Data Types and the Phylogeny of Neoaves



Simple Summary
Abstract
1. Introduction

- (1)
- (2)
2. Methods
2.1. Dataset Construction
2.2. Analysis of Base Composition
2.3. Phylogenetic Analyses
3. Results
3.1. Base Composition and the Impact of RY Coding
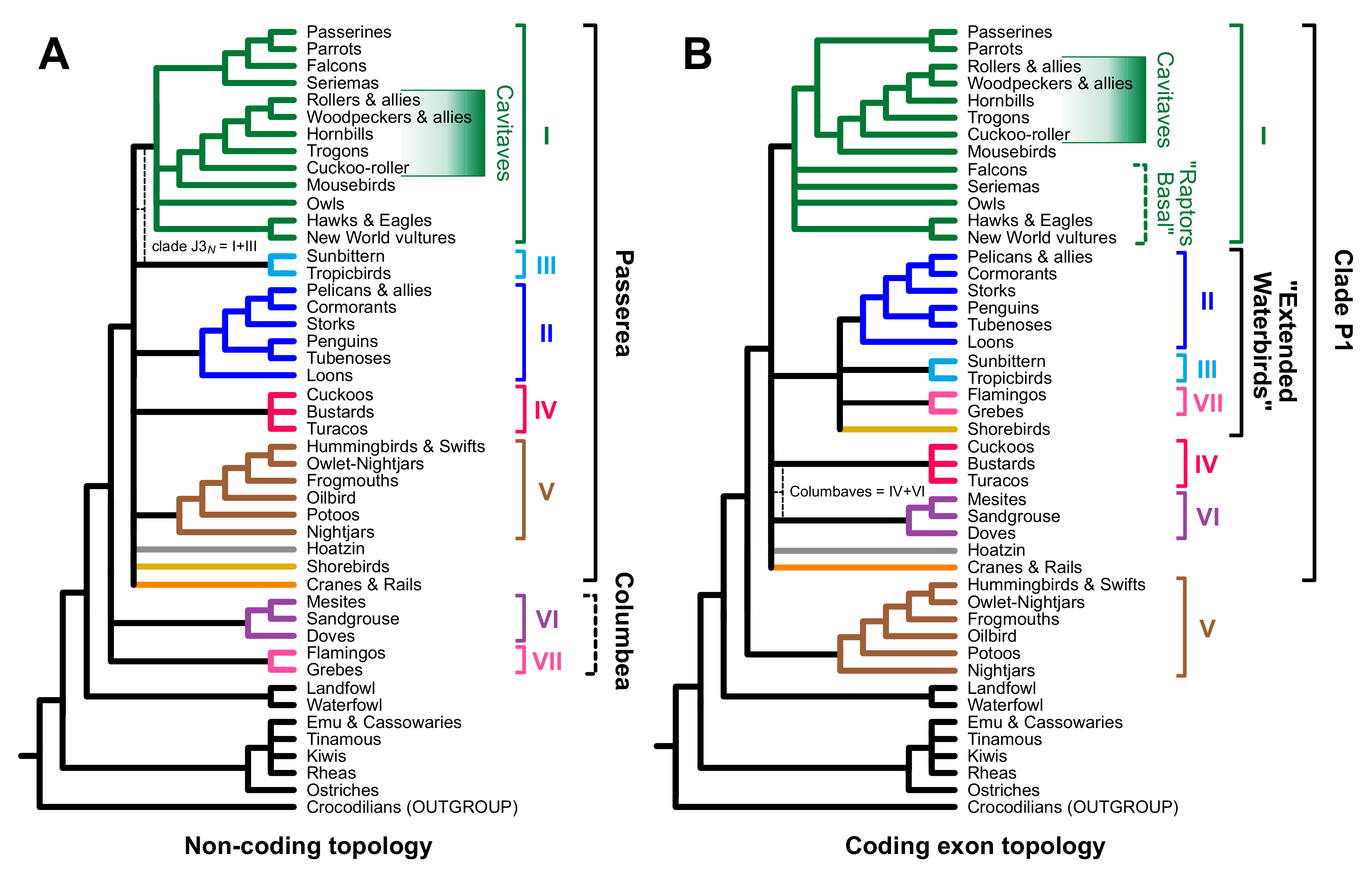
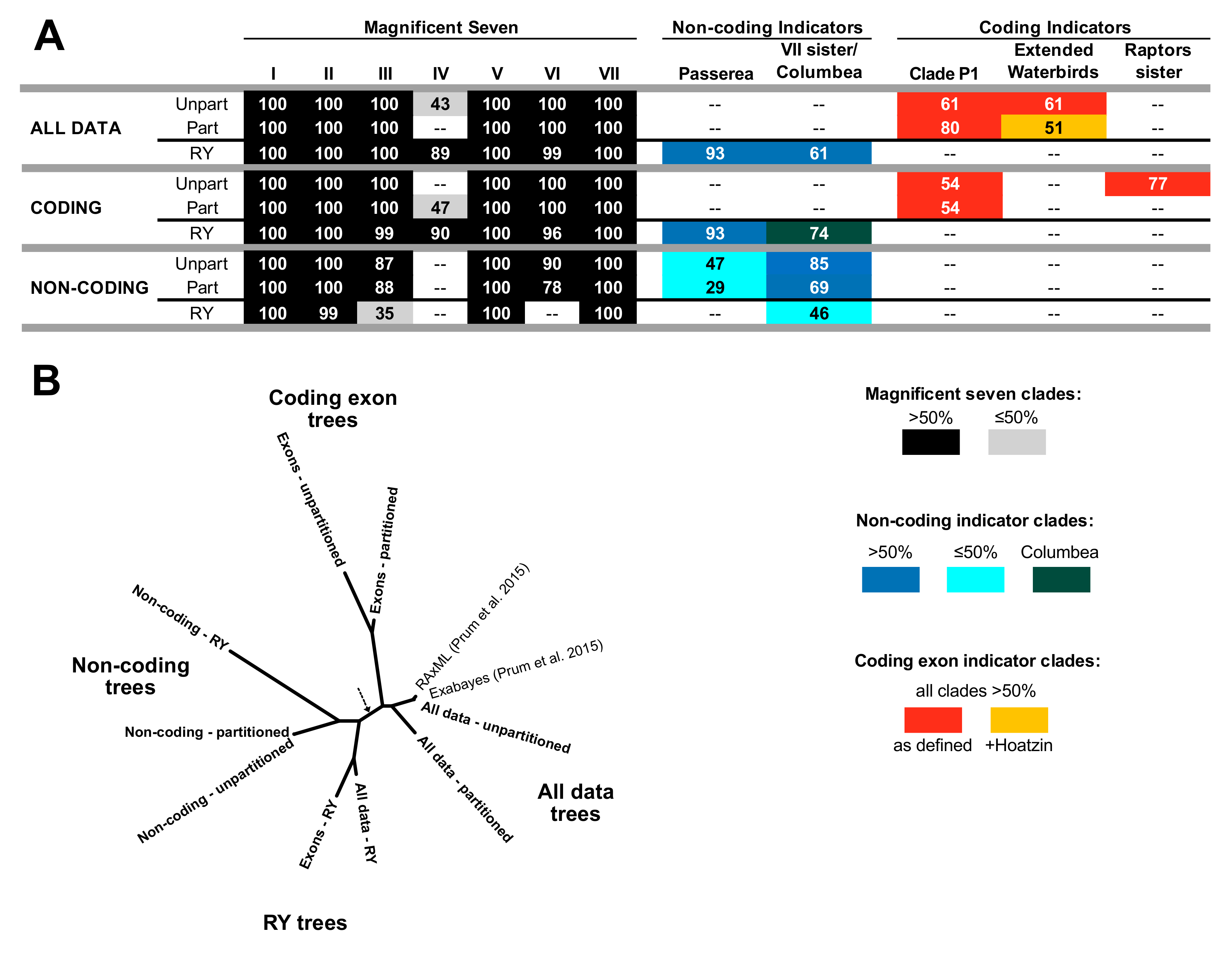
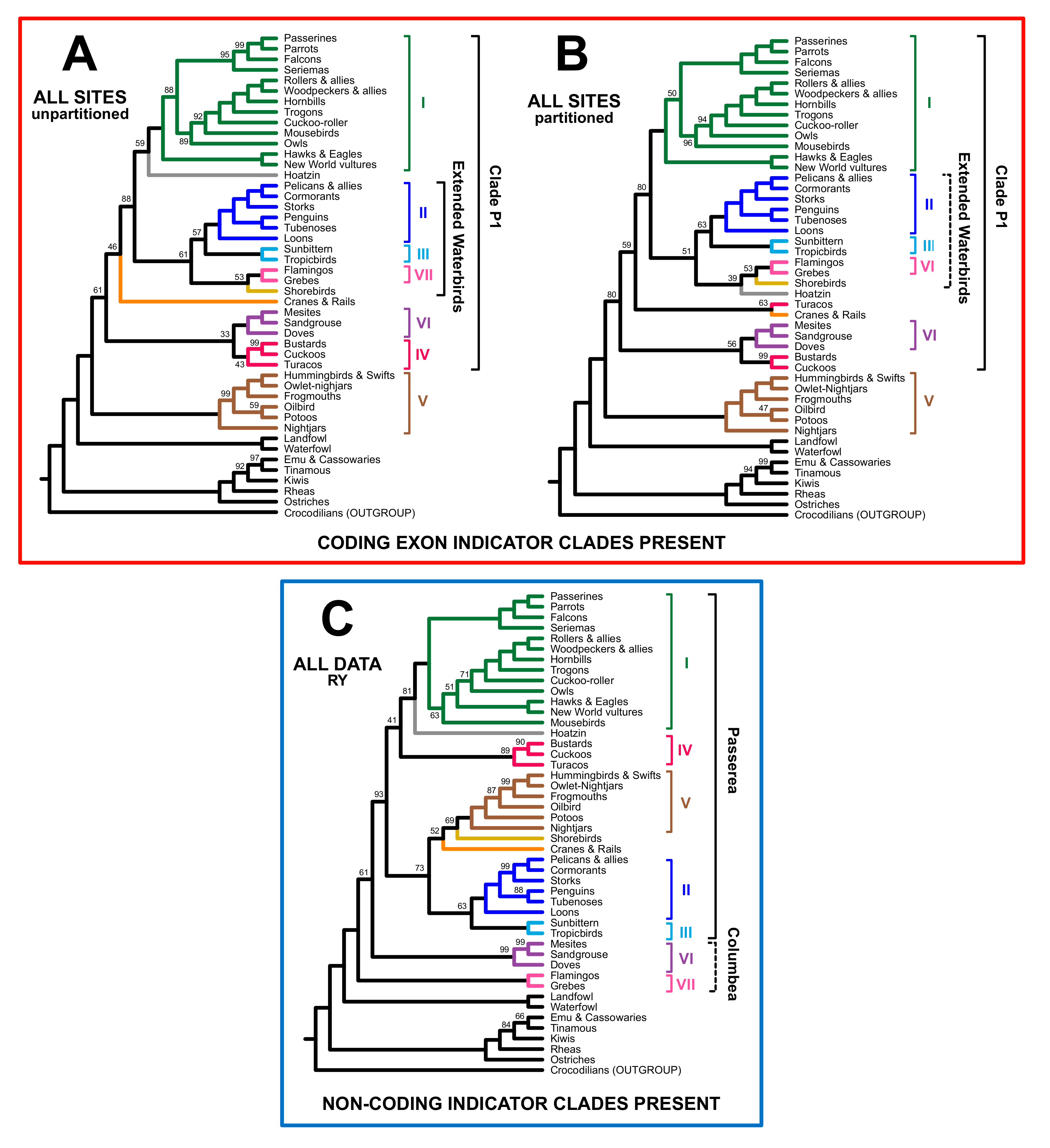
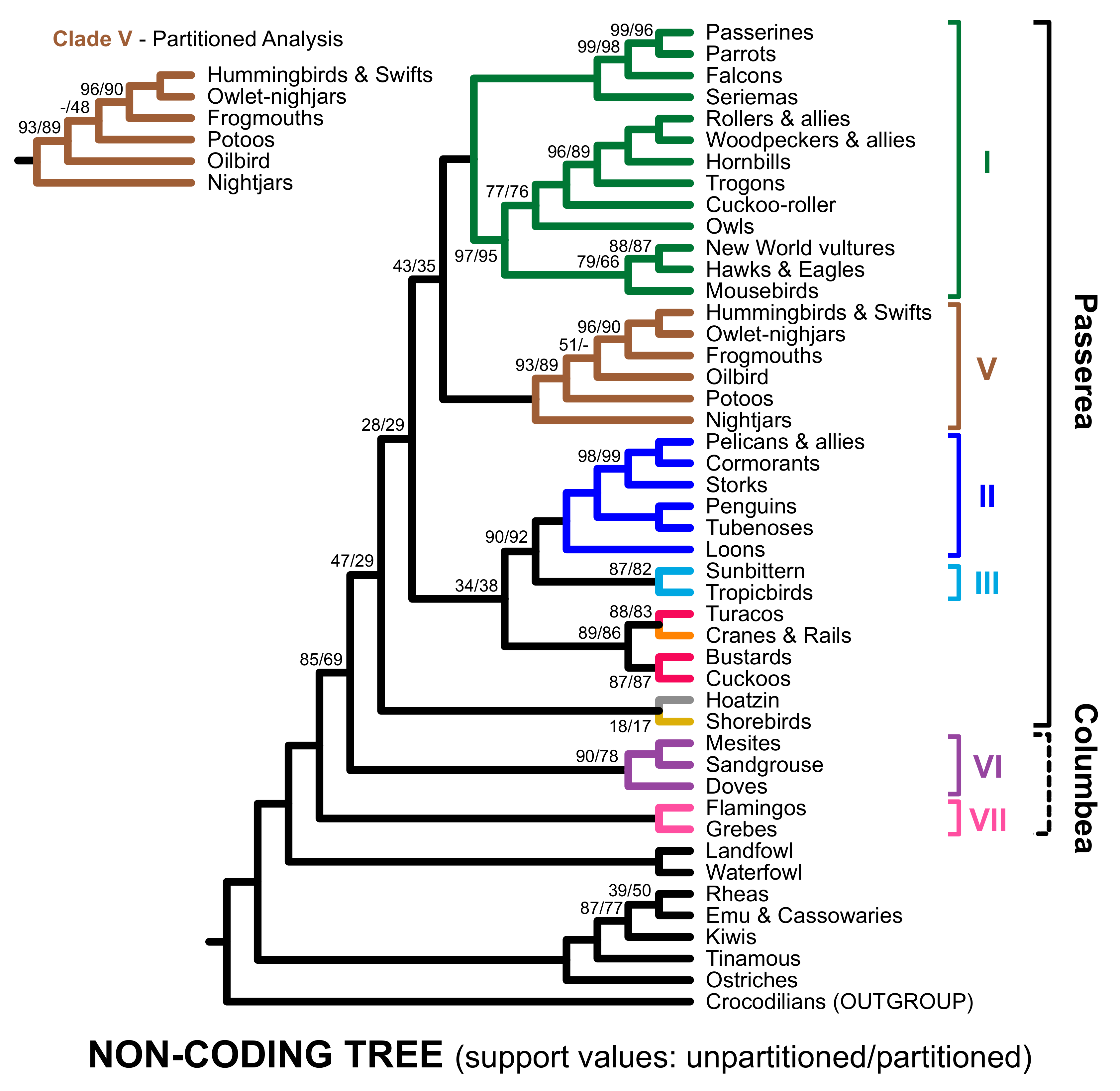
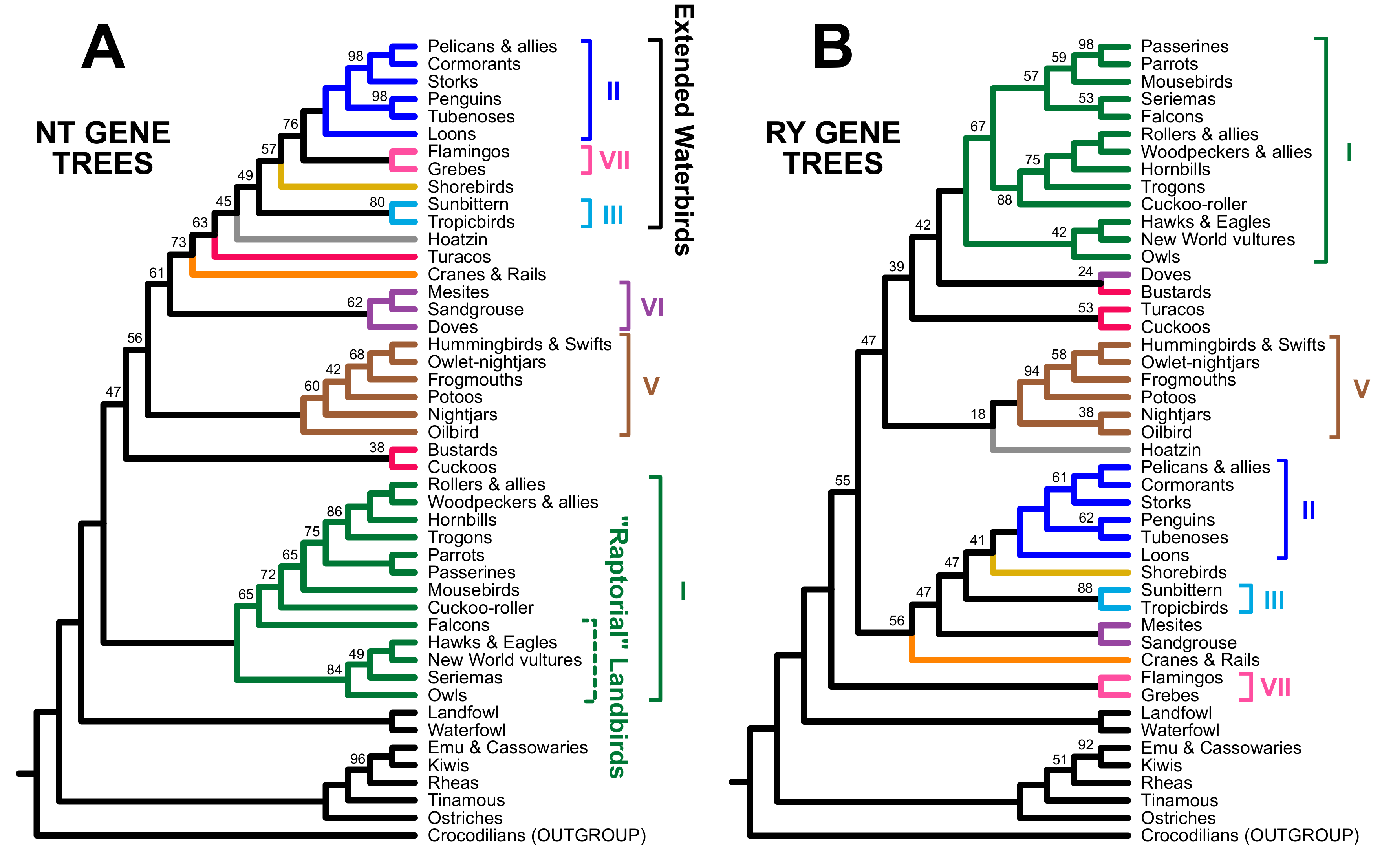
3.2. Different Data Types within the Prum Dataset Yield Distinct Topologies
3.3. RY Coding also Affects Species Tree Analyses
4. Discussion
4.1. The Role of Data Types in Phylogenomic Analyses
4.2. Implications for Avian Phylogeny
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Braun, E.L.; Cracraft, J.; Houde, P. Resolving the avian tree of life from top to bottom: The promise and potential boundaries of the phylogenomic era. In Avian Genomics in Ecology and Evolution: From the Lab into the Wild; Kraus, R.H.S., Ed.; Springer International Publishing: Cham, Switzerland, 2019; pp. 151–210. ISBN 978-3-030-16476-8. [Google Scholar]
- Groth, J.G.; Barrowclough, G.F. Basal divergences in birds and the phylogenetic utility of the nuclear RAG-1 gene. Mol. Phylogenet. Evol. 1999, 12, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Cracraft, J.; Barker, F.K.; Braun, M.; Harshman, J.; Dyke, G.J.; Feinstein, J.; Stanley, S.; Cibois, A.; Schikler, P.; Beresford, P.; et al. Phylogenetic relationships among modern birds (Neornithes): Toward an avian tree of life. In Assembling the Tree of Life; Cracraft, J., Dononghue, M.J., Eds.; Oxford University Press: Oxford, UK, 2004; pp. 468–489. [Google Scholar]
- Ericson, P.G.P.; Anderson, C.L.; Britton, T.; Elzanowski, A.; Johansson, U.S.; Källersjö, M.; Ohlson, J.I.; Parsons, T.J.; Zuccon, D.; Mayr, G. Diversification of Neoaves: Integration of molecular sequence data and fossils. Biol. Lett. 2006, 2, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Field, D.J.; Berv, J.S.; Hsiang, A.Y.; Lanfear, R.; Landis, M.J.; Dornburg, A. Timing the extant avian radiation: The rise of modern birds, and the importance of modeling molecular rate variation. In Pennaraptoran Theropod Dinosaurs: Past Progress and New Frontiers; Pittman, M., Xu, X., Eds.; Bulletin of the American Museum of Natural History 440: New York, NY, USA, 2020; pp. 159–181. [Google Scholar]
