
The Role of Structural Bioinformatics in Understanding Tumor Necrosis Factor α-Interacting Protein Mechanisms in Chronic Inflammatory Diseases: A Review


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
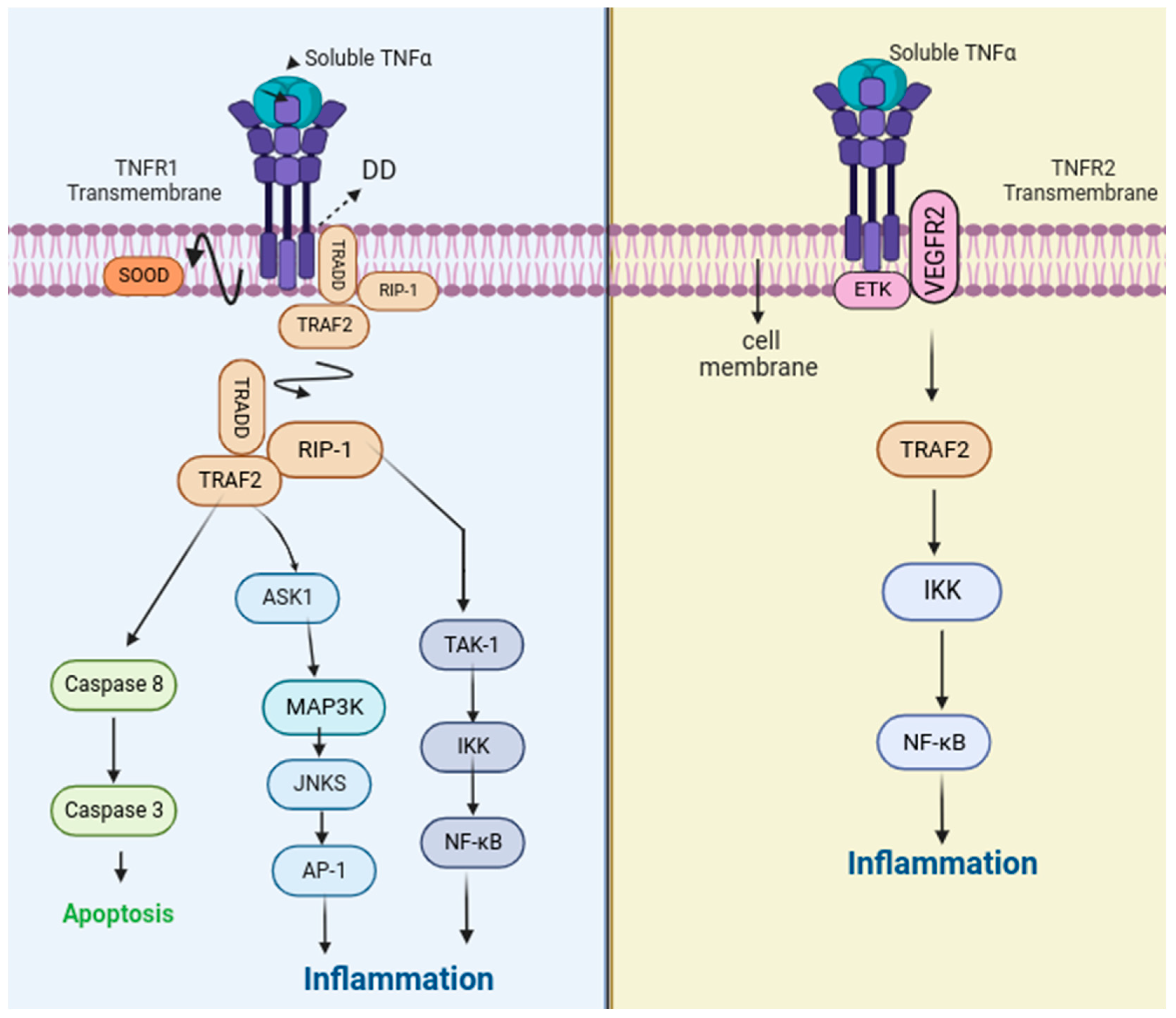
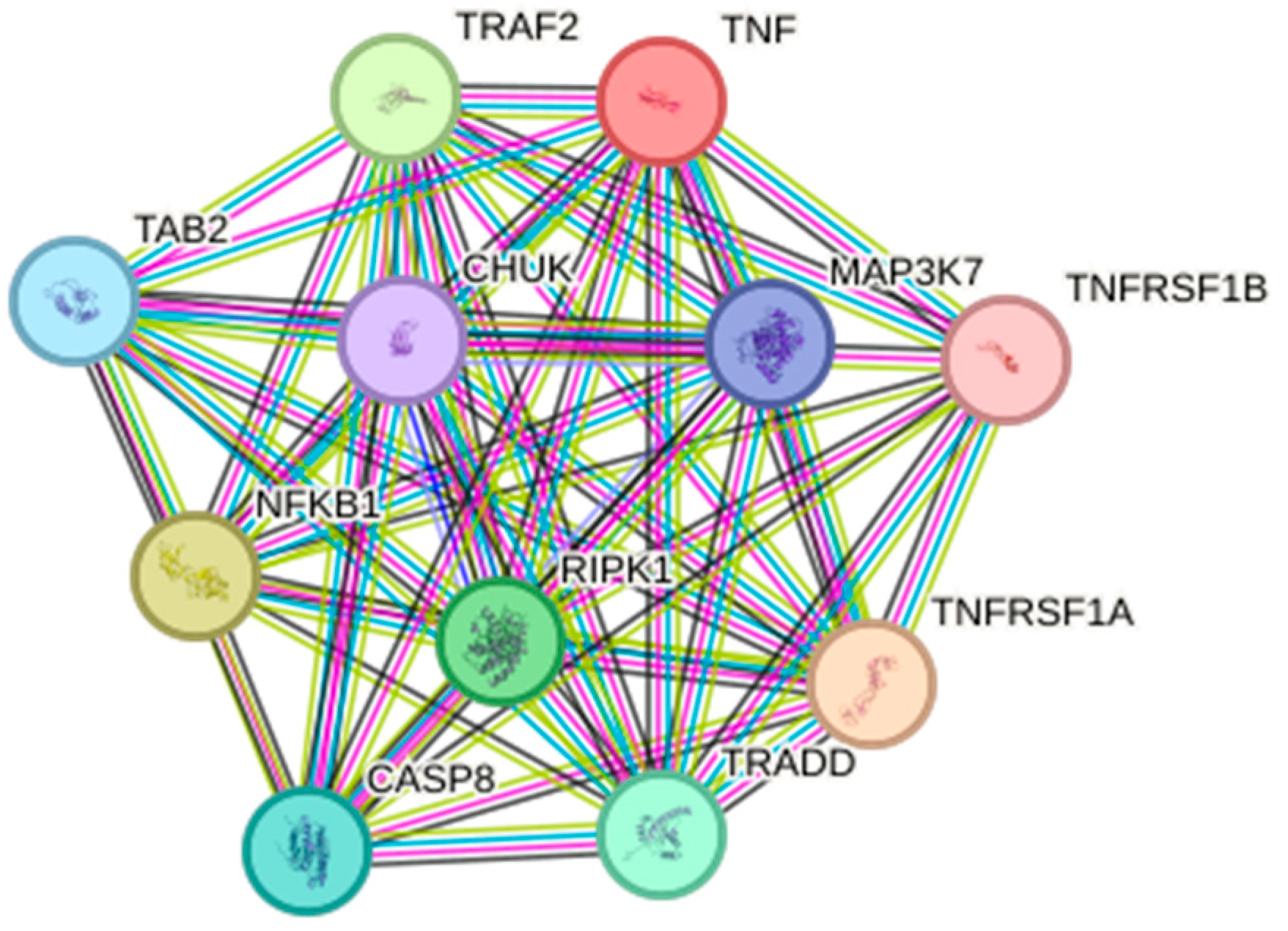
2. Function and Signaling
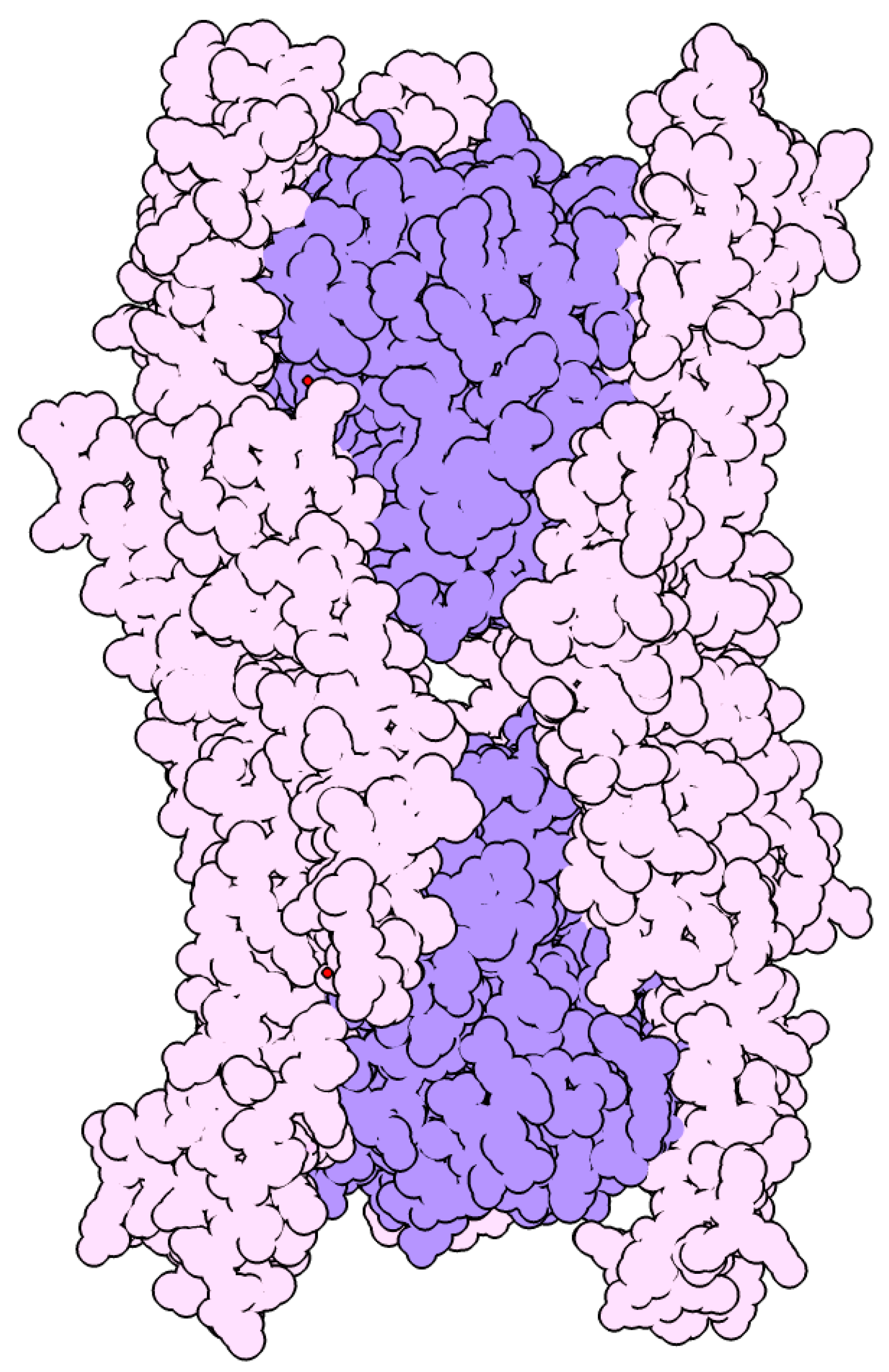
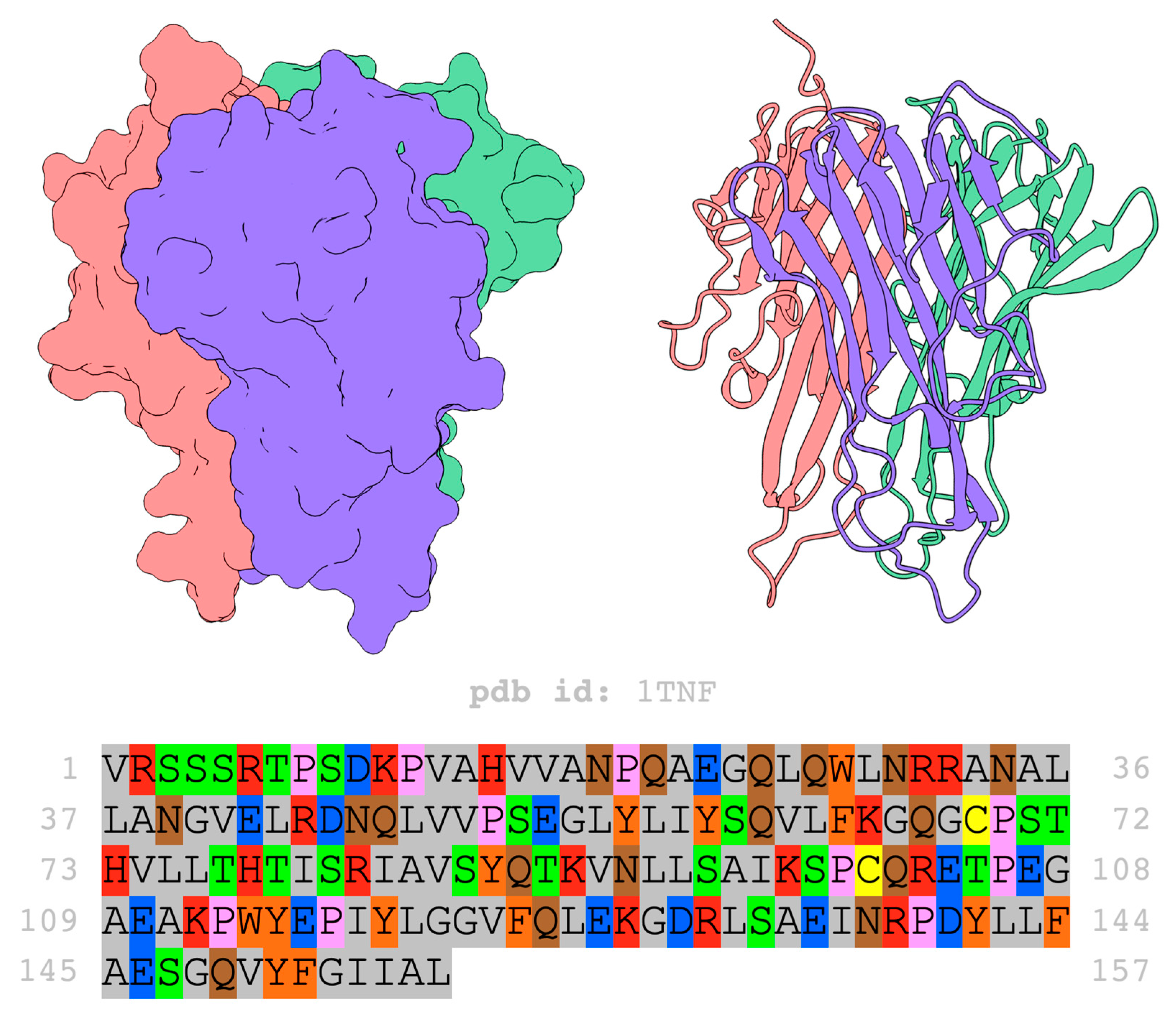
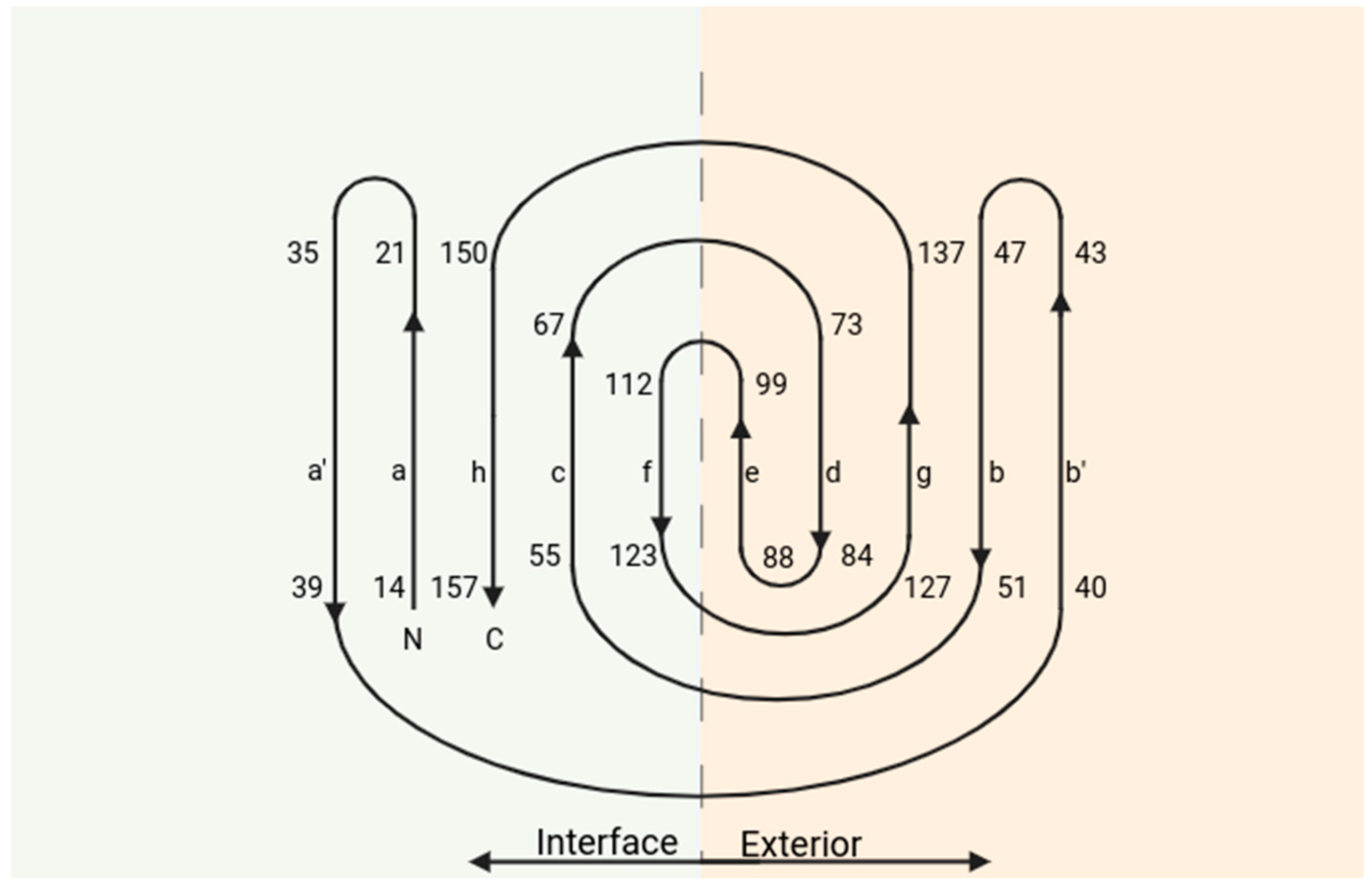
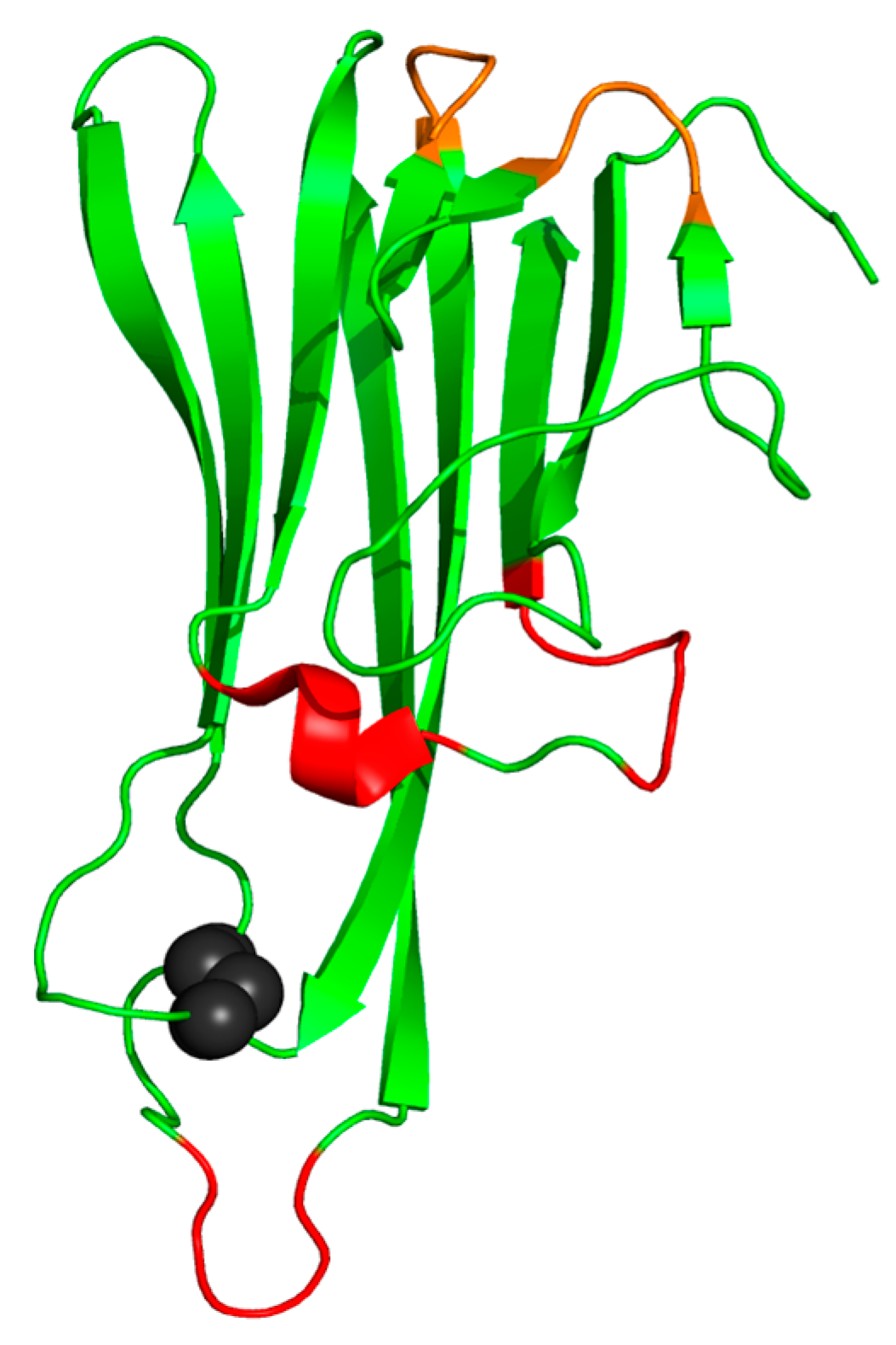
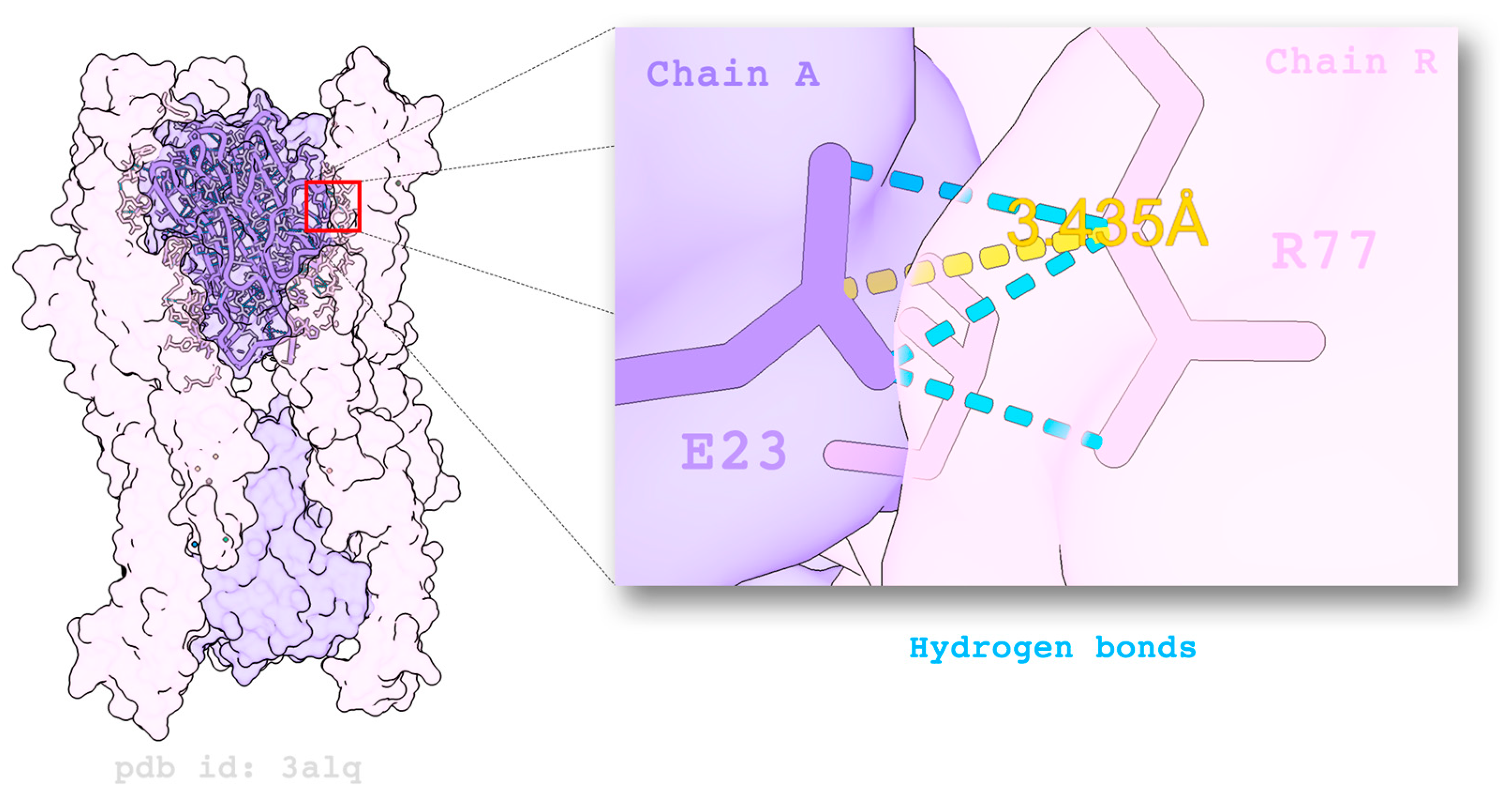
3. Structure
4. TNF-α Role in the Body’s Inflammatory Process
4.1. Rheumatoid Arthritis
4.2. Psoriasis
4.3. Crohn’s Disease
4.4. Ankylosing Spondylitis
4.5. Systemic Sclerosis
4.6. Hidradenitis Suppurativa
4.7. Vasculitis
5. TNF-α Inhibitors as Therapeutic Drugs
6. New Anti-TNF-α Agents
6.1. Ozoralizumab (TS-152)
6.2. ZINC09609430
6.3. CP-690334-01
6.4. TNF-α Inhibitory Activity Detected in Ticks
7. Limitations in TNF-α Research
8. Bioinformatics Approaches
8.1. Biological Databases
8.1.1. Sequences
8.1.2. D-Structures
8.1.3. Pathways
8.2. Protein 3D-Modeling
8.3. Assessing Intra- and Inter-Molecular Interactions
8.4. Molecular Docking and Molecular Dynamics Simulation
9. Bioinformatics Applied to Research on TNF-α Associated with Inflammatory Diseases
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Carswell, E.A.; Old, L.J.; Kassel, R.L.; Green, S.; Fiore, N.; Williamson, B. An Endotoxin-Induced Serum Factor That Causes Necrosis of Tumors. Proc. Natl. Acad. Sci. USA 1975, 72, 3666–3670. [Google Scholar] [CrossRef]
- Bradley, J. TNF-Mediated Inflammatory Disease. J. Pathol. 2008, 214, 149–160. [Google Scholar] [CrossRef]
- Eck, M.J.; Sprang, S.R. The Structure of Tumor Necrosis Factor-Alpha at 2.6 A Resolution. Implications for Receptor Binding. J. Biol. Chem. 1989, 264, 17595–17605. [Google Scholar] [CrossRef]
- Ruddle, N.H. Tumor Necrosis Factor (TNF-α) and Lymphotoxin (TNF-β). Curr. Opin. Immunol. 1992, 4, 327–332. [Google Scholar] [CrossRef]
- Pennica, D.; Hayflick, J.S.; Bringman, T.S.; Palladino, M.A.; Goeddel, D.V. Cloning and Expression in Escherichia Coli of the cDNA for Murine Tumor Necrosis Factor. Proc. Natl. Acad. Sci. USA 1985, 82, 6060–6064. [Google Scholar] [CrossRef]
- Hu, S.; Liang, S.; Guo, H.; Zhang, D.; Li, H.; Wang, X.; Yang, W.; Qian, W.; Hou, S.; Wang, H.; et al. Comparison of the Inhibition Mechanisms of Adalimumab and Infliximab in Treating Tumor Necrosis Factor α-Associated Diseases from a Molecular View. J. Biol. Chem. 2013, 288, 27059–27067. [Google Scholar] [CrossRef]
- Tracey, K.J.; Cerami, A. Tumor Necrosis Factor: A Pleiotropic Cytokine and Therapeutic Target. Annu. Rev. Med. 1994, 45, 491–503. [Google Scholar] [CrossRef]
- Torres, J.; Mehandru, S.; Colombel, J.-F.; Peyrin-Biroulet, L. Crohn’s Disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid Arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Papadakis, K.A.; Targan, S.R. Role of Cytokines in the Pathogenesis of Inflammatory Bowel Disease. Annu. Rev. Med. 2000, 51, 289–298. [Google Scholar] [CrossRef]
- Neurath, M.F. Cytokines in Inflammatory Bowel Disease. Nat. Rev. Immunol. 2014, 14, 329–342. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNF Alpha and the TNF Receptor Superfamily: Structure-Function Relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. Tumor Necrosis Factor Signaling. Cell Death Differ. 2003, 10, 45–65. [Google Scholar] [CrossRef]
- Mukai, Y.; Nakamura, T.; Yoshikawa, M.; Yoshioka, Y.; Tsunoda, S.; Nakagawa, S.; Yamagata, Y.; Tsutsumi, Y. Solution of the Structure of the TNF-TNFR2 Complex. Sci. Signal. 2010, 3, ra83. [Google Scholar] [CrossRef]
- Mukai, Y.; Nakamura, T.; Yoshioka, Y.; Shibata, H.; Abe, Y.; Nomura, T.; Taniai, M.; Ohta, T.; Nakagawa, S.; Tsunoda, S.; et al. Fast Binding Kinetics and Conserved 3D Structure Underlie the Antagonistic Activity of Mutant TNF: Useful Information for Designing Artificial Proteo-Antagonists. J. Biochem. 2009, 146, 167–172. [Google Scholar] [CrossRef]
