Abstract
Intrahepatic cholangiocarcinoma (iCCA) is a highly aggressive and heterogeneous malignancy characterized by marked resistance to standard chemotherapy and poor prognosis. While the advent of immunotherapy has revolutionized the management of several solid tumors, including melanoma, breast cancer, and non-small cell lung cancer, its efficacy in iCCA remains limited. Recent clinical trials have demonstrated the efficacy of durvalumab in combination with chemotherapy for iCCA, leading to its approval as a first-line treatment. However, overall response rates remain low, largely due to its immunosuppressive tumor immune microenvironment (TIME). The immune-cold nature of iCCA is typified by a dominant presence of immunosuppressive cell populations, including M2-polarized tumor-associated macrophages, myeloid-derived suppressor cells, and T regulatory cells. In addition, traditional biomarkers such as PD-L1 expression, tumor mutational burden, and microsatellite instability have shown limited predictive value in iCCA, highlighting the need for novel biomarkers and immunotherapeutic strategies. Emerging approaches aimed at reprogramming the TIME, including combination therapies targeting suppressive cells, stromal remodeling, and novel immune effectors like CAR-T and cancer vaccines, hold significant promise for enhancing therapeutic efficacy. This review summarizes the distinct features of iCCA TIME, key mechanisms of immune evasion, current challenges, and future directions to overcome immune resistance, with the aim of developing personalized immunotherapies to improve patient outcomes.
1. Introduction
Immunotherapy has transformed the landscape of cancer treatment, emerging as a key therapeutic approach that exploits the immune system’s innate ability to recognize and eliminate malignant cells [1]. Remarkable improvements in survival outcomes have been achieved, and immunotherapy is now established as standard of care in several malignancies, including melanoma, non-small cell lung cancer, and renal cell carcinoma [2,3,4]. However, the proportion of patients who experiences durable responses remains limited. Only about one-third of patients receiving immune checkpoint inhibitors (ICIs) achieves long-term benefit, while many either fail to respond initially or relapse after an initial response [1,5].
The rationale behind immunotherapy is to counteract the immune evasion mechanisms adopted by tumors to escape immune surveillance [6,7]. This approach promotes the activation and expansion of tumor-specific T cells, supporting sustained cytotoxic activity and long-term immunological memory. Immune checkpoint blockade (ICB) is one of the most effective strategies, targeting inhibitory molecules such as programmed cell death protein 1 (PD-1), programmed cell death ligand 1 (PD-L1), and cytotoxic T-lymphocyte protein 4 (CTLA-4) upregulated by tumors to suppress immune responses. By inhibiting these pathways, ICB restores T-cell function and enhances antitumor immunity [8].
Response to ICB is strongly influenced by the tumor immune microenvironment (TIME), whose complexity and heterogeneity significantly affects both tumor prognosis and treatment outcome [9]. Even immunologically “hot” tumors may respond poorly due to adaptive resistance. Tumors enriched in CD8+ T cells, expressing high levels of PD-L1 and chemokines that promote T cell infiltration, tend to respond better to ICB. In contrast, “cold” tumors, lacking key chemokines and innate immune activation, are characterized by poor T cell infiltration and immune exclusion [10,11].
Furthermore, the lack of reliable predictive biomarkers for ICIs response complicates patient selection. Even though high tumor mutational burden (TMB), microsatellite instability-high (MSI-H) status and high antigenicity have been associated with improved ICB response, these traits are inconsistently predictive across different tumor types [5,12]. Emerging findings suggest that additional factors, such as the composition of the TME, tumor-infiltrating lymphocytes (TILs), neutrophil–lymphocyte ratio, and the gut microbiome, may contribute to response variability, although these have yet to enter routine clinical practice [13].
Among solid tumors, intrahepatic cholangiocarcinoma (iCCA), a highly aggressive and heterogeneous malignancy originating from the biliary epithelium, emerges as one of the most difficult to eradicate and represents a significant therapeutic challenge [14]. The overall efficacy of ICIs in iCCA has been limited, underscoring the urgent need to better understand its TIME and develop novel predictive biomarkers and combination strategies to improve patient outcomes. This review provides an overview of immunotherapy in iCCA, focusing on its immune landscape, resistance mechanisms, and emerging therapeutic strategies.
2. Current Immunotherapy Landscape in iCCA
CCA is the second most frequent primary liver cancer, with a rising incidence worldwide [15]. Its silent and asymptomatic nature, particularly in early stages, combined with pronounced molecular heterogeneity, often results in delayed diagnosis [16,17]. These factors, along with its high chemoresistance, contribute to a poor overall prognosis. Surgical resection remains the only potentially curative option, but only 20–30% of patients are eligible at diagnosis [16,18,19].
Although the high heterogeneity of iCCA limits the success of standardized therapies, it also offers novel opportunities for personalized therapy. In recent years, advances in molecular profiling and immuno-oncology have indeed paved the way to new potential therapeutic approaches, with targeted therapies and immunotherapy beginning to be included in practical guidelines [19].
Concerning immunotherapy, recent clinical trials have shown the effectiveness of durvalumab, a PD-L1 inhibitor, when used in combination with chemotherapy, leading to its approval as a first-line treatment for iCCA [20]. Particularly, the TOPAZ-1 study demonstrated that addition of durvalumab to standard gemcitabine and cisplatin (GemCis) improved overall survival (OS) in patients with advanced biliary tract cancer (BTC) compared to placebo plus GemCis. Recent results from TOPAZ-1 continue to support this regimen as a first-line standard of care, showing sustained benefits for over 3 years, along with tolerable side effects [21,22]. Similarly, the KEYNOTE-966 phase III clinical trial evaluated the efficacy of pembrolizumab (anti–PD-1) combined to GemCis, further validating the modest but consistent advantages of adding ICIs to chemotherapy in unresectable or metastatic BTC [12,23]. However, ICIs have shown limited efficacy in BTC and particularly iCCA, with significant clinical responses limited to a small subgroup of patients [24,25,26].
3. Immunosuppressive Nature and Immune Cell Profile in iCCA
The TIME plays a critical role in the development, progression, and therapeutic resistance of iCCA. As observed in other solid tumors, the TIME in iCCA is predominantly immunosuppressive and limits effective antitumor immune responses. This suppression is driven by low infiltration of cytotoxic lymphocytes and the accumulation of immunoregulatory populations such as regulatory T cells (Tregs), tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), alongside cancer-associated fibroblasts (CAFs) and extensive desmoplasia, which collectively create a hostile environment for effective antitumor responses (Figure 1) [27,28,29].

Figure 1.
Immune landscape of iCCA. The TIME of iCCA is shaped by the recruitment, activation, and functional reprogramming of diverse immune cell populations. Tumor-derived chemokines and cytokines, along with innate immune evasion mechanisms (e.g., immune checkpoint upregulation, reduced antigen presentation), contribute to the polarization of tumor-associated macrophages (TAMs) toward an M2-like pro-tumor phenotype, suppression of cytotoxic CD8+ T and NK cell activity, recruitment of regulatory CD4+ T cells (Tregs), functional impairment of dendritic cells (DCs), and accumulation of immature myeloid-derived suppressor cells (MDSCs). Neutrophils display phenotypic plasticity, exhibiting both N2 (pro-tumor) and N1 (antitumor) polarization depending on context. Crosstalk among immune subsets reinforces the immunosuppressive milieu, collectively leading to an immune-cold TIME. NPM: negative prognostic marker.
Immune profiling studies consistently describe iCCA as having an “immune-cold” or “immune-excluded” phenotype, marked by limited CD8+ T cell infiltration, a dense extracellular matrix, and a predominance of immunosuppressive signaling pathways [28,30,31]. Beyond these extrinsic barriers, these tumors also exploit intrinsic immune escape mechanisms, such as the activation of inhibitory molecular pathways and tumor-specific genetic or epigenetic alterations, which reinforce the immunosuppressive TIME [32]. These features likely underlie the limited clinical efficacy of ICI when used as monotherapy in unselected iCCA patients. Moreover, the TIME of iCCA is highly heterogeneous, both in cellular composition and functional status, supporting the need for precision strategies that align therapeutic interventions with the specific immune contexture of the tumor [28].
Based on the immune composition, iCCA tumors have been classified into four immune subtypes, each associated with different immune evasion mechanisms and clinical outcomes [33]. The “immune desert” subtype, the most prevalent, shows low and inactive immune cell infiltration that correlates with poor prognosis. Conversely, the inflamed “lymphoid” subtype displays high infiltration of T lymphocytes and activation of the PD-1/PD-L1 axis, suggesting greater ICB sensibility. The “myeloid” subtype is characterized by abundant MDSCs and TAMs, contributing to a highly immunosuppressive milieu. Lastly, the “mesenchymal” subtype, rich in CAFs and desmoplasia, impairs T cell trafficking and activation [34,35,36].
In line with this classification, a recent study applied cutting-edge spatial transcriptomics combined with advanced AI-based analysis of histology slides (n = 339) to classify BTCs into three distinct immune phenotypes based on TIL profile: inflamed, immune-excluded, and immune-desert. The inflamed phenotype, enriched in CD8+ T cells and interferon gamma (IFN-γ) signaling, correlated with better responses to ICIs, longer progression-free survival (PFS) (median 4.5 vs. 1.9 months), and improved OS (median 12.6 vs. 5.1 months) compared to non-inflamed phenotypes. In contrast, immune-excluded and immune-desert phenotypes generally exhibit resistance to ICIs, likely due to the lack of T cell infiltration within the tumor core [37].
Understanding the dynamic composition of the TIME is essential for designing personalized immunotherapeutic strategies. The distinct immune phenotypes observed in iCCA suggest that immune evasion is multifactorial, involving both structural and cellular barriers. In the following sections, we will provide an in-depth overview of the major immune cell subsets within the TIME in iCCA, highlighting their roles in immune evasion and their potential as therapeutic targets.
3.1. Tumor-Associated Macrophages
TAMs constitute the most abundant infiltrating immune cell population in the iCCA microenvironment [31,38]. They exhibit remarkable plasticity and typically adopt an M2-like phenotype that promotes tumor progression and dampens antitumor immunity [28]. Their recruitment and polarization are driven by multiple tumor-derived factors, including cytokines such as interleukin (IL)-13, IL-34, and osteoactivin, all of which have been found to be elevated in patient serum [39]. Additional soluble factors like Trefoil Factor 3 (TFF3) and the polyunsaturated fatty acid 5-lipoxygenase–leukotriene B4 (ALOX5–LTB4) axis have also been implicated in driving the differentiation of monocytes toward a pro-tumoral macrophage phenotype [38,40]. Among other modulators, dickkopf-1 (DKK1) overexpression in tumor cells promotes an MHCIIlow TAM2-like program, characterized by reduced antigen-presenting capacity and increased expression of M2-associated immunosuppressive markers, thereby reinforcing a tolerogenic niche [41]. Once polarized, TAMs facilitate tumor proliferation, invasion, epithelial-to-mesenchymal transition (EMT), and resistance to therapy, partly via IL-10-mediated signal transducer and activator of transcription (STAT)3 activation and oncostatin M-driven Yes-associated protein 1–inhibitor of DNA binding 1 (YAP–ID1) signaling [38,42,43]. Importantly, TAMs are the main PD-L1-expressing population in the TIME, underscoring their direct role in immune suppression, as demonstrated in PD-L1−/− murine models [44].
Beyond tumor cell interactions, TAMs shape the immunosuppressive niche through spatial and molecular crosstalk with other stromal and immune elements. Co-localization with tumor-associated neutrophils (TANs) enhances STAT3 signaling in malignant cells via oncostatin M and IL-11, thereby enhancing tumor aggressiveness [45]. Single-cell and spatial transcriptomic analyses identified secreted phosphoprotein 1 (SPP1)+ TAMs at the invasive leading edge, where they engage CD44 on malignant cells to enhance stem-like properties, proliferation, and EMT programs, ultimately driving tumor aggressiveness and immune evasion [46]. In addition, SPP1+ TAMs cooperate with fibroblast activation protein (FAP)+ CAFs through adrenomedullin–calcitonin receptor-like receptor/receptor activity-modifying protein 1 (ADM–CALCRL/RAMP1) signaling to sustain angiogenesis and reinforce immune suppression [47]. In macrophage-rich iCCA tumors, macrophage receptor MARCO (a class A scavenger receptor)+ TAMs predominate, and transcriptional programs associated with hypoxia, angiogenesis and EMT. Their co-localization with cathepsin E+ tumor cells and activation by galectin 9–CD44 signaling reinforces immunosuppressive circuits [48].
TAMs are increasingly recognized as therapeutic targets in iCCA due to their central role in tumor maintenance and immunosuppression. High TAM infiltration and expression of markers such as signal regulatory protein alpha (SIRPα), PD-1, and MARCO are correlated with poor prognosis, early recurrence, and reduced survival [48,49]. Notably, dual expression of SIRPα and PD-1 identifies a subgroup with particularly poor outcomes, suggesting therapeutic potential for dual checkpoint blockade [49].
Despite their relevance, TAM-targeted monotherapies have encountered significant limitations. Pharmacologic inhibition of macrophage colony-stimulating factor 1 receptor (CSF1R), a key regulator of macrophage survival and differentiation, has shown limited benefit in preclinical iCCA models, likely due to compensatory mechanisms, such as the recruitment of G-MDSCs or activation of alternative pathways [31,44]. While promising results have emerged from early trials in other cancers (e.g., breast, renal, melanoma), no TAM-directed therapies have yet entered clinical testing in iCCA [50].
To overcome these limitations, combinatorial approaches are under investigation. These include CSF1R inhibitors, ALOX5 blockade, C-C chemokine receptor type 2 (CCR2) antagonists, and dual checkpoint inhibition targeting CD47-SIRPα and PD-1-PD-L1 axes [44,49,51]. Monoclonal antibodies against MARCO or agents that block soluble CD109, a factor that reprograms TAMs toward a CD73+ suppressive phenotype, are also in development [48,52]. Furthermore, TAM-derived IL-10/STAT3 and oncostatin M signaling remain attractive targets to inhibit EMT and enhance therapeutic resistance [42,43].
In summary, TAMs are central regulators of immunosuppression, immune escape, and therapeutic resistance in iCCA. While targeting these cells holds therapeutic promise, success will likely require integrated strategies that modulate both TAMs and other immunosuppressive elements in the iCCA microenvironment.
3.2. Tumor-Infiltrating Lymphocytes
TILs represent a diverse population of immune cells that migrate into tumor tissues and exert critical roles in modulating antitumor immunity. They include CD8+ cytotoxic T lymphocytes, CD4+ helper T cells, Treg, and CD20+ B cells, which dynamically interact with tumor and stromal cells to influence disease progression and treatment outcomes [28,53]. Among these, T cells constitute the predominant lymphoid population in solid tumors. Their balance between effector and regulatory phenotypes critically determines the strength and quality of antitumor immunity [28,54]. The composition, functional state, and spatial distribution of TILs, especially T cells, in the TIME have been associated with prognosis and therapeutic response, including to ICIs [54,55].
3.2.1. CD8+ Cytotoxic T Lymphocytes
CD8+ T cells are pivotal effectors of antitumor immunity, capable of recognizing and eliminating malignant cells via major histocompatibility complex (MHC) class I and the release of perforins, granzymes, and pro-inflammatory cytokines (e.g., IFN-γ, tumor necrosis factor alpha (TNF-α)) [55,56]. In iCCA, elevated CD8+ infiltration has been consistently associated with favorable clinical outcomes, including prolonged OS and reduced recurrence risk [54,57].