- Hackett, S.J.; Kimball, R.T.; Reddy, S.; Bowie, R.C.K.; Braun, E.L.; Braun, M.J.; Chojnowski, J.L.; Cox, W.A.; Han, K.-L.; Harshman, J.; et al. A phylogenomic study of birds reveals their evolutionary history. Science 2008, 320, 1763–1768. [Google Scholar] [CrossRef]
- Wang, N.; Braun, E.L.; Kimball, R.T. Testing hypotheses about the sister group of the Passeriformes using an independent 30-locus data set. Mol. Biol. Evol. 2012, 29, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Kimball, R.T.; Wang, N.; Heimer-McGinn, V.; Ferguson, C.; Braun, E.L. Identifying localized biases in large datasets: A case study using the avian tree of life. Mol. Phylogenet. Evol. 2013, 69, 1021–1032. [Google Scholar] [CrossRef]
- McCormack, J.E.; Harvey, M.G.; Faircloth, B.C.; Crawford, N.G.; Glenn, T.C.; Brumfield, R.T. A phylogeny of birds based on over 1,500 loci collected by target enrichment and high-throughput sequencing. PLoS ONE 2013, 8, e54848. [Google Scholar] [CrossRef]
- Smith, J.V.; Braun, E.L.; Kimball, R.T. Ratite nonmonophyly: Independent evidence from 40 novel loci. Syst. Biol. 2013, 62, 35–49. [Google Scholar] [CrossRef]
- Jarvis, E.D.; Mirarab, S.; Aberer, A.J.; Li, B.; Houde, P.; Li, C.; Ho, S.Y.W.; Faircloth, B.C.; Nabholz, B.; Howard, J.T.; et al. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 2014, 346, 1320–1331. [Google Scholar] [CrossRef]
- Prum, R.O.; Berv, J.S.; Dornburg, A.; Field, D.J.; Townsend, J.P.; Lemmon, E.M.; Lemmon, A.R. A comprehensive phylogeny of birds (Aves) using targeted next-generation DNA sequencing. Nature 2015, 526, 569–573. [Google Scholar] [CrossRef]
- Reddy, S.; Kimball, R.T.; Pandey, A.; Hosner, P.A.; Braun, M.J.; Hackett, S.J.; Han, K.-L.; Harshman, J.; Huddleston, C.J.; Kingston, S.; et al. Why do phylogenomic data sets yield conflicting trees? Data type influences the avian tree of life more than taxon sampling. Syst. Biol. 2017, 66, 857–879. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, S.; Yeh, C.-F.; Zhang, N.; Chen, G.; Que, P.; Dong, L.; Li, S.-H. The first set of universal nuclear protein-coding loci markers for avian phylogenetic and population genetic studies. Sci. Rep. 2018, 8, 15723. [Google Scholar] [CrossRef]
- Houde, P.; Braun, E.L.; Narula, N.; Minjares, U.; Mirarab, S. Phylogenetic signal of indels and the neoavian radiation. Diversity 2019, 11, 108. [Google Scholar] [CrossRef]