- Zhao, L.; Fu, Q.; Pan, L.; Piai, A.; Chou, J.J. The Diversity and Similarity of Transmembrane Trimerization of TNF Receptors. Front. Cell Dev. Biol. 2020, 8, 569684. [Google Scholar] [CrossRef]
- Reed, C.; Fu, Z.Q.; Wu, J.; Xue, Y.N.; Harrison, R.W.; Chen, M.J.; Weber, I.T. Crystal Structure of TNF-Alpha Mutant R31D with Greater Affinity for Receptor R1 Compared with R2. Protein Eng. 1997, 10, 1101–1107. [Google Scholar] [CrossRef][Green Version]
- Pobezinskaya, Y.L.; Liu, Z. The Role of TRADD in Death Receptor Signaling. Cell Cycle 2012, 11, 871–876. [Google Scholar] [CrossRef]
- Gaeta, M.L.; Johnson, D.R.; Kluger, M.S.; Pober, J.S. The Death Domain of Tumor Necrosis Factor Receptor 1 Is Necessary but Not Sufficient for Golgi Retention of the Receptor and Mediates Receptor Desensitization. Lab. Investig. 2000, 80, 1185–1194. [Google Scholar] [CrossRef][Green Version]
- MacEwan, D.J. TNF Ligands and Receptors—A Matter of Life and Death. Br. J. Pharmacol. 2002, 135, 855–875. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein Structure Prediction and Analysis Using the Robetta Server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef]
- Zhou, F.; Xu, X.; Wang, D.; Wu, J.; Wang, J. Identification of Novel NF-κB Transcriptional Targets in TNFα-Treated HeLa and HepG2 Cells. Cell Biol. Int. 2017, 41, 555–569. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting Modern Challenges in Visualization and Analysis. Protein Sci. Publ. Protein Soc. 2018, 27, 14. [Google Scholar] [CrossRef]
- He, M.M.; Smith, A.S.; Oslob, J.D.; Flanagan, W.M.; Braisted, A.C.; Whitty, A.; Cancilla, M.T.; Wang, J.; Lugovskoy, A.A.; Yoburn, J.C.; et al. Small-Molecule Inhibition of TNF-Alpha. Science 2005, 310, 1022–1025. [Google Scholar] [CrossRef]
- Tseng, W.-Y.; Huang, Y.-S.; Lin, H.-H.; Luo, S.-F.; McCann, F.; McNamee, K.; Clanchy, F.; Williams, R. TNFR Signalling and Its Clinical Implications. Cytokine 2018, 101, 19–25. [Google Scholar] [CrossRef]
- Mukai, Y.; Shibata, H.; Nakamura, T.; Yoshioka, Y.; Abe, Y.; Nomura, T.; Taniai, M.; Ohta, T.; Ikemizu, S.; Nakagawa, S.; et al. Structure-Function Relationship of Tumor Necrosis Factor (TNF) and Its Receptor Interaction Based on 3D Structural Analysis of a Fully Active TNFR1-Selective TNF Mutant. J. Mol. Biol. 2009, 385, 1221–1229. [Google Scholar] [CrossRef]
- Liang, S.; Dai, J.; Hou, S.; Su, L.; Zhang, D.; Guo, H.; Hu, S.; Wang, H.; Rao, Z.; Guo, Y.; et al. Structural Basis for Treating Tumor Necrosis Factor α (TNFα)-Associated Diseases with the Therapeutic Antibody Infliximab. J. Biol. Chem. 2013, 288, 13799–13807. [Google Scholar] [CrossRef]
- Huang, P.L. A Comprehensive Definition for Metabolic Syndrome. Dis. Model. Mech. 2009, 2, 231–237. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Firestein, G.S.; Budd, R.C.; Gabriel, S.E.; McInnes, I.B.; O’Dell, J.R. Kelley and Firestein’s Textbook of Rheumatology; Elsevier Health Sciences: Amsterdam, The Netherlands, 2016; ISBN 978-0-323-41494-4. [Google Scholar]
- Griffiths, C.E.; Barker, J.N. Pathogenesis and Clinical Features of Psoriasis. Lancet 2007, 370, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Suárez-Fariñas, M.; Krueger, J.G. Immunology of Psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Muñoz, F.; Dominguez-Lopez, A.; Yamamoto-Furusho, J.K. Role of Cytokines in Inflammatory Bowel Disease. World J. Gastroenterol. WJG 2008, 14, 4280–4288. [Google Scholar] [CrossRef] [PubMed]
- Braun, J.; Sieper, J. Ankylosing Spondylitis. Lancet 2007, 369, 1379–1390. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Kenna, T.; Wordsworth, B.P. Genetics of Ankylosing Spondylitis--Insights into Pathogenesis. Nat. Rev. Rheumatol. 2016, 12, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Phumethum, V.; Jamal, S.; Johnson, S.R. Biologic Therapy for Systemic Sclerosis: A Systematic Review. J. Rheumatol. 2011, 38, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Spanò, F.; Contatore, M.; Guastalla, A.; Puppo, F. Potential Use of TNF-α Inhibitors in Systemic Sclerosis. Immunotherapy 2014, 6, 283–289. [Google Scholar] [CrossRef]