However, within the tumor milieu, CD8+ T cell function is often compromised due to multiple immune evasion strategies employed by cancer cells. These include defective antigen presentation, upregulation of immunosuppressive ligands (e.g., PD-L1), and release of extracellular vesicles that alter T cell activity [56]. Single-cell transcriptomic studies have identified a subset of TAMs expressing CD5L (apoptosis inhibitor expressed by macrophages) that co-localizes with granzyme B+ CD8+ T cells. Through C-X-C motif chemokine 12–C-X-C chemokine receptor type 4 (CXCL12–CXCR4)/STAT signaling, these TAMs induce CTLA4 upregulation, leading to CD8+ T cell exhaustion and loss of cytotoxic function, independent of PD-1 [58]. Spatial analyses further revealed that tumor-intrinsic doublecortin domain-containing 2/enolase 1 (DCDC2/ENO1) activity enhances fibrinogen-like protein 1 (FGL1) expression, which binds to lymphocyte activation gene 3 (LAG-3) on CD8+ T cells and suppresses granzyme B and IFN-γ secretion, thereby further weakening their cytotoxicity and reinforcing immune evasion [59].
Phenotypic and spatial heterogeneity also shape CD8+ T cell responses in iCCA. At the invasive margin, these cells often exhibit a naive or early activated phenotype, with limited cytotoxicity and minimal exhaustion, potentially due to inadequate local antigen presentation. In contrast, the tumor core tends to harbor more exhausted and differentiated CD8+ T cells, often co-localized with immunosuppressive regulatory T cells [46]. Of relevance is the presence of a CD69+CD103+ tissue-resident memory (TRM)-like CD8+ T cell subset in iCCA tumors, which has been linked to inflamed transcriptional profiles and better response to ICIs [60].
Taken together, these findings highlight the need for deeper characterization of the phenotypic and functional states of CD8+ TILs in iCCA. Efforts to boost CD8+ T cell-mediated immunity are central to current immunotherapeutic strategies. Despite the limited efficacy of ICIs monotherapy in iCCA, combination approaches aiming to enhance CD8+ T cell infiltration, restore effector function, and reverse exhaustion are under active investigation [53,56]. These include dual targeting of myeloid-driven immunosuppression (e.g., TAMs, MDSCs), inhibition of CXCR2 to limit neutrophil recruitment, or inhibition of β-catenin signaling to improve CD8+ T cell access to the tumor [44,50]. Additionally, engineered DCs (dendritic cells) that promote antigen presentation and enhance CD8+ T cell priming have shown encouraging results in preclinical models [61].
Further clinical efforts focus on immune checkpoint inhibitors (anti-PD-1, anti-CTLA-4, anti-T-cell immunoreceptor with Ig and ITIM domains (TIGIT), which, despite limited efficacy as monotherapies, form the foundation of current combination regimens [31,62]. Notably, nivolumab with gemcitabine/cisplatin has shown an ORR of 55.6% in advanced BTC, while GOLP therapy (gemcitabine, oxaliplatin, lenvatinib, and anti-PD1) enhances CD8+ T cell activation and clonal expansion [63,64]. Adoptive cell therapies, including (chimeric antigen receptor T-cell) CAR-T targeting mucin 1 (MUC1), epidermal growth factor receptor (EGFR), glypican-3 (GPC3), or CD276 antigen (B7-H3), are under evaluation, with multiplex genome editing strategies improving efficacy in preclinical iCCA models [51,63,64]. In parallel, neoantigen-specific TIL transfer has shown promise in metastatic CCA [64].
Remodeling the TIME is equally critical to support CD8+ T cell function. Approaches include enhancing DC activity (e.g., Fms-related tyrosine kinase 3 ligand (Flt3L), polyinosinic:polycytidylic acid (poly(I:C)), depleting immunosuppressive MDSCs and TAMs (e.g., anti-CSF1R, CXCR2 blockade), and targeting CAFs with FAP-directed vaccines or CAR-T [51,65,66,67]. Bispecific inhibitors that simultaneously block PD-L1 and transforming growth factor β (TGF-β) are also being tested, offering a multi-pronged strategy to boost CD8+ T cell infiltration and cytotoxicity in iCCA [67].
3.2.2. Natural Killer Cells
Natural killer (NK) cells are innate cytotoxic lymphocytes capable of eliminating malignant or virus-infected cells without prior antigen sensitization. Unlike cytotoxic T cells, they lack T-cell receptors (TCRs) and CD3 expression, relying instead on a balance between activating and inhibitory receptors to detect cellular abnormalities. Once activated, NK cells mediate cytotoxicity through the release of perforin and granzymes, engagement of death receptors (e.g., TNF-related apoptosis-inducing ligand (TRAIL), Fas ligand (FasL)), and secretion of pro-inflammatory cytokines such as IFN-γ and TNF-α, thereby contributing to both direct tumor clearance and immune modulation [68,69].
In the liver, NK cells constitute up to 30–40% of resident lymphocytes and are thought to play a key role in immune surveillance [70]. However, in solid tumors their intratumoral density is low, and their effector functions are frequently impaired. Factors such as TGF-β, MDSCs, and altered chemokine gradients contribute to NK cell exclusion and functional exhaustion. Notably, in cancers such as renal cell carcinoma and head and neck squamous cell carcinoma, the presence of intratumoral NK cells has been associated with improved prognosis, although this effect appears to be context-dependent [69].
In iCCA the role of NK cells remains insufficiently defined. Single-cell transcriptomic analyses have revealed reduced infiltration of NK and T cells within the tumor core, consistent with an immune-excluded phenotype [31]. Although cytotoxicity-associated transcripts are detectable, these likely reflect residual or functionally exhausted NK subsets, rather than active cytotoxic cells [69]. An inverse correlation between NK cell abundance and MDSCs infiltration has been described in both preclinical models and patient samples, suggesting that MDSCs may play a key role in suppressing NK cell activity [63,69,71].
Additional mechanisms of NK cell dysfunction reinforce immune evasion in iCCA. Oliviero et al. showed that tumor cells shed the activating ligands MHC class I polypeptide-related sequence A/B (MICA/B), thereby impairing recognition by the NK cell receptor D (NKG2D) receptor and reducing NK-mediated cytotoxicity. Although circulating NK cells were found to be increased in patients, these exhibited downregulated expression of NKG2D, while tumor-infiltrating NK cells also showed reduced levels of DNAX accessory molecule 1 (DNAM-1) and CD69, indicating a functionally impaired phenotype. Importantly, treatment with the 7C6 monoclonal antibody restored IFN-γ production and NK cell cytotoxicity in preclinical models, highlighting a promising immunotherapeutic strategy [63].
Therapeutic interest in NK cells is growing, due to their inherent cytotoxicity, low risk of autoimmune toxicity, and amenability to allogeneic use or genetic modification. Multiple strategies are under evaluation to enhance NK cell antitumor function. These include ICIs targeting NK-specific receptors, such as killer-cell immunoglobulin-like receptor (KIR), NK cell receptor A (NKG2A), or TIGIT. Notably, the anti-TIGIT antibody ociperlimab is in phase II clinical trials for BTCs, including iCCA (NCT05023109) [51]. In addition to ICI-based approaches, monoclonal antibodies that block MICA/B shedding or enhance antibody-dependent cellular cytotoxicity are being explored. For instance, the 7C6 antibody has shown efficacy in restoring NK effector function in iCCA models [63]. Furthermore, adoptive transfer therapies based on allogeneic or chimeric antigen receptor-engineered NK cells are gaining traction. Chimeric antigen receptor Natural Killer cells (CAR-NK) cells targeting B7-H3 have demonstrated preclinical activity in CCA organoid models [64], and early-phase clinical trials are evaluating allogeneic NK cell infusions, alone or in combination with ICIs, in BTC [28]. Compared to CAR-T, CAR-NK therapies offer a favorable safety profile, with lower risk of cytokine release syndrome and neurotoxicity [51]. Other immunomodulatory approaches include the use of cytokine-based regimens with IL-15, IL-2, or interferon alpha (IFN-α) to enhance NK activation and proliferation. Although these strategies are still under clinical development, they represent complementary avenues to potentiate NK-mediated immunity in iCCA [51,72].
In summary, NK cells are emerging as compelling therapeutic targets in iCCA, but their function is frequently compromised by an immunosuppressive microenvironment, ligand shedding, and poor infiltration. Therapeutic interventions aimed at restoring NK cell activity, ranging from antibody-based blockade and cytokine therapy to adoptive cell transfer and CAR-NK constructs, offer promising opportunities to enhance tumor surveillance and improve outcomes in this aggressive cancer.
3.2.3. Regulatory T Cells
Tregs are a specialized subset of CD4+ T lymphocytes characterized by the expression of CD25, CTLA-4, and the transcription factor forkhead Box P3 (FOXP3) [73]. They are essential for maintaining peripheral tolerance and immune homeostasis, limiting excessive inflammation and autoimmunity. However, within the TIME, Tregs acquire highly immunosuppressive properties, dampening effector T cell responses and facilitating immune escape [74].
In iCCA, Tregs are consistently enriched in both tumor and peritumoral compartments, and their accumulation correlates with adverse clinical outcomes [31,54,74,75]. In particular, a high intratumoral FOXP3+/CD8+ T cell ratio (FCR) has been identified as a negative prognostic marker, correlating with shorter OS. In contrast, patients with a low FCR, indicative of robust CD8+ cytotoxic T cell infiltration and limited Tregs presence, tend to show more favorable prognosis [74,75]. Additionally, an elevated Treg/CD8+ ratio have been implicated in resistance to ICIs, highlighting the dominance of the Treg compartment in maintaining an immunosuppressive TIME [74].
Recent single-cell studies have revealed that intratumoral Tregs engage in extensive ligand-receptor interactions with myeloid and T cells to consolidate their suppressive phenotype [30]. For example, CD80/CD86 expressed on myeloid cells interact with CTLA4 on Tregs, inhibiting effector T cell activation. Co-stimulatory pathways, such as OX40–OX40L, also support the persistence of activated Tregs within the tumor. Moreover, inhibitory interactions such as CD200–CD200R1 and SIRPG–CD47, with ligands on intratumoral Tregs and receptors on myeloid cells, suppress inflammatory signaling and promote the establishment of an immunosuppressive niche [30,54].
While systemic Tregs depletion may restore antitumor immunity, it also carries a high risk of immune-related toxicities. Therefore, current strategies aim to selectively target tumor-infiltrating Tregs, by disrupting immunosuppressive pathways or modulating Tregs-specific mechanisms [74]. One such target is beta-galactoside-binding lectin L-14-I (LGALS1), the gene encoding galectin-1, which has been shown to be overexpressed in FOXP3+ Tregs in iCCA. High densities of galectin-1+ Tregs correlate with reduced tertiary lymphoid structures (TLS), elevated circulating galectin-1, and worse prognosis. Pharmacological blockade of galectin-1 has shown to reduce tumor burden, diminish Tregs infiltration, and enhance CD8+ T cell activity in preclinical models [76]. Another promising target is mesenchyme homeobox 1 (MEOX1), a transcription factor that reprograms Tregs toward a hyperactivated and suppressive phenotype associated with poor clinical outcome. Inhibition of MEOX1 or its downstream signaling may help restore antitumor immunity in iCCA [30]. In addition, TIGIT-expressing Tregs can interact with poliovirus receptor (PVR) on tumor cells to reinforce immune suppression. Disruption of the TIGIT-PVR axis may alleviate Treg-mediated suppression and potentiate antitumor T cell responses [77]. Additional targets under investigation include surface markers such as CD25 and C-C chemokine receptor type 8 (CCR8), as well as intracellular regulators involved in metabolic and TCR signaling pathways [73].
Combinatorial strategies that deplete or reprogram Tregs while simultaneously activating effector T cells through checkpoint blockade are currently under investigation. These approaches aim to overcome local immune suppression while minimizing systemic toxicity and may hold the key to improving the efficacy of immunotherapy in iCCA and other poorly immunogenic tumors [54,73].
3.3. Tumor-Associated Neutrophils
As the most abundant leukocytes in circulation, neutrophils are central effectors of the innate immune response. Traditionally considered short-lived cells involved in antimicrobial defense, they are now increasingly recognized as active regulators of tumor biology [78]. TANs, defined as neutrophils that infiltrate the tumor microenvironment, exhibit remarkable phenotypic plasticity and functional heterogeneity. Depending on the surrounding signals, TANs can exert either antitumor or protumor activities.
TANs may adopt an N1 phenotype, characterized by high cytotoxicity, antigen-presenting ability, and support of adaptive immunity, or into a N2 phenotype, associated with immunosuppression, angiogenesis, and metastasis progression. This functional dichotomy is shaped by tumor-derived factors: type I interferons promote N1 polarization, whereas TGF-β, typically enriched in tumors, favors the N2 phenotype and suppresses antitumor immunity. In preclinical models, TGF-β blockade restored N1 polarization and suppressed tumor growth [78,79].
In various cancers, including lung, breast, and liver tumors, increased TAN infiltration correlates with poor prognosis [79]. In iCCA, however, their role remains ambiguous and likely context dependent. A recent study showed that iCCA tumors with greater infiltration of tumor-associated endothelial cells exhibit increased neutrophil accumulation, leading to a pro-inflammatory yet immunosuppressive microenvironment associated with worse outcomes [31]. In contrast, evidence also supports a potential antitumor role for TANs. In a syngeneic murine iCCA model, Sugahara et al. demonstrated that TANs exert direct cytotoxicity via reactive oxygen species (ROS) and that their depletion accelerated tumor progression. Furthermore, enhancing TAN activity with recombinant human granulocyte colony-stimulating factor (G-CSF) promoted their maturation and synergized with gemcitabine/cisplatin chemotherapy, leading to tumor growth suppression [80]. These findings suggest that under specific immune contexts, TANs may contribute to tumor control.
Clinically, high CD15 expression, a neutrophil marker, has been associated with shorter OS and disease-free survival in both iCCA and eCCA, highlighting a negative prognostic role for TANs [81]. Additionally, the neutrophil-to-lymphocyte ratio (NLR) has consistently correlated with poor clinical outcomes in CCA, supporting its use as a non-invasive biomarker for risk stratification [82].
Given their dual role and immunomodulatory potential, TANs are gaining attention as a therapeutic target in iCCA. As monotherapy with ICIs yields limited responses (<10%), combination therapies are under active investigation [28,44]. Dual blockade of TAMs and G-MDSCs/TANs has been shown to improve PD-1 blockade efficacy and prolong survival in preclinical iCCA models [44]. More strikingly, a triple combination of tRNA (guanine-N(7)-)-methyltransferase (METTL1) depletion, CXCR2 inhibition, and PD-1 blockade nearly eradicated tumors in mice, with minimal toxicity. This approach appears to overcome resistance to anti-PD-1 therapy by reducing PMN-MDSC/TAN accumulation and restoring effector T cell function [66]. Collectively, these findings support the development of therapeutic strategies that target TANs in combination with other immunosuppressive elements of the iCCA microenvironment to enhance clinical outcomes.
3.4. Myeloid-Derived Suppressor Cells
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells pathologically activated under chronic inflammatory conditions, including cancer. Instead of differentiating into granulocytes, macrophages, or DCs, these cells remain in an immature state and are recruited to the TIME by pro-inflammatory cytokines and growth factors such as prostaglandin E2 (PGE2), G-CSF, granulocyte-macrophage colony-stimulating factor (GM-CSF), vascular endothelial growth factor (VEGF), TGF-β, and S100 family proteins, acquiring potent immunosuppressive properties [69,83]. MDSCs are broadly classified into polymorphonuclear MDSCs (PMN-MDSCs), which resemble neutrophils, and monocytic MDSCs (M-MDSCs), which resemble monocytes. A third subset, early-stage MDSCs (eMDSCs), has also been reported, though its functional significance remains less defined [69,83].