- Kuhl, H.; Frankl-Vilches, C.; Bakker, A.; Mayr, G.; Nikolaus, G.; Boerno, S.T.; Klages, S.; Timmermann, B.; Gahr, M. An unbiased molecular approach using 3’UTRs resolves the avian family-level tree of life. Mol. Biol. Evol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Suh, A. The phylogenomic forest of bird trees contains a hard polytomy at the root of Neoaves. Zool. Scr. 2016, 45, 50–62. [Google Scholar] [CrossRef]
- Jeffroy, O.; Brinkmann, H.; Delsuc, F.; Philippe, H. Phylogenomics: The beginning of incongruence? Trends Genet. 2006, 22, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.-Y.; Liang, D.; Zhang, P. Phylogenomic resolution of the phylogeny of laurasiatherian mammals: Exploring phylogenetic signals within coding and noncoding sequences. Genome Biol. Evol. 2017, 9, 1998–2012. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.O.; Hutter, C.R.; Wood, P.L.; Grismer, L.L.; Brown, R.M. Larger, unfiltered datasets are more effective at resolving phylogenetic conflict: Introns, exons, and UCEs resolve ambiguities in golden-backed frogs (Anura: Ranidae; genus Hylarana). Mol. Phylogenet. Evol. 2020, 151, 106899. [Google Scholar] [CrossRef]
- Xiao, T.-W.; Xu, Y.; Jin, L.; Liu, T.-J.; Yan, H.-F.; Ge, X.-J. Conflicting phylogenetic signals in plastomes of the tribe Laureae (Lauraceae). PeerJ 2020, 8, e10155. [Google Scholar] [CrossRef]
- Zhang, J.; Lindsey, A.R.I.; Peters, R.S.; Heraty, J.M.; Hopper, K.R.; Werren, J.H.; Martinson, E.O.; Woolley, J.B.; Yoder, M.J.; Krogmann, L. Conflicting signal in transcriptomic markers leads to a poorly resolved backbone phylogeny of chalcidoid wasps. Syst. Entomol. 2020, 45, 783–802. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Y.-H.; Jin, J.-J.; Stull, G.W.; Bruneau, A.; Cardoso, D.; De Queiroz, L.P.; Moore, M.J.; Zhang, S.-D.; Chen, S.-Y.; et al. Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Syst. Biol. 2020, 69, 613–622. [Google Scholar] [CrossRef]
- Pandey, A.; Braun, E.L. Phylogenetic analyses of sites in different protein structural environments result in distinct placements of the metazoan root. Biology 2020, 9, 64. [Google Scholar] [CrossRef] [PubMed]
- Yuri, T.; Kimball, R.T.; Harshman, J.; Bowie, R.C.K.; Braun, M.J.; Chojnowski, J.L.; Han, K.-L.; Hackett, S.J.; Huddleston, C.J.; Moore, W.S.; et al. Parsimony and model-based analyses of indels in avian nuclear genes reveal congruent and incongruent phylogenetic signals. Biology 2013, 2, 419–444. [Google Scholar] [CrossRef] [PubMed]
