | 4 | 13 | 4 | 0.0145 |
| Decrease | |||||||||
| G........... | −0.6792 | −0.7316 | −0.4849 | −0.6757 | −0.3397 | 20 | 16 | 24 | 0.0028 |
| [xyx2]...... | −0.3288 | −0.5771 | −0.4390 | −0.9101 | −0.5755 | 19 | 13 | 22 | 0.0039 |
| P...G....... | −0.0676 | −0.4900 | −0.1707 | −0.1083 | −0.3139 | 5 | 8 | 4 | 0.0085 |
| L...G....... | −0.1497 | −0.3795 | −0.1461 | −0.6752 | −0.4425 | 4 | 1 | 1 | 0.0164 |
| Y...Y....... | −0.7225 | −0.4130 | −0.6068 | −0.6308 | −0.6269 | 4 | 1 | 3 | 0.0118 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Toropova, A.P.; Toropov, A.A.; Roncaglioni, A.; Benfenati, E. Quantitative Structure–Activity Relationship Models for the Angiotensin-Converting Enzyme Inhibitory Activities of Short-Chain Peptides of Goat Milk Using Quasi-SMILES. Macromol 2024, 4, 387-400. https://doi.org/10.3390/macromol4020022
Toropova AP, Toropov AA, Roncaglioni A, Benfenati E. Quantitative Structure–Activity Relationship Models for the Angiotensin-Converting Enzyme Inhibitory Activities of Short-Chain Peptides of Goat Milk Using Quasi-SMILES. Macromol. 2024; 4(2):387-400. https://doi.org/10.3390/macromol4020022
Chicago/Turabian StyleToropova, Alla P., Andrey A. Toropov, Alessandra Roncaglioni, and Emilio Benfenati. 2024. "Quantitative Structure–Activity Relationship Models for the Angiotensin-Converting Enzyme Inhibitory Activities of Short-Chain Peptides of Goat Milk Using Quasi-SMILES" Macromol 4, no. 2: 387-400. https://doi.org/10.3390/macromol4020022
APA StyleToropova, A. P., Toropov, A. A., Roncaglioni, A., & Benfenati, E. (2024). Quantitative Structure–Activity Relationship Models for the Angiotensin-Converting Enzyme Inhibitory Activities of Short-Chain Peptides of Goat Milk Using Quasi-SMILES. Macromol, 4(2), 387-400. https://doi.org/10.3390/macromol4020022