In cancer, MDSCs accumulate in the peripheral blood, lymphoid organs, and the tumor tissues, where they contribute to tumor immune evasion. Their suppressive activity is mediated through multiple mechanisms, including depletion of amino acids critical for T cell proliferation, production of reactive oxygen species, nitric oxide, and immunosuppressive cytokines such as IL-10 and TGF-β. Additionally, MDSCs promote the recruitment and expansion of Tregs and B cells, impair DC maturation, and inhibit NK cell cytotoxicity by downregulating activating receptors and activating inhibitory checkpoints such as PD-1/PD-L1 [69,83]. M-MDSCs are considered more plastic than PMN-MDSCs and can differentiate into highly immunosuppressive TAMs, particularly under hypoxic conditions [84]. These M-MDSC-derived TAMs often acquire an M2-like phenotype, marked by expression of calcium-binding proteins S100A8/9, which are not typically expressed by monocyte-derived or tissue-resident macrophages [85].
Beyond their immunosuppressive activity, MDSCs also promote tumor progression through non-immunological mechanisms, including induction of angiogenesis, EMT, and pre-metastatic niche formation [69,83].
In iCCA, MDSCs, particularly PMN-MDSCs, are significantly expanded in circulation and enriched in tumor tissues. Elevated MDSC levels correlate with advanced disease stage, larger tumor burden, resistance to ICIs and poor clinical outcomes [66,71]. Mechanistically, tumor-derived factors such as GM-CSF, IL-6, and IL-1β drive MDSC expansion, while chemokines like CXCL1, CXCL5, and CXCL8 mediate their recruitment via CXCR2 signaling [66,71]. Beyond recruitment, tumor-derived chemokines, particularly CXCL8, also act through CXCR2 to organize suppressive niches where PMN-MDSCs interact with tumor cells, thereby restraining CD4+ and CD8+ T cell proliferation and cytotoxicity [66]. Preclinical models have also demonstrated a role for gut microbiota-derived lipopolysaccharide in amplifying MDSC infiltration through Toll-like receptor 4 (TLR4) signaling in hepatocytes, thereby contributing to immune escape and liver cancer progression [86]. This highlights a functional link between microbial dysbiosis, immune dysfunction, and tumor development in iCCA.
Given their key role in cancer progression and immune evasion, MDSCs are emerging as promising targets in iCCA. PMN-MDSCs, in particular, are associated with immunotherapy resistance and may serve as both predictive biomarkers and therapeutic targets [66,71]. Preclinical studies have shown that targeting MDSC recruitment or function, using CXCR2 antagonists, anti-Ly6G antibodies, or combined blockade of METTL1 and the CXCL8-CXCR2 axis, can enhance the efficacy of ICIs [66]. Despite encouraging results in preclinical models, direct clinical targeting of MDSCs in iCCA remains limited. Some agents such as the liver X nuclear receptor/apolipoprotein E (LXR/ApoE) agonist RGX-104 and the chemotherapeutic capecitabine may exert indirect effects on MDSCs and are under evaluation in advanced solid tumors [44]. Broader approaches, including CXCR1/2 inhibitors, anti-CXCL8 antibodies, all-trans retinoic acid, or STAT3 inhibitors, are also being tested in other malignancies [51,66]. However, the remarkable plasticity and heterogeneity of MDSCs remain major obstacles. Further investigation into the distinct roles and regulatory networks of each subset will be essential to refine MDSC-targeted therapeutic strategies in iCCA [83].
3.5. Dendritic Cells
DCs are a heterogeneous population of specialized antigen-presenting cells essential for initiating and regulating adaptive immune responses [87,88]. By capturing, processing, and presenting antigens to naive T cells, DCs orchestrate cytotoxic and helper T cell responses, thereby playing a central role in cancer immunosurveillance. Among immune cells, they serve as specialized sensors that translate innate immune signals into T cell-mediated responses [72,88].
DCs comprise functionally distinct subsets: conventional DCs (cDC1s and cDC2s), plasmacytoid DCs (pDCs), and monocyte-derived DCs (MoDCs) [88]. cDC1s specialize in cross-presenting tumor antigens to CD8+ T cells, while cDC2s primarily activate CD4+ T cells. pDCs are major producers of type I interferons, though in the tumor context they may adopt tolerogenic roles, such as promoting Treg induction. MoDCs arise under inflammatory conditions and can present antigens to both CD4+ and CD8+ T cells [88,89].
In the TME, DC function is often impaired by immunosuppressive signals such as IL-10, TGF-β, VEGF, and PGE2, which hinder their maturation, antigen-presenting capacity, and T cell activation potential. Additional mechanisms of dysfunction include tumor-derived exosomes, hypoxia-associated metabolites, and inhibitory checkpoints such as PD-L1 and T cell immunoglobulin domain and mucin domain-3 (TIM-3), all of which impair DC–T cell communication [90,91]. Spatial analyses have also shown that CAF-derived IL-6 activates STAT3 signaling in DCs, reducing MHC-II expression and promoting the emergence of indoleamine-pyrrole 2,3-dioxygenase (IDO1)+ regulatory DCs, which limit effector T cell proliferation and drive Treg expansion [67,90].
Tumor-derived exosomes, hypoxic metabolites, and immune checkpoints like PD-L1 and T cell immunoglobulin domain and mucin domain-3 (TIM-3) further suppress DC functionality and their interactions with T cells [90,91].
In iCCA, DC dysfunction is linked to immune escape and poor prognosis. Tumors with lymph node metastases exhibit reduced DC infiltration, decreased CD8+ T cell density, and downregulation of MHC-II and co-stimulatory molecules [65]. Notably, a large clinical cohort (n = 359) revealed that high peritumoral accumulation of pDCs, identified by blood dendritic cell antigen 2 (BDCA2) staining, correlates with increased tumor burden, vascular invasion, and Treg infiltration. These pDC-rich peritumoral niches were associated with lower OS and higher recurrence, especially when Tregs were also abundant [92]. These findings support the role of pDCs in establishing a tolerogenic microenvironment in iCCA.
Mechanistically, tumor-intrinsic WNT/β-catenin signaling suppresses chemokine expression (e.g., CCL4, CCL5, CXCL12), limiting DC recruitment [65]. Additionally, CAFs, especially the FAP+ subtype, have been reported to induce IDO+ regulatory DCs via IL-6/STAT3 signaling, further impairing local immunity [67,90].
Although no DC-based therapies are currently approved for iCCA, multiple strategies are under investigation. In preclinical models, administration of Flt3L and poly(I:C) expanded cDCs and reduced lymphatic metastasis. β-catenin inhibition restored DC and CD8+ T cell infiltration, suppressing tumor progression [65]. Other promising approaches include genetically engineered self-differentiated DCs silenced for TGF-β/IL-10 receptors and loaded with cAMP-dependent protein kinase type I-alpha regulatory subunit (PRKAR1A) peptides, which enhanced CD8+ cytotoxicity against iCCA cells [61]. Postoperative vaccination with autologous tumor lysate-loaded DCs combined with activated T cell transfer has also shown potential in reducing recurrence [61]. Moreover, targeting CAFs or modulating β-catenin via (protein-serine O-palmitoleoyltransferase porcupine) PORCN inhibitors may indirectly restore DC function [90]. While still experimental, these strategies highlight the therapeutic potential of reactivating DC-driven immunity in iCCA.
In summary, DCs are central regulators of antitumor immunity and represent a compelling immunotherapeutic target in iCCA. Future strategies should focus on overcoming TME-induced dysfunction, enhancing DC recruitment and antigen presentation, and integrating DC modulation into combinatorial treatment regimens.
The roles of the main immune cell populations within the TIME, together with current therapeutic strategies exploring their targeting, are summarized in Table 1.

Table 1.
Immune cell functions and therapeutic strategies within the iCCA TIME.
4. Immunotherapy Biomarkers in iCCA
Compared to other solid tumors, iCCA shows distinct molecular and immune characteristics that contribute to its limited response to immunotherapy. Overall, the response rate to ICIs in iCCA is approximately 5%, considerably lower than in HCC (~15%), non-small cell lung cancer (NSCLC) (~20%), or melanoma (~40%). Similarly, predictive biomarkers such as PD-L1 positivity, MSI-H, and high tumor mutational burden (TMB-H) are generally less prevalent in iCCA [24]. In addition, iCCA frequently harbors specific genetic alterations, including isocitrate dehydrogenase 1 (IDH1) mutations, fibroblast growth factor receptor 2 (FGFR2) fusions, and TP53 mutations, which are uncommon in other solid tumors and may serve as potential therapeutic targets or predictors of response to targeted therapies and immunotherapy [93]. These differences highlight the immunologically “cold” nature of iCCA and underscore the need for tumor-specific strategies to improve immunotherapy efficacy. Table 2 provides a comparative overview of predictive biomarkers and genetic alterations in iCCA and other solid tumors.
Table 2.
Comparative summary of predictive biomarkers and genetic alterations across iCCA and other solid tumors.
4.1. Current Predictive Biomarkers and Their Limitations
A major challenge in optimizing immunotherapy for iCCA is the identification of reliable biomarkers to predict treatment response. Even though the TIME plays a crucial role in understanding immune dynamics, molecular and cellular biomarkers are required to improve patient stratification and guide treatment decisions [115].
Among the most established are PD-L1 expression, microsatellite instability (MSI), TMB, and mismatch repair deficiency (MMR-d) (Figure 2) [24]. However, their predictive value in iCCA remains limited. PD-L1 expression is detected in approximately ~27.3% e of iCCA cases but its correlation with clinical response is controversial due to tumor heterogeneity, spatial expression variability, and different detection methods [24,116]. Similarly, MSI-H and high TMB (TMB-H), are rare in iCCA, occurring in only ~2.5% and ~4.6% of cases, respectively, thus limiting their utility as prognostic biomarkers [97].
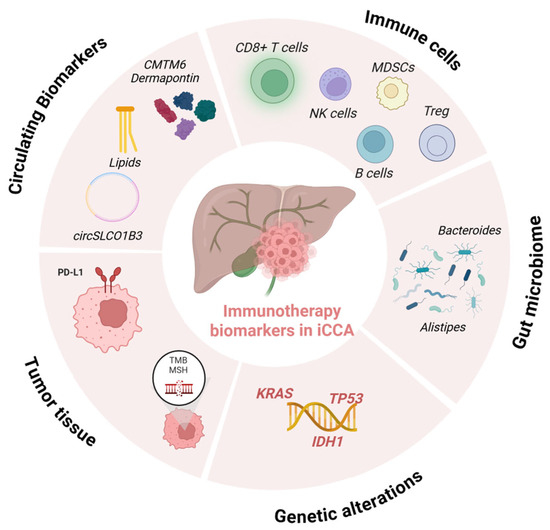
Figure 2.
Predictive biomarkers of immunotherapy response in iCCA. Validated biomarkers (e.g., MSI-H/dMMR, high TMB, PD-L1 expression) are currently used in clinical practice, while emerging biomarkers (e.g., IDH1/2 mutations, circulating biomarkers, tumor-infiltrating lymphocytes, gut microbiota composition) are under investigation in preclinical or clinical studies for their potential role in patient selection and prediction of immunotherapy efficacy (created in BioRender.com).
In iCCA, elevated TMB has been associated with improved responses to ICB, but outcomes remain variable, highlighting its limitations as a standalone predictor. Not all mutations generate immunogenic neoantigens [117], and differences in TMB measurement across sequencing platforms further complicate interpretation [118,119].
Consistent with these observations, two recent studies failed to validate PD-L1 or TMB as reliable predictive biomarkers in BTC. In one study, even though PD-L1 was expressed in a considerable subset of patients, no correlation was found with PFS or the objective response rate (ORR). Similarly, the average TMB was low (mean 4.5 mutations/Mb), with no significant difference in PFS between TMB-high and TMB-low groups [120]. Supporting this, a phase II clinical trial evaluating the efficacy of pembrolizumab in advanced BTC (NCT03110328) also found no significant correlation between PD-L1 expression, TMB, and clinical response outcomes [117].
The immunogenic potential of tumors is more accurately represented by neoantigen burden, which can result from exposure to carcinogens, oncoviral integration, APOBEC-mediated mutagenesis, or defective DNA mismatch repair [121]. A comprehensive molecular analysis revealed that approximately 6% of BTCs are hypermutated, with 2% presenting mismatch repair deficiency (MMR-d) [121,122]. This hypermutated phenotype has been associated with increased neoantigen load and enhanced T-cell recognition, supporting the rationale for the use of immunotherapy in these patients. Notably, MSI-H/MMR-d CCA represents the only BTC subgroups having FDA-approved ICI indications and have shown positive responses in clinical settings [10].
Nevertheless, the literature data on immunotherapy response predictors in iCCA are inconsistent, and no single biomarker may identify patients likely to benefit from this therapeutic approach, suggesting that a comprehensive approach, evaluating MMR, MSI, TMB, and PD-L1 in concert, could be necessary [118,119,123].
4.2. Emerging Biomarkers for Immunotherapy Response in iCCA
Due to the limited predictive value of current biomarkers, growing attention has been focused on emerging molecular and cellular features to better stratify patients for immunotherapy and overcome immunologic tolerance and resistance [13,115].
Promising prognostic factors include circulating biomarkers, such as circulating CD4+ and CD8+ T-cell ratio, levels of circulating MDSCs or Treg, soluble forms of PD-1 (sPD-1) and PD-L1 (sPD-L1), serum sCTLA-4 levels, ctDNA and CTCs, cytokines including IFN-γ, IL-6, IL-8, IL-11, and IL-2, NLR, and gut microbiota enriched in Bacteroides and Akkermansia (Figure 2) [1,124].
4.2.1. TIME-Based Biomarkers
Recent advances in multi-omic profiling have helped characterize the immune landscape of iCCA. A multi-omic analysis on 16 iCCA patients divided the tumor samples into high-immune and low-immune groups. Higher infiltration of CD8+ T-cells, CD4+ T-cells, and CD20+ B cells, along with upregulation of immune pathways, such as the TCR signaling pathway and cytokine–cytokine receptor interaction pathway, correlated with anti-PD-1 treatment better response [125]. These findings highlight the relevance of tumor-infiltrating immune cells and immune activation signatures in predicting immunotherapy response. Building on this, tumor-intrinsic immune features such as cytotoxic T-cell (CTL) markers and the tumor immune dysfunction and exclusion (TIDE) score further help in classifying CCA tumors as CTL-high or CTL-low and better predicting patients’ response to immunotherapy [126]. In particular, for CTL-high CCA tumors, TIDE can assess if T-cell dysfunction might impair immune response while, for CTL-low tumors, it can assess whether ICB may increase immune cell infiltration [103].
Furthermore, tumor-associated antigen (TAA)-specific CTL responses have been linked to improved OS in iCCA. Kida et al. showed that multiple TAA-derived CTL epitopes can effectively trigger immune responses, correlating with a significantly improved OS. Additionally, higher peripheral blood lymphocyte counts correlated with better TAA-specific immune responses, suggesting potential biomarkers for predicting immunotherapy benefit in iCCA [127].
TLS have also emerged as promising predictors of immunotherapy response in iCCA. The density and spatial distribution of TLSs within the TME strongly correlated with patient outcome and ICI efficacy. Specifically, a high concentration of intratumoral TLS has been associated with prolonged OS and enhanced ICI response, while peri-tumoral TLS correlated with poorer outcomes. Furthermore, a gene expression profile, including PAX5, TCL1A, TNFRSF13C, and CD79A, has been identified to reliably detect TLSs, offering a potential molecular tool for stratifying patients [128].