- Mayr, G. Metaves, Mirandornithes, Strisores and other novelties—A critical review of the higher-level phylogeny of neornithine birds. J. Zool. System. 2011, 49, 58–76. [Google Scholar] [CrossRef]
- Chen, A.; Field, D.J. Phylogenetic definitions for Caprimulgimorphae (Aves) and major constituent clades under the International Code of Phylogenetic Nomenclature. Senckenberg Gesellschaft für Naturforschung 2020, 70, 571–585. [Google Scholar] [CrossRef]
- Pittman, M.; O’Connor, J.; Field, D.; Turner, A.; Ma, W.; Makovicky, P.; Xu, X. Pennaraptoran Systematics. In Pennaraptoran Theropod Dinosaurs: Past Progress and New Frontiers; Pittman, M., Xu, X., Eds.; Bulletin of the American Museum of Natural History 440: New York, NY, USA, 2020; pp. 7–36. [Google Scholar]
- Pollock, D.D.; Zwickl, D.J.; McGuire, J.A.; Hillis, D.M. Increased taxon sampling is advantageous for phylogenetic inference. Syst. Biol. 2002, 51, 664–671. [Google Scholar] [CrossRef]
- Slack, K.E.; Delsuc, F.; McLenachan, P.A.; Arnason, U.; Penny, D. Resolving the root of the avian mitogenomic tree by breaking up long branches. Mol. Phylogenet. Evol. 2007, 42, 1–13. [Google Scholar] [CrossRef]
- Shen, X.-X.; Hittinger, C.T.; Rokas, A. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nat. Ecol. Evol. 2017, 1, 126. [Google Scholar] [CrossRef]
- Suh, A.; Smeds, L.; Ellegren, H. The Dynamics of incomplete lineage sorting across the ancient adaptive radiation of neoavian birds. PLoS Biol. 2015, 13, e1002224. [Google Scholar] [CrossRef]
- Ishikawa, S.A.; Inagaki, Y.; Hashimoto, T. RY-coding and non-homogeneous models can ameliorate the maximum-likelihood inferences from nucleotide sequence data with parallel compositional heterogeneity. Evol. Bioinform. 2012, 8, 357–371. [Google Scholar] [CrossRef]
- Zhang, C.; Sayyari, E.; Mirarab, S. ASTRAL-III: Increased scalability and impacts of contracting low support branches. In Comparative Genomics; Meidanis, J., Nakhleh, L., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2017; Volume 10562, pp. 53–75. ISBN 978-3-319-67978-5. [Google Scholar]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Breathnach, R.; Benoist, C.; O’Hare, K.; Gannon, F.; Chambon, P. Ovalbumin gene: Evidence for a leader sequence in mRNA and DNA sequences at the exon-intron boundaries. Proc. Natl. Acad. Sci. USA 1978, 75, 4853–4857. [Google Scholar] [CrossRef] [PubMed]