- Thyssen, J.P. Atopic Dermatitis, Filaggrin Mutations and Irritant Contact Dermatitis. Br. J. Dermatol. 2013, 168, 233–234. [Google Scholar] [CrossRef]
- Savage, K.T.; Flood, K.S.; Porter, M.L.; Kimball, A.B. TNF-α Inhibitors in the Treatment of Hidradenitis Suppurativa. Ther. Adv. Chronic Dis. 2019, 10, 2040622319851640. [Google Scholar] [CrossRef]
- Prens, E.; Deckers, I. Pathophysiology of Hidradenitis Suppurativa: An Update. J. Am. Acad. Dermatol. 2015, 73, S8–S11. [Google Scholar] [CrossRef]
- Jayne, D. The Diagnosis of Vasculitis. Best Pract. Res. Clin. Rheumatol. 2009, 23, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Sokumbi, O.; Wetter, D.A.; Makol, A.; Warrington, K.J. Vasculitis Associated with Tumor Necrosis Factor-α Inhibitors. Mayo Clin. Proc. 2012, 87, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Jarrot, P.-A.; Kaplanski, G. Anti-TNF-Alpha Therapy and Systemic Vasculitis. Mediat. Inflamm. 2014, 2014, e493593. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.; Oliver, C.; Prince, G.A.; Hemming, V.G.; Pfarr, D.S.; Wang, S.C.; Dormitzer, M.; O’Grady, J.; Koenig, S.; Tamura, J.K.; et al. Development of a Humanized Monoclonal Antibody (MEDI-493) with Potent in Vitro and in Vivo Activity against Respiratory Syncytial Virus. J. Infect. Dis. 1997, 176, 1215–1224. [Google Scholar] [CrossRef]
- Pan, A.; Gerriets, V. Etanercept. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. [Google Scholar]
- Jang, D.; Lee, A.-H.; Shin, H.-Y.; Song, H.-R.; Park, J.-H.; Kang, T.-B.; Lee, S.-R.; Yang, S.-H. The Role of Tumor Necrosis Factor Alpha (TNF-α) in Autoimmune Disease and Current TNF-α Inhibitors in Therapeutics. Int. J. Mol. Sci. 2021, 22, 2719. [Google Scholar] [CrossRef] [PubMed]
- Mease, P.J.; Ory, P.; Sharp, J.T.; Ritchlin, C.T.; Van den Bosch, F.; Wellborne, F.; Birbara, C.; Thomson, G.T.D.; Perdok, R.J.; Medich, J.; et al. Adalimumab for Long-Term Treatment of Psoriatic Arthritis: 2-Year Data from the Adalimumab Effectiveness in Psoriatic Arthritis Trial (ADEPT). Ann. Rheum. Dis. 2009, 68, 702–709. [Google Scholar] [CrossRef]
- Lee, J.U.; Shin, W.; Son, J.Y.; Yoo, K.-Y.; Heo, Y.-S. Molecular Basis for the Neutralization of Tumor Necrosis Factor α by Certolizumab Pegol in the Treatment of Inflammatory Autoimmune Diseases. Int. J. Mol. Sci. 2017, 18, 228. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.; Fleischmann, R.M.; Doyle, M.K.; Strusberg, I.; Durez, P.; Nash, P.; Amante, E.; Churchill, M.; Park, W.; Pons-Estel, B.; et al. Golimumab, a Human Anti–Tumor Necrosis Factor Monoclonal Antibody, Injected Subcutaneously Every 4 Weeks in Patients with Active Rheumatoid Arthritis Who Had Never Taken Methotrexate: 1-Year and 2-Year Clinical, Radiologic, and Physical Function Findings of a Phase III, Multicenter, Randomized, Double-Blind, Placebo-Controlled Study. Arthritis Care Res. 2013, 65, 1732–1742. [Google Scholar] [CrossRef]
- Pelechas, E.; Voulgari, P.V.; Drosos, A.A. Golimumab for Rheumatoid Arthritis. J. Clin. Med. 2019, 8, 387. [Google Scholar] [CrossRef]
- Mitoma, H.; Horiuchi, T.; Tsukamoto, H.; Ueda, N. Molecular Mechanisms of Action of Anti-TNF-α Agents—Comparison among Therapeutic TNF-α Antagonists. Cytokine 2018, 101, 56–63. [Google Scholar] [CrossRef]