4.2.2. Mutation-Based Biomarkers
Genomic alterations in BTCs significantly affect prognosis and immunotherapy response. Co-mutations in KRAS and TP53 have been linked to better outcomes in response to immunotherapy, while single KRAS mutations correlate with poor prognosis and limited benefits in iCCA [129]. Additionally, IDH1 mutations have been associated with immunosuppression. Inhibiting IDH1 may activate the immune system against cancer cells by converting an immune “cold” environment into a “hot” one, by promoting infiltration of cytotoxic T cells and increasing PD-L1 expression, as shown in preclinical models [130,131]. Based on these findings, combination strategies are being explored. An ongoing clinical trial is underway to explore the combination of IDH1 inhibitors and durvalumab in patients with unresectable or metastatic BTCs (NCT04056910) [132]. Another phase I study is investigating the safety and tolerability of ivosidenib in combination with durvalumab and gemcitabine/cisplatin as first-line therapy in patients with locally advanced, unresectable, or metastatic CCA with an IDH1 mutation. (NCT06501625).
4.2.3. Circulating Biomarkers
Circulating biomarkers such as proteins, circRNAs, and miRNAs also show promising potential in predicting immunotherapy response in CCA [115].
Among them, circSLCO1B3 (circRNA, which originates from exon 9 to exon 15 of the SLCO1B3 gene) has been shown to promote iCCA cells evasion from immune survivance by increasing PD-L1 and was correlated with poor prognosis in iCCA, suggesting its role as a biomarker for patient selection. Similarly, plasma exosomal circRNAs like circ-ADAMTS6 have been associated with immunogenic iCCA subgroups, characterized by T-cell exhaustion and neutrophil extracellular traps (NETs), which may respond well to immune checkpoint blockade [133].
Concerning proteins, PD-L1 expression was positively correlated with CMTM6 expression in CCA, suggesting better responses to anti-PD-1/PD-L1 therapy [134]. Additionally, dermatopontin (DPT) was shown to enhance immune cell infiltration in CCA by stimulating macrophages to secrete the chemokine CCL19. Moreover, high DPT levels were associated with enhanced anti-PD-1/PD-L1 immunotherapeutic responses, further supporting its role as a possible immunotherapy-related biomarker [135].
Serum lipid levels, including APOA1 and triglycerides, have also been identified as independent predictors of OS in iCCA patients receiving immunotherapy [136].
4.2.4. Gut Microbiota
Beyond tumor-intrinsic characteristics, the gut microbiota is increasingly recognized as a modulator of immunotherapy efficacy [137,138]. Mechanisms include the modulation of primary and secondary lymphoid tissue activity, the production of immunoregulatory metabolites such as short-chain fatty acids and bacterial inosine, and antigenic mimicry that can enhance T-cell–mediated tumor recognition [139,140].
Mechanistically, microbial communities enriched in taxa such as Alistipes, Bacteroides thetaiotaomicron, B. fragilis, Akkermansia muciniphila, and Faecalibacterium prausnitzii have been linked to elevated production of metabolites that enhance antigen presentation and promote CD8+ T cell activation and are frequently enriched in responders [140].
The iCCA-associated tumor microbiome has been recently investigated. Xin et al. analyzed 121 CCA tissues and 89 paired non-tumoral tissues finding different tumor microbiome composition [141]. In BTCs specifically, a prospective study using integrated metagenomic and metabolomic analysis identified 20 microbial taxa significantly associated with prolonged benefits from anti-PD-1/PD-L1 therapy. Particularly, patients enriched in Alistipes showed better survival, while taxa such as Bacilli and Lactobacillales were linked to non-response [142].
Several studies have focused on modulating gut microbiome in cancer immunotherapy [143]. The modifiable nature of the gut microbiota makes it a potential biomarker and therapeutic target. While clinical evidence in iCCA is still limited, data from other solid tumors support the rationale for probiotic- or prebiotic-based strategies aimed at enhancing immunotherapy efficacy [144,145,146,147,148].
Probiotics and prebiotics can indeed influence the immune system by enriching beneficial gut microbes and are therefore linked to an enhanced immune response. For instance, it has been highlighted that inulin supplementation in preclinical models might enhance systemic and tumor-infiltrating T cell responses, promote Th1 polarization, promote CD8+ T cell activation, and suppress tumor growth in a microbiota-dependent manner [149]. Prebiotics also elevate short-chain fatty acid (SCFA) production, decrease gut pH, and promote the growth of SCFA-producing taxa, which correlates with improved efficacy of PD-1/PD-L1 blockade [150]. A clinical trial involving liver cancer patients receiving anti-PD-1 therapy in combination with Lactobacillus rhamnosus Probio-M9 supplementation (NCT05032014) has been completed, although results have not yet been reported.
5. Novel Immunotherapeutic Approaches
Given the limited efficacy of current immunotherapies in iCCA, novel strategies are focusing on modulating the TIME to overcome the intrinsic immunosuppressive nature of this tumor. In this context, T cell-based therapies like CAR-T, TILs, cancer vaccines and combined treatment strategies have nowadays opened new insights in the management of iCCA [64,151].
5.1. CAR-T Therapy
CAR-T therapy is an innovative immunotherapeutic strategy that involves engineering patient-derived T cells to express synthetic receptors targeting specific tumor-associated antigens. While the FDA has approved six CAR-T products targeting CD19, CD22, and B-cell maturation antigen (BCMA) for hematologic cancers [152], their application in solid tumors remains limited due to several challenges. These include antigen heterogeneity, complex TMEs, difficult trafficking to the tumor sites, and potential immune-related adverse events [153]. Furthermore, T cell exhaustion can occur due to chronic antigen exposure within the TME, leading to reduced proliferative capacity and cytokine production, as well as impaired cytotoxic activity, limiting antitumor efficacy. Limited persistence of CAR-T in vivo can also compromise durable responses and long-term tumor control, particularly in hostile microenvironments characterized by immunosuppressive cells and inhibitory cytokines. Moreover, off-tumor expression of target antigens in healthy tissues may result in on-target toxicity, posing a significant safety challenge for CAR-T therapy [154,155].
Despite this, a growing number of clinical trials are now exploring the use of CAR-T on solid tumors, including iCCA [156]. For instance, carcinoembryonic antigen (CEA) is overexpressed in iCCA and other malignancies and CEA-targeted CAR-T is currently being investigated in a phase II clinical trial for CCA (NCT06043466).
In iCCA, several tumor-associated antigens have emerged as promising CAR-T targets [64]. Among these, MUC1, a glycoprotein overexpressed in iCCA and associated with worse clinical outcomes, has shown positive results. Preclinical studies using fourth-generation MUC1-specific CAR-T (CAR4) demonstrated potent tumor cell killing, accompanied by elevated secretion of pro-inflammatory cytokines such as TNF-α and IFN-γ [157,158]. These MUC1-targeted CAR-T also significantly inhibited tumor growth in iCCA xenograft models, offering compelling evidence for clinics. Based on these findings, a phase I/II clinical trial (NCT03633773) is evaluating the safety and efficacy of MUC1-directed CAR-T in patients with iCCA.
EGFR is another plausible target, whose overexpression has been reported in iCCA, and linked to a more aggressive tumor behavior [159]. A phase I clinical trial (NCT01869166) evaluating EGFR-specific CAR-T immunotherapy in patients with EGFR-positive advanced BTCs showed encouraging preliminary outcomes. Out of 17 patients, 1 achieved complete response and 10 achieved stable disease. In the infused CART-EGFR cells, enrichment of central memory T cells (Tcm) was found and linked to the clinical outcome [160]. Additionally, a case report described a patient with advanced unresectable/metastatic CCA who received EGFR-specific and CD133-specific CART sequential treatment as CART cocktail immunotherapy (NCT02541370). An 8.5-month partial response following anti-EGFR treatment, and a further 4.5-month partial response with anti-CD133 CAR-T was achieved [161].
5.2. Adoptive TIL Therapy
In addition to the CAR-T approach, TIL therapy has also emerged as a promising, personalized immunotherapeutic strategy in solid tumors, including CCA [162,163]. In particular, TIL therapy consists of isolation, ex vivo expansion, and reinfusion of autologous lymphocytes derived from tumor tissue, aiming to increase the number of tumor-reactive T cells [151].
Although limited clinical data are currently available, early case reports in iCCA have shown encouraging benefits. For instance, one patient experienced long-term remission (>3 years) after infusion of CD3-activated TILs enriched against tumor lysate [164]. Another case report described tumor regression and prolonged OS after the infusion of CD4+ TILs specifically recognizing an ERBB2-interacting protein [165]. This patient was enrolled in an ongoing phase II trial (NCT01174121) investigating the use of TIL therapy in various solid tumors, including CCA. The detailed results of this trial are still pending [166].
Several clinical trials are currently exploring TIL therapy in CCA. A Phase II study (NCT03801083) is evaluating TIL transfer in metastatic BTCs using lymphodepletion followed by high-dose IL-2. Another trial (NCT05088190) is testing PD-1–positive TILs combined with anti–PD-L1 and IL-2 iCCA. Additionally, a Phase II trial (NCT03820310) is assessing Tcm therapy after radical resection of iCCA, aiming to improve survival outcomes.
Beyond T cells, recent findings suggest that B cells may also play a role in shaping TIME in iCCA [167]. Indeed, high levels of B cell, although rare in liver tumors, are associated with better overall and RFS in some CCA patients, potentially by enhancing the cytotoxic activity CD8+ T cell [168,169]. These include modulating TLS formation, mainly composed of CD20+ B cells and CD8+ T cells, enhancing B cell antigen presentation, or depleting immunosuppressive B cell subsets such as Bregs. One therapeutic approach is the administration of CD40–CD40L pathway agonists that have shown potential in preclinical models, boosting antigen presentation by B cells and DCs [170,171]. In murine models of iCCA, the combination of anti-CD40 with anti-PD-1 and chemotherapy significantly inhibited tumor growth and prolonged survival. Based on these results, clinical trials investigating CD40 agonists such as CDX-1140 in primary liver cancers are currently ongoing (NCT05849480) [172,173].
5.3. Cancer Vaccines
Vaccine-based immunotherapy is increasingly being explored in iCCA as a promising approach to stimulate and enhance tumor-specific immune responses [174,175]. The high prevalence of recurring driver mutations such as TP53, FGFR2 fusions, IDH1, TERT, BAP1, and BRAF makes neoantigen vaccines particularly attractive in iCCA, providing tumor-specific targets that may be exploited for personalized vaccine strategies [64].
Several vaccine platforms are currently being investigated, including cell-based vaccines (e.g., DC vaccines), peptide/protein-based vaccines, and nucleic acid-based vaccines such as mRNA vaccines [176]. Among these, DC vaccines stimulate CTL responses against tumor cells by ex vivo loading of autologous antigen-presenting cells with tumor peptides or lysates [177].
Recent evidence suggests a two-step immunotherapeutic strategy: the induction of neoantigen-specific T cells and the maintenance of their effector function to avoid exhaustion [178]. In this context, the study by Kotera et al. [179] in patients with resected iCCA represents an early example of such a combined approach. Particularly, the co-administration of a DC-based vaccine with anti-CD3-activated T lymphocyte infusion demonstrated a favorable safety profile. Particularly, the treatment was associated with increased total lymphocytes, particularly CD8+ T cells, and reduced Tregs (FoxP3+), leading to improved RFS and OS [179].
In addition, a retrospective study in patients with unresectable or recurrent BTCs, including iCCA, showed that the combination of DC vaccination with gemcitabine was associated with stable disease and increased OS compared to DC vaccination alone, suggesting potential synergistic effects between chemotherapy and immunotherapy [180].
Similarly, MUC1-loaded DC vaccines promoted antitumor immunity in iCCA patients, producing a good clinical response [181]. In the same retrospective study, another DC-based vaccine targeting Wilms tumor 1 (WT1) demonstrated a disease control rate in the small cohort of advanced CCA patients, with the greatest clinical benefit observed in those who also received chemotherapy and maintained good nutritional status [181].
MUC1 targeting showed encouraging results also in peptide-based vaccines with good safety and immunogenicity in patients with BTCs [182]. Additionally, multi-peptide vaccine approaches targeting antigens such as LY6K, DEPDC1, IMP3, and TTK have shown the induction of robust CTL responses and early evidence of improved PFS and OS [183].
mRNA vaccines are also under investigation in iCCA [184]. Transcriptomic analyses of CCA tumors identified an “immune desert” subtype (IS2) characterized by poor immune infiltration, which might benefit the most from vaccination strategies stimulating immune responses. Candidate antigens such as CD247, FCGR1A, and TRRAP have been proposed as targets for mRNA vaccine development [185]. Table 3 summarizes the main emerging immunotherapy approaches being studied in iCCA.
Table 3.
Emerging immunotherapeutic strategies in iCCA.
6. Conclusions
iCCA comprises a group of molecularly heterogeneous tumors with an immunologically “cold” microenvironment. It is characterized by low neoantigen load and poor response to current therapies. While ICIs combined with chemotherapy have been approved as first-line treatment, clinical benefit is restricted to a small subset of patients. This underlies the need for more effective and tailored approaches. Advances in understanding tumor immunobiology and molecular profiling have enabled the development of personalized strategies targeting tumor-specific antigens and neoantigens. However, despite promising preliminary clinical results, these therapies face considerable challenges and are not yet part of standard therapy for iCCA. Emerging approaches such as adoptive T cell transfer, cancer vaccines and modulation of the TIME offer potential to overcome immune resistance and broaden the spectrum of responsive patients. Ongoing research and clinical studies are essential to optimize combination therapies, identify reliable biomarkers, and translate these advances into stable and broadly effective management of iCCA.