- Maddison, D.R.; Swofford, D.L.; Maddison, W.P. NEXUS: An extensible file format for systematic information. Syst. Biol. 1997, 46, 590–621. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed]
- Chernomor, O.; von Haeseler, A.; Minh, B.Q. Terrace Aware data structure for phylogenomic inference from supermatrices. Syst. Biol. 2016, 65, 997–1008. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. Ufboot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Neyman, J. Molecular studies of evolution: A source of novel statistical problems. In Molecular Studies of Evolution: A Source of Novel Statistical Problems; Gupta, S.S., Yackel, J., Eds.; New York Academic Press: New York, NY, USA, 1971; pp. 1–27. [Google Scholar]
- Farris, J.S. A probability model for inferring evolutionary trees. Syst. Biol. 1973, 22, 250–256. [Google Scholar] [CrossRef]
- Cavender, J.A. Taxonomy with confidence. Math. Biosci. 1978, 40, 271–280. [Google Scholar] [CrossRef]
- Sayyari, E.; Mirarab, S. Fast coalescent-based computation of local branch support from quartet frequencies. Mol. Biol. Evol. 2016, 33, 1654–1668. [Google Scholar] [CrossRef]
- Bogdanowicz, D.; Giaro, K. Matching split distance for unrooted binary phylogenetic trees. IEEE/ACM Trans. Comput. Biol. Bioinform. 2011, 9, 150–160. [Google Scholar] [CrossRef]
- Lin, Y.; Rajan, V.; Moret, B.M.E. A metric for phylogenetic trees based on matching. IEEE/ACM Trans. Comput. Biol. Bioinform. 2012, 9, 1014–1022. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Suh, A.; Paus, M.; Kiefmann, M.; Churakov, G.; Franke, F.A.; Brosius, J.; Kriegs, J.O.; Schmitz, J. Mesozoic retroposons reveal parrots as the closest living relatives of passerine birds. Nat. Commun. 2011, 2, 443. [Google Scholar] [CrossRef] [PubMed]
- Tiley, G.P.; Pandey, A.; Kimball, R.T.; Braun, E.L.; Burleigh, J.G. Whole genome phylogeny of Gallus: Introgression and data-type effects. Avian Res. 2020, 11, 7. [Google Scholar] [CrossRef]
- Pamilo, P.; Nei, M. Relationships between gene trees and species trees. Mol. Biol. Evol. 1988, 5, 568–583. [Google Scholar] [CrossRef]
- Maddison, W.P. Gene trees in species trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Hill, W.G.; Robertson, A. The effect of linkage on limits to artificial selection. Genet. Res. 1966, 8, 269. [Google Scholar] [CrossRef]