- Kaltsonoudis, E.; Zikou, A.K.; Voulgari, P.V.; Konitsiotis, S.; Argyropoulou, M.I.; Drosos, A.A. Neurological Adverse Events in Patients Receiving Anti-TNF Therapy: A Prospective Imaging and Electrophysiological Study. Arthritis Res. Ther. 2014, 16, R125. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H. Clinical Pharmacokinetics of Etanercept: A Fully Humanized Soluble Recombinant Tumor Necrosis Factor Receptor Fusion Protein. J. Clin. Pharmacol. 2005, 45, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Dixon, W.G.; Watson, K.; Lunt, M.; Hyrich, K.L.; Silman, A.J.; Symmons, D.P.M. Rates of Serious Infection, Including Site-Specific and Bacterial Intracellular Infection, in Rheumatoid Arthritis Patients Receiving Anti-Tumor Necrosis Factor Therapy: Results from the British Society for Rheumatology Biologics Register. Arthritis Rheum. 2006, 54, 2368–2376. [Google Scholar] [CrossRef]
- Keam, S.J. Ozoralizumab: First Approval. Drugs 2023, 83, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y. Ozoralizumab: First Nanobody® Therapeutic for Rheumatoid Arthritis. Expert Opin. Biol. Ther. 2023, 23, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Saddala, M.S.; Huang, H. Identification of Novel Inhibitors for TNFα, TNFR1 and TNFα-TNFR1 Complex Using Pharmacophore-Based Approaches. J. Transl. Med. 2019, 17, 215. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef]
- Kwak, M.S.; Lee, H.H.; Cha, J.M.; Shin, H.P.; Jeon, J.W.; Yoon, J.Y. Novel Candidate Drugs in Anti-Tumor Necrosis Factor Refractory Crohn’s Diseases: In Silico Study for Drug Repositioning. Sci. Rep. 2020, 10, 10708. [Google Scholar] [CrossRef]
- Barrett, T.; Suzek, T.O.; Troup, D.B.; Wilhite, S.E.; Ngau, W.-C.; Ledoux, P.; Rudnev, D.; Lash, A.E.; Fujibuchi, W.; Edgar, R. NCBI GEO: Mining Millions of Expression Profiles—Database and Tools. Nucleic Acids Res. 2005, 33, D562–D566. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Kuhn, M.; Simonovic, M.; Roth, A.; Minguez, P.; Doerks, T.; Stark, M.; Muller, J.; Bork, P.; et al. The STRING Database in 2011: Functional Interaction Networks of Proteins, Globally Integrated and Scored. Nucleic Acids Res. 2011, 39, D561–D568. [Google Scholar] [CrossRef]
- Koník, P.; Slavíková, V.; Salát, J.; Reznícková, J.; Dvoroznáková, E.; Kopecký, J. Anti-Tumour Necrosis Factor-Alpha Activity in Ixodes Ricinus Saliva. Parasite Immunol. 2006, 28, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Kazimírová, M.; Štibrániová, I. Tick Salivary Compounds: Their Role in Modulation of Host Defences and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2013, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Šimo, L.; Kazimirova, M.; Richardson, J.; Bonnet, S.I. The Essential Role of Tick Salivary Glands and Saliva in Tick Feeding and Pathogen Transmission. Front. Cell. Infect. Microbiol. 2017, 7, 281. [Google Scholar] [CrossRef] [PubMed]
- Rezkova, M.; Kopecky, J. Anti-Tumour Necrosis Factor Activity in Saliva of Various Tick Species and Its Appearance during the Feeding Period. Folia Parasitol. 2017, 64, 32. [Google Scholar] [CrossRef] [PubMed]
- Papp, K.A.; Tyring, S.; Lahfa, M.; Prinz, J.; Griffiths, C.E.M.; Nakanishi, A.M.; Zitnik, R.; van de Kerkhof, P.C.M.; Melvin, L. Etanercept Psoriasis Study Group A Global Phase III Randomized Controlled Trial of Etanercept in Psoriasis: Safety, Efficacy, and Effect of Dose Reduction. Br. J. Dermatol. 2005, 152, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Weinblatt, M.E.; Keystone, E.C.; Furst, D.E.; Moreland, L.W.; Weisman, M.H.; Birbara, C.A.; Teoh, L.A.; Fischkoff, S.A.; Chartash, E.K. Adalimumab, a Fully Human Anti-Tumor Necrosis Factor Alpha Monoclonal Antibody, for the Treatment of Rheumatoid Arthritis in Patients Taking Concomitant Methotrexate: The ARMADA Trial. Arthritis Rheum. 2003, 48, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Verri, W.A., Jr.; Cunha, T.M.; Poole, S.; Ferreira, S.H.; Cunha, F.Q. Cytokine Inhibitors and Pain Control. Rev. Bras. Reumatol. 2007, 47, 341–353. [Google Scholar] [CrossRef]
- Charles, P.; Elliott, M.J.; Davis, D.; Potter, A.; Kalden, J.R.; Antoni, C.; Breedveld, F.C.; Smolen, J.S.; Eberl, G.; deWoody, K.; et al. Regulation of Cytokines, Cytokine Inhibitors, and Acute-Phase Proteins Following Anti-TNF-Alpha Therapy in Rheumatoid Arthritis. J. Immunol. 1999, 163, 1521–1528. [Google Scholar] [CrossRef]