Author Contributions
Writing—original draft preparation, N.P., E.S.-L., F.M. and A.G.; writing—review and editing, A.G. and F.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research and the APC were funded by PRIN 2022, grant number 2022PNZ9KP (coordinator Stefania Cannito, PI University of Florence Alessandra Gentilini).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| ADAMTS6 | A Disintegrin and Metalloproteinase with Thrombospondin Motifs 6 |
| ADM | Adrenomedullin |
| ALOX5 | Polyunsaturated fatty acid 5-lipoxygenase |
| APOBEC | Apolipoprotein B mRNA Editing Catalytic Polypeptide-like |
| APOA1 | Apolipoprotein A1 |
| ApoE | Apolipoprotein E |
| B7-H3 | CD276 antigen |
| BCMA | B-cell Maturation Antigen |
| BDCA1 | Blood Dendritic Cell Antigen 2 |
| BTC | Biliary Tract Cancer |
| CAF(s) | Cancer-associated fibroblast(s) |
| CALCRL | Calcitonin receptor-like receptor |
| CAR-NK | Chimeric Antigen Receptor Natural Killer cells |
| CAR-T | Chimeric Antigen Receptor T-cell |
| CCA | Cholangiocarcinoma |
| CCR2/8 | C-C chemokine receptor type 2/8 |
| cDCs | Conventional Dendritic cells |
| CEA | Carcinoembryonic Antigen |
| CMTM6 | CKLF-like MARVEL transmembrane domain-containing protein 6 |
| CSF1R | Macrophage Colony-Stimulating Factor 1 receptor |
| CTL | Cytotoxic T Lymphocyte |
| CTLA-4 | Cytotoxic T-lymphocyte protein 4 |
| CXCL | C-X-C motif chemokine |
| CXCR | C-X-C chemokine receptor type |
| DC(s) | Dendritic Cell(s) |
| DCDC2 | Doublecortin domain-containing 2 |
| DPT | Dermatopontin |
| eMDSCs | Early-stage Myeloid-derived suppressor cells |
| EGFR | Epidermal Growth Factor Receptor |
| EMT | Epithelial-to-mesenchymal transition |
| ENO1 | Enolase 1 |
| FAP | Fibroblast activation protein |
| FasL | Fas ligand |
| FCR | FOXP3+/CD8+ T cell ratio |
| FGFR2 | Fibroblast Growth Factor Receptor 2 |
| FGL1 | Fibrinogen-like protein 1 |
| Flt3L | Fms-related tyrosine kinase 3 ligand |
| FOXP3 | Forkhead Box P3 |
| G-CSF | Granulocyte Colony-Stimulating Factor |
| GemCis | Gemcitabine plus Cisplatin |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| GPC3 | Glypican-3 |
| HCC | Hepatocellular Carcinoma |
| ICB | Immune Checkpoint Blockade |
| iCCA | Intrahepatic cholangiocarcinoma |
| ICI(s) | Immune Checkpoint Inhibitor(s) |
| IDH1/2 | Isocitrate Dehydrogenase 1/2 |
| IDO | Indoleamine-pyrrole 2,3-dioxygenase |
| IFN- α/γ | Interferon alpha/gamma |
| IL | Interleukin |
| KIR | Killer-cell immunoglobulin-like receptor |
| KRAS | Kirsten Rat Sarcoma Viral Oncogene Homolog |
| LAG-3 | Lymphocyte activation gene 3 |
| LGALS1 | Beta-galactoside-binding lectin L-14-I |
| LTB4 | Leukotriene B4 |
| LXR | Liver X nuclear receptor |
| LY6K | Lymphocyte Antigen 6 Complex Locus K |
| M-MDSCs | Monocytic Myeloid-derived suppressor cells |
| MDSC(s) | Myeloid-derived suppressor cell(s) |
| MEOX1 | Mesenchyme homeobox 1 |
| METTL1 | tRNA (guanine-N(7)-)-methyltransferase |
| MHC | Major Histocompatibility Complex |
| MICA/B | MHC class I polypeptide-related sequence A/B |
| MMR-d | Mismatch Repair Deficiency |
| MoDCs | Monocyte-derived Dendritic cells |
| MUC1 | Mucin 1 |
| MSI-H | Microsatellite Instability-High |
| NETs | Neutrophil Extracellular Traps |
| NK | Natural Killer |
| NKG2A/D | NK cell receptor A/D |
| NLR | Neutrophil-to-Lymphocyte Ratio |
| NSCLC | Non-Small Cell Lung Cancer |
| ORR | Objective Response Rate |
| OS | Overall Survival |
| PD-1 | Programmed Cell Death Protein 1 |
| PD-L1 | Programmed Death Ligand 1 |
| pDCs | Plasmacytoid Dendritic cells |
| PFS | Progression-Free Survival |
| PGE2 | Prostaglandin E2 |
| PMN-MDSCs | Polymorphonuclear Myeloid-derived suppressor cells |
| poly(I:C) | Polyinosinic:Polycytidylic acid |
| PORCN | Protein-serine O-palmitoleoyltransferase porcupine |
| PVR | Poliovirus receptor |
| RAMP1 | Receptor activity-modifying protein 1 |
| RFS | Relapse-Free Survival |
| SCFA | Short-Chain Fatty Acid |
| SIRPα | Signal Regulatory Protein alpha |
| SPP1 | Secreted Phosphoprotein 1 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| TAA | Tumor-Associated Antigen |
| TAM(s) | Tumor-associated macrophage(s) |
| TAN(s) | Tumor-associated neutrophil(s) |
| Tcm | Central Memory T-cell |
| TCR(s) | T-cell receptor(s) |
| TERT | Telomerase Reverse Transcriptase |
| TFF3 | Trefoil Factor 3 |
| TGF-β | Transforming growth factor β |
| TIDE | Tumor Immune Dysfunction and Exclusion |
| TIGIT | T-cell immunoreceptor with Ig and ITIM domains |
| TIL(s) | Tumor-Infiltrating Lymphocyte(s) |
| TIM-3 | T cell Immunoglobulin Domain and Mucin Domain-3 |
| TIME | Tumor Immune Microenvironment |
| TLR4 | Toll-like receptor 4 |
| TLS | Tertiary Lymphoid Structures |
| TMB | Tumor Mutational Burden |
| TMB-H | High Tumor Mutational Burden |
| TNF-α | Tumor necrosis factor alpha |
| TP53 | Tumor protein 53 |
| TRAIL | TNF-related apoptosis-inducing ligand |
| Treg(s) | Regulatory T cell(s) |
| PRKAR1A | cAMP-dependent protein kinase type I-alpha regulatory subunit |
| TRM | Tissue-resident memory |
| TRRAP | Transformation/Transcription Domain-Associated Protein |
| VEGF | Vascular Endothelial Growth Factor |
| WT1 | Wilms Tumor 1 |
References
- Catalano:, M.; Iannone, L.F.; Nesi, G.; Nobili, S.; Mini, E.; Roviello, G. Immunotherapy-related biomarkers: Confirmations and uncertainties. Crit. Rev. Oncol. Hematol. 2023, 192, 104135. [Google Scholar] [CrossRef]
- Sussman, T.A.; Ott, P.A. Adjuvant immunotherapy for melanoma patients: Progress and opportunities. ESMO Open 2024, 9, 102962. [Google Scholar] [CrossRef]
- Reck, M.; Remon, J.; Hellmann, M.D. First-Line Immunotherapy for Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2022, 40, 586–597. [Google Scholar] [CrossRef]
- Voss, M.H.; Motzer, R.J. Adjuvant Immunotherapy for Kidney Cancer—A New Strategy with New Challenges. N. Engl. J. Med. 2024, 390, 1432–1433. [Google Scholar]
- Kim, J.; Maharjan, R.; Park, J. Current Trends and Innovative Approaches in Cancer Immunotherapy. AAPS PharmSciTech 2024, 25, 168. [Google Scholar] [CrossRef]
- Desai, R.; Coxon, A.T.; Dunn, G.P. Therapeutic applications of the cancer immunoediting hypothesis. Semin. Cancer Biol. 2022, 78, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Galassi, C.; Chan, T.A.; Vitale, I.; Galluzzi, L. The hallmarks of cancer immune evasion. Cancer Cell 2024, 42, 1825–1863. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.C.; Duffy, C.R.; Allison, J.P. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov. 2018, 8, 1069–1086. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef]
- Loeuillard, E.; Conboy, C.B.; Gores, G.J.; Ilyas, S.I. Immunobiology of cholangiocarcinoma. JHEP Rep. 2019, 1, 297–311. [Google Scholar] [CrossRef]
- Woo, S.R.; Corrales, L.; Gajewski, T.F. Innate immune recognition of cancer. Annu. Rev. Immunol. 2015, 33, 445–474. [Google Scholar] [CrossRef]
- Almhanna, K. Immune checkpoint inhibitors in combination with chemotherapy for patients with biliary tract cancer: What did we learn from TOPAZ-1 and KEYNOTE-966. Transl. Cancer Res. 2024, 13, 22–24. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Moris, D.; Palta, M.; Kim, C.; Allen, P.J.; Morse, M.A.; Lidsky, M.E. Advances in the treatment of intrahepatic cholangiocarcinoma: An overview of the current and future therapeutic landscape for clinicians. CA Cancer J. Clin. 2023, 73, 198–222. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Tavolari, S.; Brandi, G. Cholangiocarcinoma: Epidemiology and risk factors. Liver Int. 2019, 39 (Suppl. S1), 19–31. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL-ILCA Clinical Practice Guidelines on the management of intrahepatic cholangiocarcinoma. J. Hepatol. 2023, 79, 181–208. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Olaizola, P.; Paiva, N.A.; Olaizola, I.; Agirre-Lizaso, A.; Landa, A.; Bujanda, L.; Perugorria, M.J.; Banales, J.M. Pathogenesis of Cholangiocarcinoma. Annu. Rev. Pathol. 2021, 16, 433–463. [Google Scholar] [CrossRef]
- Elvevi, A.; Laffusa, A.; Scaravaglio, M.; Rossi, R.E.; Longarini, R.; Stagno, A.M.; Cristoferi, L.; Ciaccio, A.; Cortinovis, D.L.; Invernizzi, P.; et al. Clinical treatment of cholangiocarcinoma: An updated comprehensive review. Ann. Hepatol. 2022, 27, 100737. [Google Scholar] [CrossRef]
- Speckart, J.; Rasmusen, V.; Talib, Z.; GnanaDev, D.A.; Rahnemai-Azar, A.A. Emerging Therapies in Management of Cholangiocarcinoma. Cancers 2024, 16, 613. [Google Scholar] [CrossRef]
- Wang, J.; Xu, Y.; Hong, B.; Hou, Q.; Chen, W.; Zhang, W.; Zheng, W. PD-1/PD-L1 inhibitors plus chemotherapy versus chemotherapy alone as the first line treatment for advanced biliary tract cancer: A pooled analysis of KEYNOTE-966 and TOPAZ-1 trails. World J. Surg. Oncol. 2025, 23, 228. [Google Scholar] [CrossRef]
- Oh, D.Y.; He, A.R.; Bouattour, M.; Okusaka, T.; Qin, S.; Chen, L.T.; Kitano, M.; Lee, C.K.; Kim, J.W.; Chen, M.H.; et al. Durvalumab or placebo plus gemcitabine and cisplatin in participants with advanced biliary tract cancer (TOPAZ-1): Updated overall survival from a randomised phase 3 study. Lancet Gastroenterol. Hepatol. 2024, 9, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Oh, D.Y.; He, A.R.; Bouattour, M.; Okusaka, T.; Qin, S.; Chen, L.T.; Kitano, M.; Lee, C.K.; Kim, J.W.; Chen, M.H.; et al. Durvalumab plus chemotherapy in advanced biliary tract cancer: 3-year overall survival update from the phase III TOPAZ-1 study. J. Hepatol. 2025, 83, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Yoo, C.; Ueno, M.; Klümpen, H.J.; Kelley, R.K.; Vogel, A.; Furuse, J.; Ren, Z.; Yau, T.; Chan, S.L.; Ozaka, M.; et al. Health-related quality of life in participants with advanced biliary tract cancer from the randomized phase III KEYNOTE-966 study. J. Hepatol. 2025, 83, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.; Ricci, A.D.; Brandi, G. PD-L1, TMB, MSI, and Other Predictors of Response to Immune Checkpoint Inhibitors in Biliary Tract Cancer. Cancers 2021, 13, 558. [Google Scholar] [CrossRef]
- Lo, J.H.; Agarwal, R.; Goff, L.W.; Heumann, T.R. Immunotherapy in Biliary Tract Cancers: Current Standard-of-Care and Emerging Strategies. Cancers 2023, 15, 3312. [Google Scholar] [CrossRef]
- Vogel, A.; Bathon, M.; Saborowski, A. Immunotherapies in clinical development for biliary tract cancer. Expert. Opin. Investig. Drugs 2021, 30, 351–363. [Google Scholar] [CrossRef]
- Cammarota, A.; Balsano, R.; Pressiani, T.; Bozzarelli, S.; Rimassa, L.; Lleo, A. The Immune-Genomics of Cholangiocarcinoma: A Biological Footprint to Develop Novel Immunotherapies. Cancers 2025, 17, 272. [Google Scholar] [CrossRef]
- Greten, T.F.; Schwabe, R.; Bardeesy, N.; Ma, L.; Goyal, L.; Kelley, R.K.; Wang, X.W. Immunology and immunotherapy of cholangiocarcinoma. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 349–365. [Google Scholar] [CrossRef]
- Fabris, L.; Sato, K.; Alpini, G.; Strazzabosco, M. The Tumor Microenvironment in Cholangiocarcinoma Progression. Hepatology 2021, 73 (Suppl. S1), 75–85. [Google Scholar] [CrossRef]
- Alvisi, G.; Termanini, A.; Soldani, C.; Portale, F.; Carriero, R.; Pilipow, K.; Costa, G.; Polidoro, M.; Franceschini, B.; Malenica, I.; et al. Multimodal single-cell profiling of intrahepatic cholangiocarcinoma defines hyperactivated Tregs as a potential therapeutic target. J. Hepatol. 2022, 77, 1359–1372. [Google Scholar] [CrossRef]
- Jiang, S.; Lu, H.; Pan, Y.; Yang, A.; Aikemu, A.; Li, H.; Hao, R.; Huang, Q.; Qi, X.; Tao, Z.; et al. Characterization of the distinct immune microenvironments between hepatocellular carcinoma and intrahepatic cholangiocarcinoma. Cancer Lett. 2024, 588, 216799. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.L.; Valle, J.W.; Ilyas, S.I. Immunobiology of cholangiocarcinoma. J. Hepatol. 2023, 79, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Peng, L.; Dong, L.; Liu, D.; Ma, J.; Lin, J.; Chen, X.; Lin, P.; Song, G.; Zhang, M.; et al. Geospatial Immune Heterogeneity Reflects the Diverse Tumor-Immune Interactions in Intrahepatic Cholangiocarcinoma. Cancer Discov. 2022, 12, 2350–2371. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Peng, L.; Dong, L.; Liu, D.; Ma, J.; Lin, J.; Chen, X.; Lin, P.; Song, G.; Zhang, M.; et al. FAP Promotes Immunosuppression by Cancer-Associated Fibroblasts in the Tumor Microenvironment via STAT3-CCL2 Signaling. Cancer Res. 2016, 76, 4124–4135. [Google Scholar]