- Nikolaev, S.; Montoya-Burgos, J.I.; Margulies, E.H.; NISC Comparative Sequencing Program; Rougemont, J.; Nyffeler, B.; Antonarakis, S.E. Early history of mammals is elucidated with the ENCODE multiple species sequencing data. PLoS Genet. 2007, 3, e2. [Google Scholar] [CrossRef]
- Gillung, J.P.; Winterton, S.L.; Bayless, K.M.; Khouri, Z.; Borowiec, M.L.; Yeates, D.; Kimsey, L.S.; Misof, B.; Shin, S.; Zhou, X.; et al. Anchored phylogenomics unravels the evolution of spider flies (Diptera, Acroceridae) and reveals discordance between nucleotides and amino acids. Mol. Phylogenet. Evol. 2018, 128, 233–245. [Google Scholar] [CrossRef]
- Vasilikopoulos, A.; Balke, M.; Beutel, R.G.; Donath, A.; Podsiadlowski, L.; Pflug, J.M.; Waterhouse, R.M.; Meusemann, K.; Peters, R.S.; Escalona, H.E.; et al. Phylogenomics of the superfamily Dytiscoidea (Coleoptera: Adephaga) with an evaluation of phylogenetic conflict and systematic error. Mol. Phylogenet. Evol. 2019, 135, 270–285. [Google Scholar] [CrossRef]
- Vasilikopoulos, A.; Gustafson, G.T.; Balke, M.; Niehuis, O.; Beutel, R.G.; Misof, B. Resolving the phylogenetic position of Hygrobiidae (Coleoptera: Adephaga) requires objective statistical tests and exhaustive phylogenetic methodology: A response to Cai et al. (2020). Mol. Phylogenet. Evol. 2020, in press, corrected proof. [Google Scholar] [CrossRef]
- Braun, E.L.; Kimball, R.T. Examining Basal avian divergences with mitochondrial sequences: Model complexity, taxon sampling, and sequence length. Syst. Biol. 2002, 51, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Delsuc, F.; Phillips, M.J.; Penny, D. Comment on “Hexapod origins: Monophyletic or paraphyletic?”. Science 2003, 301, 1482. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Phillips, M.J.; Penny, D. The root of the mammalian tree inferred from whole mitochondrial genomes. Mol. Phylogenet. Evol. 2003, 28, 171–185. [Google Scholar] [CrossRef]
- Vera-Ruiz, V.A.; Lau, K.W.; Robinson, J.; Jermiin, L.S. Statistical tests to identify appropriate types of nucleotide sequence recoding in molecular phylogenetics. BMC Bioinform. 2014, 15 (Suppl. S2), S8. [Google Scholar] [CrossRef]
- Galtier, N.; Gouy, M. Inferring pattern and process: Maximum-likelihood implementation of a nonhomogeneous model of DNA sequence evolution for phylogenetic analysis. Mol. Biol. Evol. 1998, 15, 871–879. [Google Scholar] [CrossRef]