- Chadwick, L.; Zhao, S.; Mysler, E.; Moots, R.J. Review of Biosimilar Trials and Data on Etanercept in Rheumatoid Arthritis. Curr. Rheumatol. Rep. 2018, 20, 84. [Google Scholar] [CrossRef]
- Prabhakar, S. Translational Research Challenges: Finding the Right Animal Models. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2012, 60, 1141–1146. [Google Scholar] [CrossRef]
- Byng-Maddick, R.; Turner, C.T.; Pollara, G.; Ellis, M.; Guppy, N.J.; Bell, L.C.K.; Ehrenstein, M.R.; Noursadeghi, M. Tumor Necrosis Factor (TNF) Bioactivity at the Site of an Acute Cell-Mediated Immune Response Is Preserved in Rheumatoid Arthritis Patients Responding to Anti-TNF Therapy. Front. Immunol. 2017, 8, 932. [Google Scholar] [CrossRef]
- The UniProt Consortium UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The Protein Families Database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.J.; Bento, A.P.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A Large-Scale Bioactivity Database for Drug Discovery. Nucleic Acids Res. 2012, 40, D1100–D1107. [Google Scholar] [CrossRef]
- Schievella, A.R.; Chen, J.H.; Graham, J.R.; Lin, L.-L. MADD, a Novel Death Domain Protein That Interacts with the Type 1 Tumor Necrosis Factor Receptor and Activates Mitogen-Activated Protein Kinase*. J. Biol. Chem. 1997, 272, 12069–12075. [Google Scholar] [CrossRef]
- Schneeberger, P.E.; Kortüm, F.; Korenke, G.C.; Alawi, M.; Santer, R.; Woidy, M.; Buhas, D.; Fox, S.; Juusola, J.; Alfadhel, M.; et al. Biallelic MADD Variants Cause a Phenotypic Spectrum Ranging from Developmental Delay to a Multisystem Disorder. Brain 2020, 143, 2437–2453. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Watanabe, M.; Nakamaru, Y.; Takagi, D.; Takahashi, H.; Fukuda, S.; Hatakeyama, S. TRIM39 Negatively Regulates the NFκB-Mediated Signaling Pathway through Stabilization of Cactin. Cell. Mol. Life Sci. 2016, 73, 1085–1101. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-H.; Han, S.-Y.; Azam, T.; Yoon, D.-Y.; Dinarello, C.A. Interleukin-32: A Cytokine and Inducer of TNFα. Immunity 2005, 22, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.S.-H.; Lee, H.-M.; Yang, F.; Che, C.-M.; Wong, C.C.L.; Abagyan, R.; Leung, C.-H.; Ma, D.-L. Structure-Based Discovery of Natural Product-Like TNF-α Inhibitors. Angew. Chem. Int. Ed. Engl. 2010, 49, 2860–2864. [Google Scholar] [CrossRef]
- Byla, P.; Andersen, M.H.; Holtet, T.L.; Jacobsen, H.; Munch, M.; Gad, H.H.; Thøgersen, H.C.; Hartmann, R. Selection of a Novel and Highly Specific Tumor Necrosis Factor Alpha (TNFalpha) Antagonist: Insight from the Crystal Structure of the Antagonist-TNFalpha Complex. J. Biol. Chem. 2010, 285, 12096–12100. [Google Scholar] [CrossRef]
- Beirnaert, E.; Desmyter, A.; Spinelli, S.; Lauwereys, M.; Aarden, L.; Dreier, T.; Loris, R.; Silence, K.; Pollet, C.; Cambillau, C.; et al. Bivalent Llama Single-Domain Antibody Fragments against Tumor Necrosis Factor Have Picomolar Potencies Due to Intramolecular Interactions. Front. Immunol. 2017, 8, 867. [Google Scholar] [CrossRef]
- Yang, Z.; West, A.P.; Bjorkman, P.J. Crystal Structure of TNFalpha Complexed with a Poxvirus MHC-Related TNF Binding Protein. Nat. Struct. Mol. Biol. 2009, 16, 1189–1191. [Google Scholar] [CrossRef]
- Cha, S.S.; Kim, J.S.; Cho, H.S.; Shin, N.K.; Jeong, W.; Shin, H.C.; Kim, Y.J.; Hahn, J.H.; Oh, B.H. High Resolution Crystal Structure of a Human Tumor Necrosis Factor-Alpha Mutant with Low Systemic Toxicity. J. Biol. Chem. 1998, 273, 2153–2160. [Google Scholar] [CrossRef]
- Shibata, H.; Yoshioka, Y.; Ohkawa, A.; Minowa, K.; Mukai, Y.; Abe, Y.; Taniai, M.; Nomura, T.; Kayamuro, H.; Nabeshi, H.; et al. Creation and X-Ray Structure Analysis of the Tumor Necrosis Factor Receptor-1-Selective Mutant of a Tumor Necrosis Factor-Alpha Antagonist. J. Biol. Chem. 2008, 283, 998–1007. [Google Scholar] [CrossRef]