- Martin-Serrano, M.A.; Kepecs, B.; Torres-Martin, M.; Bramel, E.R.; Haber, P.K.; Merritt, E.; Rialdi, A.; Param, N.J.; Maeda, M.; Lindblad, K.E.; et al. Novel microenvironment-based classification of intrahepatic cholangiocarcinoma with therapeutic implications. Gut 2023, 72, 736–748. [Google Scholar] [CrossRef]
- Job, S.; Rapoud, D.; Dos Santos, A.; Gonzalez, P.; Desterke, C.; Pascal, G.; Elarouci, N.; Ayadi, M.; Adam, R.; Azoulay, D.; et al. Identification of Four Immune Subtypes Characterized by Distinct Composition and Functions of Tumor Microenvironment in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 72, 965–981. [Google Scholar] [CrossRef]
- Bang, Y.H.; Lee, C.K.; Bang, K.; Kim, H.D.; Kim, K.P.; Jeong, J.H.; Park, I.; Ryoo, B.Y.; Lee, D.K.; Choi, H.J.; et al. Artificial Intelligence-Powered Spatial Analysis of Tumor-Infiltrating Lymphocytes as a Potential Biomarker for Immune Checkpoint Inhibitors in Patients with Biliary Tract Cancer. Clin. Cancer Res. 2024, 30, 4635–4643. [Google Scholar] [CrossRef]
- Dong, Z.R.; Zhang, M.Y.; Qu, L.X.; Zou, J.; Yang, Y.H.; Ma, Y.L.; Yang, C.C.; Cao, X.L.; Wang, L.Y.; Zhang, X.L.; et al. Spatial resolved transcriptomics reveals distinct cross-talk between cancer cells and tumor-associated macrophages in intrahepatic cholangiocarcinoma. Biomark. Res. 2024, 12, 100. [Google Scholar] [CrossRef]
- Raggi, C.; Correnti, M.; Sica, A.; Andersen, J.B.; Cardinale, V.; Alvaro, D.; Chiorino, G.; Forti, E.; Glaser, S.; Alpini, G.; et al. Cholangiocarcinoma stem-like subset shapes tumor-initiating niche by educating associated macrophages. J. Hepatol. 2017, 66, 102–115. [Google Scholar] [CrossRef]
- Chen, J.; Tang, Y.; Qin, D.; Yu, X.; Tong, H.; Tang, C.; Tang, Z. ALOX5 acts as a key role in regulating the immune microenvironment in intrahepatic cholangiocarcinoma, recruiting tumor-associated macrophages through PI3K pathway. J. Transl. Med. 2023, 21, 923. [Google Scholar] [CrossRef]
- Jarman, E.J.; Horcas-Lopez, M.; Waddell, S.H.; MacMaster, S.; Gournopanos, K.; Soong, D.Y.H.; Musialik, K.I.; Tsokkou, P.; Ng, M.E.; Cambridge, W.A.; et al. DKK1 drives immune suppressive phenotypes in intrahepatic cholangiocarcinoma and can be targeted with anti-DKK1 therapeutic DKN-01. Liver International. 2023, 43, 208–220. [Google Scholar] [CrossRef]
- Yuan, H.; Lin, Z.; Liu, Y.; Jiang, Y.; Liu, K.; Tu, M.; Yao, N.; Qu, C.; Hong, J. Intrahepatic cholangiocarcinoma induced M2-polarized tumor-associated macrophages facilitate tumor growth and invasiveness. Cancer Cell Int. 2020, 20, 586. [Google Scholar] [CrossRef]
- Guo, Y.; Miao, S.; Jin, Y.; Li, Q.; Wang, Y.; Zhang, X.; Li, J. Tumor-associated macrophages contribute to cholangiocarcinoma progression and chemoresistance through activation of ID1. Ann. Hepatol. 2025, 30, 101773. [Google Scholar] [CrossRef] [PubMed]
- Loeuillard, E.; Yang, J.; Buckarma, E.; Wang, J.; Liu, Y.; Conboy, C.; Pavelko, K.D.; Li, Y.; O’Brien, D.; Wang, C.; et al. Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J. Clin. Investig. 2020, 130, 5380–5396. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, P.; Sun, R.; Li, J.; Hu, Z.; Xin, H.; Luo, C.; Zhou, J.; Fan, J.; Zhou, S. Tumor-associated neutrophils and macrophages interaction contributes to intrahepatic cholangiocarcinoma progression by activating STAT3. J. Immunother. Cancer 2021, 9, e001946. [Google Scholar] [CrossRef] [PubMed]
- Zuyin, L.; Zhao, L.; Qian, C.; Changkun, Z.; Delin, M.; Jialing, H.; Zhuomiaoyu, C.; Yuzi, L.; Jiaxi, Z.; Jie, G.; et al. Single-Cell and Spatial Transcriptomics Delineate the Microstructure and Immune Landscape of Intrahepatic Cholangiocarcinoma in the Leading-Edge Area. Adv. Sci. 2025, 12, e2412740. [Google Scholar] [CrossRef]
- Long, F.; Zhong, W.; Zhao, F.; Xu, Y.; Hu, X.; Jia, G.; Huang, L.; Yi, K.; Wang, N.; Si, H.; et al. DAB2 + macrophages support FAP + fibroblasts in shaping tumor barrier and inducing poor clinical outcomes in liver cancer. Theranostics 2024, 14, 4822–4843. [Google Scholar] [CrossRef]
- Fan, G.; Tao, C.; Li, L.; Xie, T.; Tang, L.; Han, X.; Shi, Y. The co-location of MARCO+ tumor-associated macrophages and CTSE+ tumor cells determined the poor prognosis in intrahepatic cholangiocarcinoma. Hepatology 2025, 82, 25–41. [Google Scholar] [CrossRef]
- Yang, H.; Yan, M.; Li, W.; Xu, L. SIRPα and PD1 expression on tumor-associated macrophage predict prognosis of intrahepatic cholangiocarcinoma. J. Transl. Med. 2022, 20, 140. [Google Scholar] [CrossRef]
- Chen, F.; Sheng, J.; Li, X.; Gao, Z.; Hu, L.; Chen, M.; Fei, J.; Song, Z. Tumor-associated macrophages: Orchestrators of cholangiocarcinoma progression. Front. Immunol. 2024, 15, 1451474. [Google Scholar] [CrossRef]
- Yi, M.; Li, T.; Niu, M.; Mei, Q.; Zhao, B.; Chu, Q.; Dai, Z.; Wu, K. Exploiting innate immunity for cancer immunotherapy. Mol. Cancer 2023, 22, 187. [Google Scholar] [CrossRef]
- Cui, T.; Sun, L.; Guo, X.; Cheng, C.; Zhang, N.; Zhou, S.; Chu, Q.; Xing, C.; Liang, S.; Liu, Y.; et al. Tumor-derived CD109 orchestrates reprogramming of tumor-associated macrophages to dampen immune response. J. Hepatol. 2025, 83, 946–958. [Google Scholar]
- Dai, Y.; Dong, C.; Wang, Z.; Zhou, Y.; Wang, Y.; Hao, Y.; Chen, P.; Liang, C.; Li, G. Infiltrating T lymphocytes and tumor microenvironment within cholangiocarcinoma: Immune heterogeneity, intercellular communication, immune checkpoints. Front. Immunol. 2024, 15, 1482291. [Google Scholar] [CrossRef]
- Hua, S.; Gu, X.; Jin, H.; Zhang, X.; Liu, Q.; Yang, J. Tumor-infiltrating T lymphocytes: A promising immunotherapeutic target for preventing immune escape in cholangiocarcinoma. Biomed. Pharmacother. 2024, 177, 117080. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Teng, M.W.L.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yu, D.; Qian, H.; Shi, Y.; Tao, Z. CD8+ T cell-based cancer immunotherapy. J. Transl. Med. 2024, 22, 394. [Google Scholar] [CrossRef] [PubMed]
- Vigano, L.; Soldani, C.; Franceschini, B.; Cimino, M.; Lleo, A.; Donadon, M.; Roncalli, M.; Aghemo, A.; Di Tommaso, L.; Torzilli, G. Tumor-Infiltrating Lymphocytes and Macrophages in Intrahepatic Cholangiocellular Carcinoma. Impact on Prognosis after Complete Surgery. J. Gastrointest. Surg. 2019, 23, 2216–2224. [Google Scholar] [CrossRef]
- Lu, J.C.; Wu, L.L.; Sun, Y.N.; Huang, X.Y.; Gao, C.; Guo, X.J.; Zeng, H.Y.; Qu, X.D.; Chen, Y.; Wu, D.; et al. Macro CD5L+ deteriorates CD8+T cells exhaustion and impairs combination of Gemcitabine-Oxaliplatin-Lenvatinib-anti-PD1 therapy in intrahepatic cholangiocarcinoma. Nat. Commun. 2024, 15, 621. [Google Scholar] [CrossRef]
- Wan, W.; Li, Y.; Sun, W.; Cheng, Z.; Ma, F.; Shen, S.; Liu, H.; Zhang, J. The DCDC2/ENO1 axis promotes tumor progression and immune evasion in intrahepatic cholangiocarcinoma via activating FGL1-LAG3 checkpoint. J. Exp. Clin. Cancer Res. 2025, 44, 177. [Google Scholar] [CrossRef]
- Kim, H.D.; Jeong, S.; Park, S.; Lee, Y.J.; Ju, Y.S.; Kim, D.; Song, G.W.; Lee, J.H.; Kim, S.Y.; Shin, J.; et al. Implication of CD69+ CD103+ tissue-resident-like CD8+ T cells as a potential immunotherapeutic target for cholangiocarcinoma. Liver Int. 2021, 41, 764–776. [Google Scholar] [CrossRef]
- Panya, A.; Thepmalee, C.; Sawasdee, N.; Sujjitjoon, J.; Phanthaphol, N.; Junking, M.; Wongkham, S.; Yenchitsomanus, P.T. Cytotoxic activity of effector T cells against cholangiocarcinoma is enhanced by self-differentiated monocyte-derived dendritic cells. Cancer Immunol. Immunother. 2018, 67, 1579–1588. [Google Scholar] [CrossRef]
- Oliveira, G.; Wu, C.J. Dynamics and specificities of T cells in cancer immunotherapy. Nat. Rev. Cancer Nat. Res. 2023, 23, 295–316. [Google Scholar] [CrossRef]
- Oliviero, B.; Varchetta, S.; Mele, D.; Pessino, G.; Maiello, R.; Falleni, M.; Tosi, D.; Donadon, M.; Soldani, C.; Franceschini, B.; et al. MICA/B-targeted antibody promotes NK cell–driven tumor immunity in patients with intrahepatic cholangiocarcinoma. Oncoimmunology 2022, 11, 2035919. [Google Scholar] [CrossRef]
- Li, D.; Andaloori, L.; Crowe, M.; Lin, S.; Hong, J.; Zaidi, N.; Ho, M. Development of CAR-T Therapies and Personalized Vaccines for the Treatment of Cholangiocarcinoma: Current Progress, Mechanisms of Action, and Challenges. Am. J. Pathol. 2025, 195, 453–469. [Google Scholar] [CrossRef]
- Sun, B.Y.; Wang, Z.T.; Chen, K.Z.; Song, Y.; Wu, J.F.; Zhang, D.; Sun, G.Q.; Zhou, J.; Fan, J.; Hu, B.; et al. Mobilization and activation of tumor-infiltrating dendritic cells inhibits lymph node metastasis in intrahepatic cholangiocarcinoma. Cell Death Discov. 2024, 10, 304. [Google Scholar] [CrossRef]
- Liu, H.; Zeng, X.; Ren, X.; Zhang, Y.; Huang, M.; Tan, L.; Dai, Z.; Lai, J.; Xie, W.; Chen, Z.; et al. Targeting tumour-intrinsic N7-methylguanosine tRNA modification inhibits MDSC recruitment and improves anti-PD-1 efficacy. Gut 2023, 72, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Ielpo, S.; Barberini, F.; Dabbagh Moghaddam, F.; Pesce, S.; Cencioni, C.; Spallotta, F.; De Ninno, A.; Businaro, L.; Marcenaro, E.; Bei, R.; et al. Crosstalk and communication of cancer-associated fibroblasts with natural killer and dendritic cells: New frontiers and unveiled opportunities for cancer immunotherapy. Cancer Treat. Rev. 2024, 131, 102843. [Google Scholar] [CrossRef] [PubMed]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural killer cells: Development, maturation, and clinical utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Sharabi, A. Targeting myeloid-derived suppressor cells to enhance natural killer cell-based immunotherapy. Pharmacol. Ther. 2022, 235, 108114. [Google Scholar] [CrossRef]
- Rimassa, L.; Personeni, N.; Aghemo, A.; Lleo, A. The immune milieu of cholangiocarcinoma: From molecular pathogenesis to precision medicine. J. Autoimmun. 2019, 100, 17–26. [Google Scholar] [CrossRef]
- Ma, C.; Zhang, Q.; Greten, T.F. MDSCs in liver cancer: A critical tumor-promoting player and a potential therapeutic target. Cell Immunol. 2021, 361, 104295. [Google Scholar] [CrossRef]
- Han, J.; Wang, H. Cytokine-overexpressing dendritic cells for cancer immunotherapy. Exp. Mol. Med. 2024, 56, 2559–2568. [Google Scholar] [CrossRef] [PubMed]
- Tay, C.; Tanaka, A.; Sakaguchi, S. Tumor-infiltrating regulatory T cells as targets of cancer immunotherapy. Cancer Cell 2023, 41, 450–465. [Google Scholar] [CrossRef] [PubMed]
- Imianowski, C.J.; Chen, Q.; Workman, C.J.; Vignali, D.A.A. Regulatory T cells in the tumour microenvironment. Nat. Rev. Cancer 2025, 25, 703–722. [Google Scholar] [CrossRef] [PubMed]
- Konishi, D.; Umeda, Y.; Yoshida, K.; Shigeyasu, K.; Yano, S.; Toji, T.; Takeda, S.; Yoshida, R.; Yasui, K.; Fuji, T.; et al. Regulatory T cells induce a suppressive immune milieu and promote lymph node metastasis in intrahepatic cholangiocarcinoma. Br. J. Cancer 2022, 127, 757–765. [Google Scholar] [CrossRef]
- Nie, J.; Zhang, S.; Guo, Y.; Liu, C.; Shi, J.; Wu, H.; Na, R.; Liang, Y.; Yu, S.; Quan, F.; et al. Mapping of the T-cell Landscape of Biliary Tract Cancer Unravels Anatomic Subtype-Specific Heterogeneity. Cancer Res. 2025, 85, 704–722. [Google Scholar] [CrossRef]
- Zhang, M.; Yang, H.; Wan, L.; Wang, Z.; Wang, H.; Ge, C.; Liu, Y.; Hao, Y.; Zhang, D.; Shi, G.; et al. Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol. 2020, 73, 1118–1130. [Google Scholar] [CrossRef]
- Gao, X.; Xu, M.; Xiao, H.; Han, Z.; Wang, Z.; Sun, G.; Zhang, D.; Shuangjian, Q.; Ren, N.; Zhou, C.; et al. Tumor-associated neutrophils: A complex role in cancer. Clin. Immunol. 2025, 280, 110558. [Google Scholar] [CrossRef]
- Zhou, Y.; Shen, G.; Zhou, X.; Li, J. Therapeutic potential of tumor-associated neutrophils: Dual role and phenotypic plasticity. Signal Transduct. Target. Ther. 2025, 10, 178. [Google Scholar] [CrossRef]
- Sugahara, O.; Koga, D.; Oka, T.; Sugiyama, S.; Wada, R.; Higa, T.; Nakayama, K.I. Antitumor Activity of Tumor-Infiltrating Neutrophils Revealed by a Syngeneic Mouse Model of Cholangiocarcinoma. Cancer Sci. 2025, 116, 2457–2470. [Google Scholar] [CrossRef]
- Mao, Z.Y.; Zhu, G.Q.; Xiong, M.; Ren, L.; Bai, L. Prognostic value of neutrophil distribution in cholangiocarcinoma. World J. Gastroenterol. 2015, 21, 4961–4968. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Heij, L.R.; Czigany, Z.; Dahl, E.; Dulk, M.D.; Lang, S.A.; Ulmer, T.F.; Neumann, U.P.; Bednarsch, J. The prognostic value of neutrophil-to-lymphocyte ratio in cholangiocarcinoma: A systematic review and meta-analysis. Sci. Rep. 2022, 12, 12691. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Shi, H.; Zhang, B.; Ou, X.; Ma, Q.; Chen, Y.; Shu, P.; Li, D.; Wang, Y. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct. Target. Ther. 2021, 6, 362. [Google Scholar] [CrossRef] [PubMed]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1α regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Kwak, T.; Wang, F.; Deng, H.; Condamine, T.; Kumar, V.; Perego, M.; Kossenkov, A.; Montaner, L.J.; Xu, X.; Xu, W.; et al. Distinct Populations of Immune-Suppressive Macrophages Differentiate from Monocytic Myeloid-Derived Suppressor Cells in Cancer. Cell Rep. 2020, 33, 108571. [Google Scholar] [CrossRef]