- Hannaford, N.E.; Heaps, S.E.; Nye, T.M.; Williams, T.A.; Embley, T.M. Incorporating compositional heterogeneity into Lie Markov models for phylogenetic inference. arXiv 2020, arXiv:2007.08511. [Google Scholar]
- Blair, C.; Murphy, R.W. Recent trends in molecular phylogenetic analysis: Where to next? J. Hered. 2011, 102, 130–138. [Google Scholar] [CrossRef]
- Kainer, D.; Lanfear, R. The effects of partitioning on phylogenetic inference. Mol. Biol. Evol. 2015, 32, 1611–1627. [Google Scholar] [CrossRef]
- Patel, S.; Kimball, R.T.; Braun, E.L. Error in phylogenetic estimation for bushes in the tree of life. J. Phylogenet. Evol. Biol. 2013, 1, 110. [Google Scholar] [CrossRef]
- Roch, S.; Warnow, T. On the robustness to gene tree estimation error (or lack thereof) of coalescent-based species tree methods. Syst. Biol. 2015, 64, 663–676. [Google Scholar] [CrossRef]
- Springer, M.S.; Gatesy, J. The gene tree delusion. Mol. Phylogenet. Evol. 2016, 94, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Ericson, P.G.P. Evolution of terrestrial birds in three continents: Biogeography and parallel radiations. J. Biogeogr. 2012, 39, 813–824. [Google Scholar] [CrossRef]
- Houde, P.; Braun, E.L.; Zhou, L. Deep-time demographic inference suggests ecological release as driver of neoavian adaptive radiation. Diversity 2020, 12, 164. [Google Scholar] [CrossRef]
- Claramunt, S.; Cracraft, J. A new time tree reveals Earth history’s imprint on the evolution of modern birds. Sci. Adv. 2015, 1, e1501005. [Google Scholar] [CrossRef] [PubMed]
- Cracraft, J.; Houde, P.; Ho, S.Y.W.; Mindell, D.P.; Fjeldså, J.; Lindow, B.; Edwards, S.V.; Rahbek, C.; Mirarab, S.; Warnow, T.; et al. Response to Comment on “Whole-genome analyses resolve early branches in the tree of life of modern birds”. Science 2015, 349, 1460. [Google Scholar] [CrossRef] [PubMed]
- Kimball, R.T.; Oliveros, C.H.; Wang, N.; White, N.D.; Barker, F.K.; Field, D.J.; Ksepka, D.T.; Chesser, R.T.; Moyle, R.G.; Braun, M.J.; et al. A phylogenomic supertree of birds. Diversity 2019, 11, 109. [Google Scholar] [CrossRef]
- Xu, B.; Yang, Z. Challenges in species tree estimation under the multispecies coalescent model. Genetics 2016, 204, 1353–1368. [Google Scholar] [CrossRef]
- Zhang, G.; Li, C.; Li, Q.; Li, B.; Larkin, D.M.; Lee, C.; Storz, J.F.; Antunes, A.; Greenwold, M.J.; Meredith, R.W.; et al. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 2014, 346, 1311–1320. [Google Scholar] [CrossRef]