- London, N.; Movshovitz-Attias, D.; Schueler-Furman, O. The Structural Basis of Peptide-Protein Binding Strategies. Structure 2010, 18, 188–199. [Google Scholar] [CrossRef]
- Vanhee, P.; Reumers, J.; Stricher, F.; Baeten, L.; Serrano, L.; Schymkowitz, J.; Rousseau, F. PepX: A Structural Database of Non-Redundant Protein–Peptide Complexes. Nucleic Acids Res. 2010, 38, D545–D551. [Google Scholar] [CrossRef]
- Martins, P.; Mariano, D.; Carvalho, F.C.; Bastos, L.L.; Moraes, L.; Paixão, V.; Cardoso de Melo-Minardi, R. Propedia v2.3: A Novel Representation Approach for the Peptide-Protein Interaction Database Using Graph-Based Structural Signatures. Front. Bioinform. 2023, 3, 1103103. [Google Scholar] [CrossRef]
- Martins, P.M.; Santos, L.H.; Mariano, D.; Queiroz, F.C.; Bastos, L.L.; Gomes, I.d.S.; Fischer, P.H.C.; Rocha, R.E.O.; Silveira, S.A.; de Lima, L.H.F.; et al. Propedia: A Database for Protein–Peptide Identification Based on a Hybrid Clustering Algorithm. BMC Bioinform. 2021, 22, 1. [Google Scholar] [CrossRef]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for Taxonomy-Based Analysis of Pathways and Genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Yang, L.; Zhang, Z.; Wang, B.; Feng, B.; Liu, Z. Identification of Targets for Subsequent Treatment of Crohn’s Disease Patients After Failure of Anti-TNF Therapy. J. Inflamm. Res. 2023, 16, 4617–4631. [Google Scholar] [CrossRef] [PubMed]
- Jie, K.; Feng, W.; Boxiang, Z.; Maofeng, G.; Jianbin, Z.; Zhaoxuan, L.; Yangyi, Z.; Liang, C.; Haobo, S.; Wensheng, L.; et al. Identification of Pathways and Key Genes in Venous Remodeling After Arteriovenous Fistula by Bioinformatics Analysis. Front. Physiol. 2020, 11, 565240. [Google Scholar] [CrossRef]
- Liu, Y.; Duan, Y.; Li, Y. Integrated Gene Expression Profiling Analysis Reveals Probable Molecular Mechanism and Candidate Biomarker in Anti-TNFα Non-Response IBD Patients. J. Inflamm. Res. 2020, 13, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein-Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Polley, S.; Passos, D.O.; Huang, D.-B.; Mulero, M.C.; Mazumder, A.; Biswas, T.; Verma, I.M.; Lyumkis, D.; Ghosh, G. Structural Basis for the Activation of IKK1/α. Cell Rep. 2016, 17, 1907–1914. [Google Scholar] [CrossRef]
- Li, Y.; Okatsu, K.; Fukai, S.; Sato, Y. Structural Basis for Specific Recognition of K6-Linked Polyubiquitin Chains by the TAB2 NZF Domain. Biophys. J. 2021, 120, 3355–3362. [Google Scholar] [CrossRef]
- Halder, D.; Das, S.; Joseph, A.; Jeyaprakash, R.S. Molecular Docking and Dynamics Approach to in Silico Drug Repurposing for Inflammatory Bowels Disease by Targeting TNF Alpha. J. Biomol. Struct. Dyn. 2023, 41, 3462–3475. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI Gene Expression and Hybridization Array Data Repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Barrett, T.; Edgar, R. [19] Gene Expression Omnibus: Microarray Data Storage, Submission, Retrieval, and Analysis. In Methods in Enzymology; DNA Microarrays, Part B: Databases and Statistics; Academic Press: Cambridge, MA, USA, 2006; Volume 411, pp. 352–369. [Google Scholar]
- Fiser, A.; Sali, A. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate Prediction of Protein Structures and Interactions Using a Three-Track Neural Network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Sali, A. Comparative Protein Structure Modeling of Genes and Genomes. Annu. Rev. Biophys. Biomol. Struct. 2000, 29, 291–325. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.-Y.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Protein Sci. 2007, Chapter 2, Unit 2.9. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved Protein Structure Prediction Using Potentials from Deep Learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Thornton, J.M.; Laskowski, R.A.; Borkakoti, N. AlphaFold Heralds a Data-Driven Revolution in Biology and Medicine. Nat. Med. 2021, 27, 1666–1669. [Google Scholar] [CrossRef]