- Zhang, Q.; Ma, C.; Duan, Y.; Heinrich, B.; Rosato, U.; Diggs, L.P.; Ma, L.; Roy, S.; Fu, Q.; Brown, Z.J.; et al. Gut Microbiome Directs Hepatocytes to Recruit MDSCs and Promote Cholangiocarcinoma. Cancer Discov. 2021, 11, 1248–1267. [Google Scholar] [CrossRef]
- Takagi, S.; Miyagawa, S.; Ichikawa, E.; Soeda, J.; Miwa, S.; Miyagawa, Y.; Iijima, S.; Noike, T.; Kobayashi, A.; Kawasaki, S. Dendritic cells, T-cell infiltration, and grp94 expression in cholangiocellular carcinoma. Hum. Pathol. 2004, 35, 881–886. [Google Scholar] [CrossRef]
- Mazzoccoli, L.; Liu, B. Dendritic Cells in Shaping Anti-Tumor T Cell Response. Cancers 2024, 16, 2211. [Google Scholar] [CrossRef]
- Mentucci, F.M.; Ferrara, M.G.; Ercole, A.; Rumie Vittar, N.B.; Lamberti, M.J. Interplay between cancer-associated fibroblasts and dendritic cells: Implications for tumor immunity. Front. Immunol. 2025, 16, 1515390. [Google Scholar] [CrossRef]
- Chen, J.; Duan, Y.; Che, J.; Zhu, J. Dysfunction of dendritic cells in tumor microenvironment and immunotherapy. Cancer Commun. 2024, 44, 1047–1070. [Google Scholar] [CrossRef]
- Hansen, F.J.; David, P.; Weber, G.F. The Multifaceted Functionality of Plasmacytoid Dendritic Cells in Gastrointestinal Cancers: A Potential Therapeutic Target? Cancers 2024, 16, 2216. [Google Scholar] [CrossRef]
- Hu, Z.Q.; Zhou, Z.J.; Luo, C.B.; Xin, H.Y.; Li, J.; Yu, S.Y.; Zhou, S.L. Peritumoral plasmacytoid dendritic cells predict a poor prognosis for intrahepatic cholangiocarcinoma after curative resection. Cancer Cell Int. 2020, 20, 582. [Google Scholar] [CrossRef]
- Kendall, T.; Verheij, J.; Gaudio, E.; Evert, M.; Guido, M.; Goeppert, B.; Carpino, G. Anatomical, histomorphological and molecular classification of cholangiocarcinoma. Liver Int. 2019, 39, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Chen, D.; Zhao, B.; Ren, L.; Huang, R.; Feng, B.; Chen, H. The predictive value of PD-L1 expression in patients with advanced hepatocellular carcinoma treated with PD-1/PD-L1 inhibitors: A systematic review and meta-analysis. Cancer Med. 2023, 12, 9282–9292. [Google Scholar] [CrossRef]
- Ngo, P.; Cooper, W.A.; Wade, S.; Fong, K.M.; Canfell, K.; Karikios, D.; Weber, M. Why PD-L1 expression varies between studies of lung cancer: Results from a Bayesian meta-analysis. Sci. Rep. 2025, 15, 4166. [Google Scholar] [CrossRef] [PubMed]
- Kaunitz, G.J.; Cottrell, T.R.; Lilo, M.; Muthappan, V.; Esandrio, J.; Berry, S.; Xu, H.; Ogurtsova, A.; Anders, R.A.; Fischer, A.H.; et al. Melanoma subtypes demonstrate distinct PD-L1 expression profiles. Lab. Investig. 2017, 97, 1063–1071. [Google Scholar] [CrossRef]
- Frega, G.; Cossio, F.P.; Banales, J.M.; Cardinale, V.; Macias, R.I.R.; Braconi, C.; Lamarca, A. Lacking Immunotherapy Biomarkers for Biliary Tract Cancer: A Comprehensive Systematic Literature Review and Meta-Analysis. Cells 2023, 12, 2098. [Google Scholar] [CrossRef] [PubMed]
- Kubecek, O.; Trojanova, P.; Molnarova, V.; Kopecky, J. Microsatellite instability as a predictive factor for immunotherapy in malignant melanoma. Med. Hypotheses 2016, 93, 74–76. [Google Scholar] [CrossRef]
- Akagi, K.; Oki, E.; Taniguchi, H.; Nakatani, K.; Aoki, D.; Kuwata, T.; Yoshino, T. Real-world data on microsatellite instability status in various unresectable or metastatic solid tumors. Cancer Sci. 2021, 112, 1105–1113. [Google Scholar] [CrossRef]
- Tian, J.; Wang, H.; Lu, C.; Liu, L.; Zhang, X.; Xie, Y.; Li, R.; Lv, X.; Fu, D.; Zhang, L.; et al. Genomic characteristics and prognosis of lung cancer patients with MSI-H: A cohort study. Lung Cancer 2023, 181, 107255. [Google Scholar] [CrossRef]
- Shao, C.; Li, G.; Huang, L.; Pruitt, S.; Castellanos, E.; Frampton, G.; Carson, K.R.; Snow, T.; Singal, G.; Fabrizio, D.; et al. Prevalence of High Tumor Mutational Burden and Association with Survival in Patients with Less Common Solid Tumors. JAMA Netw Open 2020, 3, e2025109. [Google Scholar] [CrossRef]
- Gabbia, D.; De Martin, S. Tumor Mutational Burden for Predicting Prognosis and Therapy Outcome of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2023, 24, 3441. [Google Scholar] [CrossRef]
- Jung, J.; Heo, Y.J.; Park, S. High tumor mutational burden predicts favorable response to anti-PD-(L)1 therapy in patients with solid tumor: A real-world pan-tumor analysis. J. Immunother. Cancer 2023, 11, e006454. [Google Scholar] [CrossRef] [PubMed]
- Doi, N.; Ino, Y.; Fuse, M.; Esaki, M.; Shimada, K.; Hiraoka, N. Correlation of Vein-Rich Tumor Microenvironment of Intrahepatic Cholangiocarcinoma with Tertiary Lymphoid Structures and Patient Outcome. Mod. Pathol. 2024, 37, 100401. [Google Scholar] [CrossRef] [PubMed]
- Karapetyan, L.; Li, A.; Vargas De Stefano, D.; Abushukair, H.M.; Al-Bzour, A.N.; Knight, A.; Layding, C.; Wang, H.; Xu, J.; Yao, J.; et al. Differences in the pathological, transcriptomic, and prognostic implications of lymphoid structures between primary and metastatic cutaneous melanomas. J. Immunother. Cancer 2024, 12, e009231. [Google Scholar] [CrossRef] [PubMed]
- Elfving, H.; Yu, H.; Fessehatsion, K.K.; Brunnström, H.; Botling, J.; Gulyas, M.; Backman, M.; Lindberg, A.; Strell, C.; Micke, P. Spatial distribution of tertiary lymphoid structures in the molecular and clinical context of non-small cell lung cancer. Cell. Oncol. 2025, 48, 801–813. [Google Scholar] [CrossRef]
- Li, H.; Liu, H.; Fu, H.; Li, J.; Xu, L.; Wang, G.; Wu, H. Peritumoral Tertiary Lymphoid Structures Correlate With Protective Immunity and Improved Prognosis in Patients with Hepatocellular Carcinoma. Front. Immunol. 2021, 12, 648812. [Google Scholar] [CrossRef]
- Dong, L.; Lu, D.; Chen, R.; Lin, Y.; Zhu, H.; Zhang, Z.; Cai, S.; Cui, P.; Song, G.; Rao, D.; et al. Proteogenomic characterization identifies clinically relevant subgroups of intrahepatic cholangiocarcinoma. Cancer Cell. 2022, 40, 70–87.e15. [Google Scholar] [CrossRef]
- Lee, J.H.; Shin, D.H.; Park, W.Y.; Shin, N.; Kim, A.; Lee, H.J.; Kim, Y.K.; Choi, K.U.; Kim, J.Y.; Yang, Y.I.; et al. IDH1 R132C mutation is detected in clear cell hepatocellular carcinoma by pyrosequencing. World J. Surg. Oncol. 2017, 15, 82. [Google Scholar] [CrossRef]
- Shibata, T.; Kokubu, A.; Miyamoto, M.; Sasajima, Y.; Yamazaki, N. Mutant IDH1 confers an in vivo growth in a melanoma cell line with BRAF mutation. Am. J. Pathol. 2011, 178, 1395–1402. [Google Scholar] [CrossRef]
- Vogel, A.; Segatto, O.; Stenzinger, A.; Saborowski, A. FGFR2 Inhibition in Cholangiocarcinoma. Annu. Rev. Med. 2023, 74, 293–306. [Google Scholar] [CrossRef] [PubMed]
- Bold, N.; Buyanbat, K.; Enkhtuya, A.; Myagmar, N.; Batbayar, G.; Sandag, Z.; Damdinbazar, D.; Oyunbat, N.; Boldbaatar, T.; Enkhbaatar, A.; et al. High-Frequency Mutations in TP53, AXIN1, CTNNB1, and KRAS, and Polymorphisms in JAK1 Genes Among Mongolian HCC Patients. Cancer Rep. 2025, 8, e70227. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.K.H.; Skoulidis, F.; Kerr, K.M.; Ahn, M.J.; Kapp, J.R.; Soares, F.A.; Yatabe, Y. KRAS G12C in advanced NSCLC: Prevalence, co-mutations, and testing. Lung Cancer 2023, 184, 107293. [Google Scholar] [CrossRef]
- Rabbie, R.; Ferguson, P.; Wong, K.; Couturier, D.L.; Moran, U.; Turner, C.; Emanuel, P.; Haas, K.; Saunus, J.M.; Davidson, M.R.; et al. The mutational landscape of melanoma brain metastases presenting as the first visceral site of recurrence. Br. J. Cancer. 2021, 124, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Bousou, T.E.; Sarantis, P.; Anastasiou, I.A.; Trifylli, E.M.; Liapopoulos, D.; Korakaki, D.; Koustas, E.; Katsimpoulas, M.; Karamouzis, M.V. Biomarkers for the Evaluation of Immunotherapy in Patients with Cholangiocarcinoma. Cancers 2025, 17, 555. [Google Scholar] [CrossRef]
- Weinberg, B.A.; Xiu, J.; Lindberg, M.R.; Shields, A.F.; Hwang, J.J.; Poorman, K.; Salem, M.E.; Pishvaian, M.J.; Holcombe, R.F.; Marshall, J.L.; et al. Molecular profiling of biliary cancers reveals distinct molecular alterations and potential therapeutic targets. J. Gastrointest. Oncol. 2019, 10, 652–662. [Google Scholar] [CrossRef]
- Kim, R.; Park, J.K.; Kwon, M.; An, M.; Hong, J.Y.; Park, J.O.; Lim, S.H.; Kim, S.T. Comprehensive molecular characterization to predict immunotherapy response in advanced biliary tract cancer: A phase II trial of pembrolizumab. Oncol. Res. 2025, 33, 57–65. [Google Scholar]
- Chan, T.A.; Yarchoan, M.; Jaffee, E.; Swanton, C.; Quezada, S.A.; Stenzinger, A.; Peters, S. Development of tumor mutation burden as an immunotherapy biomarker: Utility for the oncology clinic. Ann. Oncol. 2019, 30, 44–56. [Google Scholar] [CrossRef]
- Jardim, D.L.; Goodman, A.; de Melo Gagliato, D.; Kurzrock, R. The Challenges of Tumor Mutational Burden as an Immunotherapy Biomarker. Cancer Cell. 2021, 39, 154–173. [Google Scholar] [CrossRef]
- Zhang, Y.; Gou, M. Combined Chemotherapy-Immunotherapy for Advanced Biliary Tract Cancer (BTC): A Clinical, Genomic, and Biomarker Analysis. J. Gastrointest. Cancer. 2025, 56, 90. [Google Scholar] [CrossRef]
- Nakamura, H.; Arai, Y.; Totoki, Y.; Shirota, T.; Elzawahry, A.; Kato, M.; Hama, N.; Hosoda, F.; Urushidate, T.; Ohashi, S.; et al. Genomic spectra of biliary tract cancer. Nat. Genet. 2015, 47, 1003–1010. [Google Scholar] [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef]
- Ricci, A.D.; Rizzo, A.; Brandi, G. Immunotherapy in Biliary Tract Cancer: Worthy of a Second Look. Cancer Control. 2020, 27, 1073274820948047. [Google Scholar] [CrossRef]
- Lin, G.; Liu, Y.; Li, S.; Mao, Y.; Wang, J.; Shuang, Z.; Chen, J.; Li, S. Elevated neutrophil-to-lymphocyte ratio is an independent poor prognostic factor in patients with intrahepatic cholangiocarcinoma. Oncotarget 2016, 7, 50963–50971. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xie, Y.; Cai, Y.; Hu, H.; He, M.; Liu, L.; Liao, C.; Wang, Y.; Wang, J.; Ren, X.; et al. Multiomic Analysis Reveals Comprehensive Tumor Heterogeneity and Distinct Immune Subtypes in Multifocal Intrahepatic Cholangiocarcinoma. Clin. Cancer Res. 2022, 28, 1896–1910. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, Y.; Tan, Z.; Song, Y.; Chen, K.; Liu, S.; Peng, C.; Chen, X. Machine Learning-based Macrophage Signature for Predicting Prognosis and Immunotherapy Benefits in Cholangiocarcinoma. Curr. Med. Chem. 2024, 32, 8945–8958. [Google Scholar] [CrossRef] [PubMed]
- Kida, A.; Mizukoshi, E.; Tamai, T.; Terashima, T.; Kitahara, M.; Arai, K.; Yamashita, T.; Fushimi, K.; Honda, M.; Kaneko, S. Immune responses against tumour-associated antigen-derived cytotoxic T lymphocyte epitopes in cholangiocarcinoma patients. Liver Int. 2018, 38, 2040–2050. [Google Scholar]
- Shang, T.; Jiang, T.; Lu, T.; Wang, H.; Cui, X.; Pan, Y.; Xu, M.; Pei, M.; Ding, Z.; Feng, X.; et al. Tertiary lymphoid structures predict the prognosis and immunotherapy response of cholangiocarcinoma. Front. Immunol. 2023, 14, 1166497. [Google Scholar] [CrossRef]
- Chen, X.; Wang, D.; Liu, J.; Qiu, J.; Zhou, J.; Ying, J.; Shi, Y.; Wang, Z.; Lou, H.; Cui, J.; et al. Genomic alterations in biliary tract cancer predict prognosis and immunotherapy outcomes. J. Immunother. Cancer 2021, 9, e003214. [Google Scholar] [CrossRef]
- Wu, M.J.; Shi, L.; Dubrot, J.; Merritt, J.; Vijay, V.; Wei, T.Y.; Kessler, E.; Olander, K.E.; Adil, R.; Pankaj, A.; et al. Mutant IDH Inhibits IFNγ-TET2 Signaling to Promote Immunoevasion and Tumor Maintenance in Cholangiocarcinoma. Cancer Discov. 2022, 12, 812–835. [Google Scholar]
- Aguado-Fraile, E.; Tassinari, A.; Ishii, Y.; Sigel, C.; Lowery, M.A.; Goyal, L.; Gliser, C.; Jiang, L.; Pandya, S.S.; Wu, B.; et al. Molecular and morphological changes induced by ivosidenib correlate with efficacy in mutant-IDH1 cholangiocarcinoma. Future Oncol. 2021, 17, 2057–2074. [Google Scholar] [CrossRef]