- Stiller, J.; Zhang, G. Comparative phylogenomics, a stepping stone for bird biodiversity studies. Diversity 2019, 11, 115. [Google Scholar] [CrossRef]
- Feng, S.; Stiller, J.; Deng, Y.; Armstrong, J.; Fang, Q.; Reeve, A.H.; Xie, D.; Chen, G.; Guo, C.; Faircloth, B.C.; et al. Dense sampling of bird diversity increases power of comparative genomics. Nature 2020, 587, 252–257. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Type | Sites | %A | %C | %G | %T | %GC |
|---|---|---|---|---|---|---|
| Coding data | ||||||
| Exon c1 | Invariant | 29.6 | 21.1 | 31.4 | 17.9 | 52.5 |
| Informative | 31.0 | 26.4 | 24.7 | 17.9 | 51.1 | |
| Exon c2 | Invariant | 33.3 | 23.7 | 15.9 | 27.0 | 39.6 |
| Informative | 31.0 | 29.1 | 22.4 | 17.5 | 51.5 | |
| Exon c3 | Invariant | 17.0 | 21.6 | 43.4 | 17.9 | 65.0 |
| Informative | 23.8 | 26.0 | 22.4 | 27.8 | 48.4 | |
| Non-coding data | ||||||
| Intron | Invariant | 29.0 | 14.6 | 21.7 | 34.7 | 36.3 |
| Informative | 26.4 | 19.9 | 20.5 | 33.2 | 40.4 | |
| UTR | Invariant | 26.6 | 16.4 | 20.3 | 36.6 | 36.7 |
| Informative | 29.1 | 19.4 | 22.0 | 29.5 | 41.4 | |
| Intergenic | Invariant | 31.3 | 18.8 | 22.0 | 27.8 | 40.8 |
| Informative | 30.2 | 20.8 | 21.3 | 27.6 | 42.1 |
| RY (AG-CT) | SW (GC-AT) | KM (GT-AC) | ||||
|---|---|---|---|---|---|---|
| Data Type | Inf Sites 1 | Variation 2 | Inf Sites 1 | Variation 2 | Inf Sites 1 | Variation 2 |
| Coding exons | ||||||
| Exon c1 | 11,374 | 0.39% | 23,325 | 1.29% | 21,170 | 0.37% |
| Exon c2 | 6958 | 0.32% | 13,774 | 1.17% | 14,531 | 0.35% |
| Exon c3 | 37,673 | 0.56% | 87,299 | 2.56% | 87,755 | 0.32% |
| Non-coding data | ||||||
| Introns | 22,510 | 0.25% | 31,014 | 0.73% | 30,780 | 0.29% |
| UTRs | 7654 | 0.40% | 12,522 | 1.23% | 12,383 | 0.32% |
| Intergenic | 310 | 1.90% | 648 | 3.24% | 638 | 1.28% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Braun, E.L.; Kimball, R.T. Data Types and the Phylogeny of Neoaves. Birds 2021, 2, 1-22. https://doi.org/10.3390/birds2010001
Braun EL, Kimball RT. Data Types and the Phylogeny of Neoaves. Birds. 2021; 2(1):1-22. https://doi.org/10.3390/birds2010001
Chicago/Turabian StyleBraun, Edward L., and Rebecca T. Kimball. 2021. "Data Types and the Phylogeny of Neoaves" Birds 2, no. 1: 1-22. https://doi.org/10.3390/birds2010001
APA StyleBraun, E. L., & Kimball, R. T. (2021). Data Types and the Phylogeny of Neoaves. Birds, 2(1), 1-22. https://doi.org/10.3390/birds2010001