- Erba, F.; Paola, L.D.; Venere, A.D.; Mastrangelo, E.; Cossu, F.; Mei, G.; Minicozzi, V. Head or Tail? A Molecular Dynamics Approach to the Complex Structure of TNF-Associated Factor TRAF2. Biomol. Concepts 2023, 14, 20220031. [Google Scholar] [CrossRef]
- Esposito, L.; Balasco, N.; Smaldone, G.; Berisio, R.; Ruggiero, A.; Vitagliano, L. AlphaFold-Predicted Structures of KCTD Proteins Unravel Previously Undetected Relationships among the Members of the Family. Biomolecules 2021, 11, 1862. [Google Scholar] [CrossRef]
- Martins, P.M.; Mayrink, V.D.; Silveira, S.d.A.; da Silveira, C.H.; de Lima, L.H.F.; de Melo-Minardi, R.C. How to Compute Protein Residue Contacts More Accurately? In Proceedings of the 33rd Annual ACM Symposium on Applied Computing, New York, NY, USA, 9–13 April 2018; Association for Computing Machinery: New York, NY, USA; pp. 60–67. [Google Scholar]
- Jubb, H.C.; Higueruelo, A.P.; Ochoa-Montaño, B.; Pitt, W.R.; Ascher, D.B.; Blundell, T.L. Arpeggio: A Web Server for Calculating and Visualising Interatomic Interactions in Protein Structures. J. Mol. Biol. 2017, 429, 365–371. [Google Scholar] [CrossRef]
- Pimentel, V.; Mariano, D.; Cantão, L.X.S.; Bastos, L.L.; Fischer, P.; de Lima, L.H.F.; Fassio, A.V.; de Melo-Minardi, R.C. VTR: A Web Tool for Identifying Analogous Contacts on Protein Structures and Their Complexes. Front. Bioinform. 2021, 1, 730350. [Google Scholar] [CrossRef]
- Dos Santos, V.P.; Rodrigues, A.; Dutra, G.; Bastos, L.; Mariano, D.; Mendonça, J.G.; Lobo, Y.J.G.; Mendes, E.; Maia, G.; Machado, K.D.S.; et al. E-Volve: Understanding the Impact of Mutations in SARS-CoV-2 Variants Spike Protein on Antibodies and ACE2 Affinity through Patterns of Chemical Interactions at Protein Interfaces. PeerJ 2022, 10, e13099. [Google Scholar] [CrossRef]
- Fassio, A.V.; Martins, P.M.; Guimarães, S.d.S.; Junior, S.S.A.; Ribeiro, V.S.; de Melo-Minardi, R.C.; Silveira, S.d.A. Vermont: A Multi-Perspective Visual Interactive Platform for Mutational Analysis. BMC Bioinform. 2017, 18, 403. [Google Scholar] [CrossRef]
- Silveira, S.A.; Fassio, A.V.; Gonçalves-Almeida, V.M.; de Lima, E.B.; Barcelos, Y.T.; Aburjaile, F.F.; Rodrigues, L.M.; Meira Jr, W.; de Melo-Minardi, R.C. VERMONT: Visualizing Mutations and Their Effects on Protein Physicochemical and Topological Property Conservation. BMC Proc. 2014, 8, S4. [Google Scholar] [CrossRef]
- Fassio, A.V.; Santos, L.H.; Silveira, S.A.; Ferreira, R.S.; de Melo-Minardi, R.C. nAPOLI: A Graph-Based Strategy to Detect and Visualize Conserved Protein-Ligand Interactions in Large-Scale. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 17, 1317–1328. [Google Scholar] [CrossRef]
- Fassio, A.V.; Shub, L.; Ponzoni, L.; McKinley, J.; O’Meara, M.J.; Ferreira, R.S.; Keiser, M.J.; de Melo Minardi, R.C. Prioritizing Virtual Screening with Interpretable Interaction Fingerprints. J. Chem. Inf. Model. 2022, 62, 4300–4318. [Google Scholar] [CrossRef]
- Pires, D.E.; de Melo-Minardi, R.C.; dos Santos, M.A.; da Silveira, C.H.; Santoro, M.M.; Meira, W. Cutoff Scanning Matrix (CSM): Structural Classification and Function Prediction by Protein Inter-Residue Distance Patterns. BMC Genom. 2011, 12, S12. [Google Scholar] [CrossRef]
- Pires, D.E.V.; de Melo-Minardi, R.C.; da Silveira, C.H.; Campos, F.F.; Meira, W., Jr. aCSM: Noise-Free Graph-Based Signatures to Large-Scale Receptor-Based Ligand Prediction. Bioinformatics 2013, 29, 855–861. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. mCSM: Predicting the Effects of Mutations in Proteins Using Graph-Based Signatures. Bioinformatics 2014, 30, 335–342. [Google Scholar] [CrossRef]