- Uson Junior, P.L.S.; Borad, M.J. Clinical Utility of Ivosidenib in the Treatment of IDH1-Mutant Cholangiocarcinoma: Evidence To Date. Cancer Manag. Res. 2023, 15, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, G.; Wu, Z.; Dong, Y.; Shi, Y.; Yang, F.; Chen, X.; Wang, J.; Du, S.; Xu, H.; et al. Exosomal circ-PTPN22 and circ-ADAMTS6 mark T cell exhaustion and neutrophil extracellular traps in Asian intrahepatic cholangiocarcinoma. Mol. Ther. Nucleic Acids 2023, 31, 151–163. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, M.; Pu, H.; Guo, S.; Zhang, S.; Wang, Y. Prognostic Implications of Pan-Cancer CMTM6 Expression and Its Relationship with the Immune Microenvironment. Front. Oncol. 2020, 10, 585961. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Li, S.; Liu, K.; Fan, R.; Liu, F.; Zhang, H.; Liu, D.; Shen, D. Downregulation of dermatopontin in cholangiocarcinoma cells suppresses CCL19 secretion of macrophages and immune infiltration. J. Cancer Res. Clin. Oncol. 2024, 150, 66. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhang, D.; Sima, X.; Fu, Y.; Zeng, H.; Hu, Z.; Hou, J.; Pan, Y.; Zhang, Y.; Zhou, Z.; et al. Levels of pretreatment serum lipids predict responses to PD-1 inhibitor treatment in advanced intrahepatic cholangiocarcinoma. Int. Immunopharmacol. 2023, 115, 109687. [Google Scholar] [CrossRef] [PubMed]
- Park, E.M.; Chelvanambi, M.; Bhutiani, N.; Kroemer, G.; Zitvogel, L.; Wargo, J.A. Targeting the gut and tumor microbiota in cancer. Nat. Med. 2022, 28, 690–703. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The microbiome and human cancer. Science 2021, 371, eabc4552. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, J.; Xia, Q. Role of gut microbiome in cancer immunotherapy: From predictive biomarker to therapeutic target. Exp. Hematol. Oncol. 2023, 12, 84. [Google Scholar] [CrossRef]
- Xin, H.Y.; Zou, J.X.; Sun, R.Q.; Hu, Z.Q.; Chen, Z.; Luo, C.B.; Zhou, Z.J.; Wang, P.C.; Li, J.; Yu, S.Y.; et al. Characterization of tumor microbiome and associations with prognosis in intrahepatic cholangiocarcinoma. J. Gastroenterol. 2024, 59, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Wang, Y.; Zhu, R.; Wang, S.; Xue, J.; Zhang, D.; Lan, Z.; Zhang, C.; Liang, Y.; Zhang, N.; et al. Gut microbiota and metabolites signatures of clinical response in anti-PD-1/PD-L1 based immunotherapy of biliary tract cancer. Biomark. Res. 2024, 12, 56. [Google Scholar] [CrossRef] [PubMed]
- Roussel, E.; Brasse-Lagnel, C.; Tuech, J.J.; Montialoux, H.; Papet, E.; Tortajada, P.; Bekri, S.; Schwarz, L. Influence of Probiotics Administration Before Liver Resection in Patients with Liver Disease: A Randomized Controlled Trial. World J. Surg. 2022, 46, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Ke, Y.; Liu, Q.; Yang, J.; Liu, F.; Xu, R.; Zhou, H.; Chen, A.; Xiao, J.; Meng, F.; et al. Engineered Lactococcus lactis secreting Flt3L and OX40 ligand for in situ vaccination-based cancer immunotherapy. Nat. Commun. 2022, 13, 7466. [Google Scholar] [CrossRef]
- Singh, A.; Alexander, S.G.; Martin, S. Gut microbiome homeostasis and the future of probiotics in cancer immunotherapy. Front. Immunol. 2023, 14, 1114499. [Google Scholar] [CrossRef]
- Spencer, C.N.; McQuade, J.L.; Gopalakrishnan, V.; McCulloch, J.A.; Vetizou, M.; Cogdill, A.P.; Khan, M.A.W.; Zhang, X.; White, M.G.; Peterson, C.B.; et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science 2021, 374, 1632–1640. [Google Scholar] [CrossRef]
- Kang, X.; Liu, C.; Ding, Y.; Ni, Y.; Ji, F.; Lau, H.C.H.; Jiang, L.; Sung, J.J.; Wong, S.H.; Yu, J.; et al. Roseburia intestinalis generated butyrate boosts anti-PD-1 efficacy in colorectal cancer by activating cytotoxic CD8+ T cells. Gut 2023, 72, 2112–2122. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, M.; Liu, X.; Chai, M.; Diao, L.; Ma, L.; Nie, S.; Xu, M.; Wang, Y.; Mo, F.; et al. Probiotics formulation and cancer nanovaccines show synergistic effect in immunotherapy and prevention of colon cancer. iScience 2023, 26, 107167. [Google Scholar] [CrossRef]
- Boucher, E.; Plazy, C.; Richard, M.L.; Suau, A.; Mangin, I.; Cornet, M.; Aldebert, D.; Toussaint, B.; Hannani, D. Inulin prebiotic reinforces host cancer immunosurveillance via ɣδ T cell activation. Front. Immunol. 2023, 14, 1104224. [Google Scholar] [CrossRef]
- Huang, J.; Gong, C.; Zhou, A. Modulation of gut microbiota: A novel approach to enhancing the effects of immune checkpoint inhibitors. Ther. Adv. Med. Oncol. 2023, 15, 17588359231204854. [Google Scholar] [CrossRef]
- Dadgar, N.; Arunachalam, A.K.; Hong, H.; Phoon, Y.P.; Arpi-Palacios, J.E.; Uysal, M.; Wehrle, C.J.; Aucejo, F.; Ma, W.W.; Melenhorst, J.J.; et al. Advancing Cholangiocarcinoma Care: Insights and Innovations in T Cell Therapy. Cancers 2024, 16, 3232. [Google Scholar] [CrossRef]
- Castellarin, M.; Watanabe, K.; June, C.H.; Kloss, C.C.; Posey, A.D. Driving cars to the clinic for solid tumors. Gene Ther. 2018, 25, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Li, X.; Wang, X.; Cheng, L.; Li, Z.; Zhang, C.; Ye, Z.; Qian, Q. Current Progress in CAR-T Cell Therapy for Solid Tumors. Int. J. Biol. Sci. 2019, 15, 2548–2560. [Google Scholar] [CrossRef] [PubMed]
- Amorós-Pérez, B.; Rivas-Pardo, B.; Gómez del Moral, M.; Subiza, J.L.; Martínez-Naves, E. State of the Art in CAR-T Cell Therapy for Solid Tumors: Is There a Sweeter Future? Cells 2024, 13, 725. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Uslu, U.; June, C.H. Beyond the blood: Expanding CAR T cell therapy to solid tumors. Nat. Biotechnol. 2025, 43, 506–515. [Google Scholar] [CrossRef]
- Supimon, K.; Sangsuwannukul, T.; Sujjitjoon, J.; Phanthaphol, N.; Chieochansin, T.; Poungvarin, N.; Wongkham, S.; Junking, M.; Yenchitsomanus, P.T. Anti-mucin 1 chimeric antigen receptor T cells for adoptive T cell therapy of cholangiocarcinoma. Sci. Rep. 2021, 11, 6276. [Google Scholar] [CrossRef]
- Mao, L.; Su, S.; Li, J.; Yu, S.; Gong, Y.; Chen, C.; Hu, Z.; Huang, X. Development of Engineered CAR T Cells Targeting Tumor-Associated Glycoforms of MUC1 for the Treatment of Intrahepatic Cholangiocarcinoma. J. Immunother. 2023, 46, 89–95. [Google Scholar] [CrossRef]
- Gomes, R.V.; Rodrigues, M.Â.; Rodrigues, J.B.S.R.; Vidigal, P.T.; Damasceno, K.A.; Lima, H.A.; Gomes, D.A.; Machado, C.J.; Resende, V. Expression of epidermal growth factor receptor (EGFR) in cholangiocarcinomas: Predictive factors and survival. Rev. Col. Bras. Cir. 2018, 45, e1826. [Google Scholar] [CrossRef]
- Guo, Y.; Feng, K.; Liu, Y.; Wu, Z.; Dai, H.; Yang, Q.; Wang, Y.; Jia, H.; Han, W. Phase I Study of Chimeric Antigen Receptor-Modified T Cells in Patients with EGFR-Positive Advanced Biliary Tract Cancers. Clin. Cancer Res. 2018, 24, 1277–1286. [Google Scholar] [CrossRef]
- Feng, K.C.; Guo, Y.L.; Liu, Y.; Dai, H.R.; Wang, Y.; Lv, H.Y.; Huang, J.H.; Yang, Q.M.; Han, W.D. Cocktail treatment with EGFR-specific and CD133-specific chimeric antigen receptor-modified T cells in a patient with advanced cholangiocarcinoma. J. Hematol. Oncol. 2017, 10, 4. [Google Scholar] [CrossRef]
- Paijens, S.T.; Vledder, A.; de Bruyn, M.; Nijman, H.W. Tumor-infiltrating lymphocytes in the immunotherapy era. Cell. Mol. Immunol. 2021, 18, 842–859. [Google Scholar] [CrossRef]
- Kumar, A.; Watkins, R.; Vilgelm, A.E. Cell Therapy with TILs: Training and Taming T Cells to Fight Cancer. Front. Immunol. 2021, 12, 690499. [Google Scholar] [CrossRef]
- Higuchi, R.; Yamamoto, M.; Hatori, T.; Shimizu, K.; Imai, K.; Takasaki, K. Intrahepatic cholangiocarcinoma with lymph node metastasis successfully treated by immunotherapy with CD3-activated T cells and dendritic cells after surgery: Report of a case. Surg. Today 2006, 36, 559–562. [Google Scholar] [CrossRef]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef]
- Amhis, N.; Carignan, J.; Tai, L.H. Transforming pancreaticobiliary cancer treatment: Exploring the frontiers of adoptive cell therapy and cancer vaccines. Mol. Ther. Oncol. 2024, 32, 200825. [Google Scholar] [CrossRef]
- Ding, G.Y.; Ma, J.Q.; Yun, J.P.; Chen, X.; Ling, Y.; Zhang, S.; Shi, J.Y.; Chang, Y.Q.; Ji, Y.; Wang, X.Y.; et al. Distribution and density of tertiary lymphoid structures predict clinical outcome in intrahepatic cholangiocarcinoma. J. Hepatol. 2022, 76, 608–618. [Google Scholar] [CrossRef]
- Fridman, W.H.; Petitprez, F.; Meylan, M.; Chen, T.W.; Sun, C.M.; Roumenina, L.T.; Sautès-Fridman, C. B cells and cancer: To B or not to B? J. Exp. Med. 2021, 218, e20200851. [Google Scholar] [CrossRef] [PubMed]
- Fridman, W.H.; Meylan, M.; Petitprez, F.; Sun, C.M.; Italiano, A.; Sautès-Fridman, C. B cells and tertiary lymphoid structures as determinants of tumour immune contexture and clinical outcome. Nat. Rev. Clin. Oncol. 2022, 19, 441–457. [Google Scholar] [CrossRef] [PubMed]
- Vonderheide, R.H.; Glennie, M.J. Agonistic CD40 antibodies and cancer therapy. Clin. Cancer Res. 2013, 19, 1035–1043. [Google Scholar] [CrossRef]
- Casolino, R.; Braconi, C. CD40-agonist: A new avenue for immunotherapy combinations in cholangiocarcinoma. J. Hepatol. 2021, 74, 1021–1024. [Google Scholar] [CrossRef] [PubMed]
- Kverneland, A.H.; Chamberlain, C.A.; Borch, T.H.; Nielsen, M.; Mørk, S.K.; Kjeldsen, J.W.; Lorentzen, C.L.; Jørgensen, L.P.; Riis, L.B.; Yde, C.W.; et al. Adoptive cell therapy with tumor-infiltrating lymphocytes supported by checkpoint inhibition across multiple solid cancer types. J. Immunother. Cancer 2021, 9, e003499. [Google Scholar] [CrossRef] [PubMed]
- Vitale, L.A.; Thomas, L.J.; He, L.Z.; O’Neill, T.; Widger, J.; Crocker, A.; Sundarapandiyan, K.; Storey, J.R.; Forsberg, E.M.; Weidlick, J.; et al. Development of CDX-1140, an agonist CD40 antibody for cancer immunotherapy. Cancer Immunol. Immunother. 2019, 68, 233–245. [Google Scholar] [PubMed]
- Hochnadel, I.; Kossatz-Boehlert, U.; Jedicke, N.; Lenzen, H.; Manns, M.P.; Yevsa, T. Cancer vaccines and immunotherapeutic approaches in hepatobiliary and pancreatic cancers. Hum. Vaccin. Immunother. 2017, 13, 2931–2952. [Google Scholar] [CrossRef][Green Version]
- Zhao, L.M.; Shi, A.D.; Yang, Y.; Liu, Z.L.; Hu, X.Q.; Shu, L.Z.; Tang, Y.C.; Zhang, Z.L. Advances in molecular and cell therapy for immunotherapy of cholangiocarcinoma. Front. Oncol. 2023, 13, 1140103. [Google Scholar] [CrossRef]
- Igarashi, Y.; Sasada, T. Cancer Vaccines: Toward the Next Breakthrough in Cancer Immunotherapy. J. Immunol. Res. 2020, 2020, 5825401. [Google Scholar] [CrossRef]
- Kamigaki, T.; Takimoto, R.; Okada, S.; Ibe, H.; Oguma, E.; Goto, S. Personalized Dendritic-cell-based Vaccines Targeting Cancer Neoantigens. Anticancer Res. 2024, 44, 3713–3724. [Google Scholar] [CrossRef]
- Popovic, A.; Jaffee, E.M.; Zaidi, N. Emerging strategies for combination checkpoint modulators in cancer immunotherapy. J. Clin. Investig. 2018, 128, 3209–3218. [Google Scholar] [CrossRef]
- Kotera, Y.; Kougen, Y.; Aruga, A.; Yamamoto, M. Dendritic cell vaccine for intrahepatic cholangio cellular carcinoma--a study of relationship between immuno-reaction and clinical outcome. Gan To Kagaku Ryoho 2009, 36, 1964–1966. [Google Scholar]
- Shimizu, K.; Kotera, Y.; Aruga, A.; Takeshita, N.; Takasaki, K.; Yamamoto, M. Clinical utilization of postoperative dendritic cell vaccine plus activated T-cell transfer in patients with intrahepatic cholangiocarcinoma. J. Hepatobiliary Pancreat. Sci. 2012, 19, 171–178. [Google Scholar] [CrossRef]
- Kobayashi, M.; Sakabe, T.; Abe, H.; Tanii, M.; Takahashi, H.; Chiba, A.; Yanagida, E.; Shibamoto, Y.; Ogasawara, M.; Tsujitani, S.; et al. Dendritic cell-based immunotherapy targeting synthesized peptides for advanced biliary tract cancer. J. Gastrointest. Surg. 2013, 17, 1609–1617. [Google Scholar] [CrossRef]
- Yamamoto, K.; Ueno, T.; Kawaoka, T.; Hazama, S.; Fukui, M.; Suehiro, Y.; Hamanaka, Y.; Ikematsu, Y.; Imai, K.; Oka, M.; et al. MUC1 peptide vaccination in patients with advanced pancreas or biliary tract cancer. Anticancer Res. 2005, 25, 3575–3579. [Google Scholar]
- Aruga, A.; Takeshita, N.; Kotera, Y.; Okuyama, R.; Matsushita, N.; Ohta, T.; Takeda, K.; Yamamoto, M. Long-term Vaccination with Multiple Peptides Derived from Cancer-Testis Antigens Can Maintain a Specific T-cell Response and Achieve Disease Stability in Advanced Biliary Tract Cancer. Clin. Cancer Res. 2013, 19, 2224–2231. [Google Scholar] [CrossRef]
- Tang, T.Y.; Huang, X.; Zhang, G.; Lu, M.H.; Liang, T.B. mRNA vaccine development for cholangiocarcinoma: A precise pipeline. Mil. Med. Res. 2022, 9, 40. [Google Scholar] [CrossRef]
- Huang, X.; Tang, T.; Zhang, G.; Liang, T. Identification of tumor antigens and immune subtypes of cholangiocarcinoma for mRNA vaccine development. Mol. Cancer 2021, 20, 50. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).