- Mariano, D.C.B.; Santos, L.H.; Machado, K.d.S.; Werhli, A.V.; de Lima, L.H.F.; de Melo-Minardi, R.C. A Computational Method to Propose Mutations in Enzymes Based on Structural Signature Variation (SSV). Int. J. Mol. Sci. 2019, 20, 333. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B. CSM-Lig: A Web Server for Assessing and Comparing Protein–Small Molecule Affinities. Nucleic Acids Res. 2016, 44, W557–W561. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. NNScore 2.0: A Neural-Network Receptor-Ligand Scoring Function. J. Chem. Inf. Model. 2011, 51, 2897–2903. [Google Scholar] [CrossRef]
- Le Grand, S.; Götz, A.W.; Walker, R.C. SPFP: Speed without Compromise—A Mixed Precision Model for GPU Accelerated Molecular Dynamics Simulations. Comput. Phys. Commun. 2013, 184, 374–380. [Google Scholar] [CrossRef]
- Khan, M.A.-A.-K.; Turjya, R.R.; Islam, A.B.M.M.K. Computational Engineering the Binding Affinity of Adalimumab Monoclonal Antibody for Designing Potential Biosimilar Candidate. J. Mol. Graph. Model. 2021, 102, 107774. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, G.; Mahrosh, H.S.; Arif, R. In Silico Characterization of Growth Differentiation Factors as Inhibitors of TNF-Alpha and IL-6 in Immune-Mediated Inflammatory Disease Rheumatoid Arthritis. BioMed Res. Int. 2021, 2021, e5538535. [Google Scholar] [CrossRef]
- Hong, S.-T.; Su, Y.-C.; Wang, Y.-J.; Cheng, T.-L.; Wang, Y.-T. Anti-TNF Alpha Antibody Humira with pH-Dependent Binding Characteristics: A Constant-pH Molecular Dynamics, Gaussian Accelerated Molecular Dynamics, and In Vitro Study. Biomolecules 2021, 11, 334. [Google Scholar] [CrossRef]
- Abraham, E. Computational Design of Variant TNF Molecules: A Novel Methodology for Inhibition of Proinflammatory Cascades. Sci. STKE 2003, 2003, pe51. [Google Scholar] [CrossRef]
- Agnihotri, P.; Deka, H.; Chakraborty, D.; Monu; Saquib, M.; Kumar, U.; Biswas, S. Anti-Inflammatory Potential of Selective Small Compounds by Targeting TNF-α & NF-kB Signaling: A Comprehensive Molecular Docking and Simulation Study. J. Biomol. Struct. Dyn. 2023, 41, 13815–13828. [Google Scholar] [CrossRef] [PubMed]
- Pierri, C.L.; Bossis, F.; Punzi, G.; De Grassi, A.; Cetrone, M.; Parisi, G.; Tricarico, D. Molecular Modeling of Antibodies for the Treatment of TNFα-Related Immunological Diseases. Pharmacol. Res. Perspect. 2016, 4, e00197. [Google Scholar] [CrossRef] [PubMed]
- Abechi, S.E.; Ejeh, S.; Abduljelil, A. In Silico Screening of Potential Tumor Necrosis Factor Alpha (TNF-α) Inhibitors through Molecular Modeling, Molecular Docking, and Pharmacokinetics Evaluations. Sci. Afr. 2023, 21, e01830. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bastos, L.L.; Mariano, D.; Lemos, R.P.; Bialves, T.S.; Oliveira, C.J.F.; de Melo-Minardi, R.C. The Role of Structural Bioinformatics in Understanding Tumor Necrosis Factor α-Interacting Protein Mechanisms in Chronic Inflammatory Diseases: A Review. Immuno 2024, 4, 14-42. https://doi.org/10.3390/immuno4010002
Bastos LL, Mariano D, Lemos RP, Bialves TS, Oliveira CJF, de Melo-Minardi RC. The Role of Structural Bioinformatics in Understanding Tumor Necrosis Factor α-Interacting Protein Mechanisms in Chronic Inflammatory Diseases: A Review. Immuno. 2024; 4(1):14-42. https://doi.org/10.3390/immuno4010002
Chicago/Turabian StyleBastos, Luana Luiza, Diego Mariano, Rafael Pereira Lemos, Tatiane Senna Bialves, Carlo Jose Freire Oliveira, and Raquel C. de Melo-Minardi. 2024. "The Role of Structural Bioinformatics in Understanding Tumor Necrosis Factor α-Interacting Protein Mechanisms in Chronic Inflammatory Diseases: A Review" Immuno 4, no. 1: 14-42. https://doi.org/10.3390/immuno4010002
APA StyleBastos, L. L., Mariano, D., Lemos, R. P., Bialves, T. S., Oliveira, C. J. F., & de Melo-Minardi, R. C. (2024). The Role of Structural Bioinformatics in Understanding Tumor Necrosis Factor α-Interacting Protein Mechanisms in Chronic Inflammatory Diseases: A Review. Immuno, 4(1), 14-42. https://doi.org/10.3390/immuno4010002