Abstract
Prolonged survival and durable responses in several late-stage cancers such as melanoma and lung cancer have been made possible with the use of immune checkpoint inhibitors targeting the programmed cell-death protein 1 (PD-1) or its ligand PD-L1. While it is prudent to focus on the unprecedented and durable clinical responses, there are subsets of cancer patients that do not respond to immunotherapies or respond early and then relapse later. Many pathways of resistance have been characterized, and more continue to be uncovered. To overcome the development of resistance, an in-depth investigation is necessary to identify alternative immune receptors and signals with the overarching goal of expanding treatment options for those with demonstrated resistance to PD1 checkpoint immunotherapy. In this mini-review, we will discuss the mechanisms by which tumors exhibit resistance to anti-PD-1/PD-L1 immunotherapy and explore strategies to overcome such resistances.
1. Introduction
Immune checkpoint inhibition is a promising mode of immunotherapy that has shown incredible clinical efficacy in the treatment of several types of solid and liquid tumors. Immune checkpoint inhibitors (ICIs) are monoclonal antibodies that physically interfere with the transduction of inhibitory signals to activated T-cells, thereby potentiating a robust anti-tumor immune response [1]. Of the nine ICIs that have received FDA approval for clinical use, seven target the PD-1/PD-L1 immune checkpoint–an approach that has shown to be strikingly efficacious while exhibiting acceptable patient tolerability profiles [2,3,4].
Programmed cell death protein 1 (PD-1) is a transmembrane receptor protein first identified in 1992 during a screen for apoptosis-associated molecules by now-Nobel-laureate Tasuku Honjo and others at Kyoto University. Its function remained unknown until 1999, when the same group of researchers used murine models to show that PD-1 is an important negative regulator of immune responses and that its deficiency leads to the development of organ-specific autoimmunity [5,6]. PD-1′s ligand, PD-L1, was identified later that same year alongside experimental evidence confirming that the PD-1/PD-L1 interaction is indeed what mediates self-tolerance through its transduction of inhibitory signals to activated T cells [7,8]. PD-L1 expression plays a critical role in preventing autoimmunity; however, some cancers co-opt this immune checkpoint and overexpress PD-L1 to induce immune tolerance locally within the tumor microenvironment (TME), thereby allowing the tumor cells to evade elimination by the immune system [1,9,10,11,12].
Inhibition of the PD-1/PD-L1 immune checkpoint with monoclonal antibody inhibitors has revolutionized the therapeutic landscape of several cancers, including melanoma, non-small cell lung cancer, renal cell carcinoma, and urothelial cancers [13]. However, despite the robust response rates, prolonged overall survival, and prolonged progression-free survival, resistance to anti-PD-1/PD-L1 therapy is a significant challenge, as only a minority of patients (20–30%) are estimated to experience a positive response to PD-1/PD-L1 blockade therapy, while the rest experience cancer progression due to either primary or acquired resistance [14,15]. Further, while it has been observed that the clinical efficacy of PD-1/PD-L1 inhibitors correlates positively with the tumor mutational burden (TMB) and PD-L1 expression, the lack of uniformity in the expression-dependent effect suggests that there are additional mechanisms at play. The purpose of this mini-review is to elucidate the mechanisms underlying resistance to PD-1/PD-L1 checkpoint inhibition therapy and investigate emerging strategies for overcoming such resistance.
2. PD-1/PD-L1 Expression and Function
PD-1 is a transmembrane receptor protein that is expressed on the surface of activated T-cells and functions in attenuating the immune response by inhibiting the activity of effector T-cells [16]. Upon binding its ligand, PD-L1, the tyrosine residues on the cytoplasmic portion of the PD-1 protein become phosphorylated and bind Src homology region 2 domain-containing phosphatase-1 and 2 (SHP-1 and SHP-2), which proceed to dephosphorylate nearby signaling molecules, causing a profound downregulation in pathways responsible for T-cell metabolism, cytokine synthesis, proliferation, and survival, in addition to interfering with transcription factors, such as GATA-3, T-bet, and Eomes, that are implicated in T-cell effector functions [17,18].
Its ligand, PD-L1, is ubiquitously expressed on cells throughout the body, although its expression is differential. In certain tissues, such as the retinal pigment epithelium, pancreatic islets, and neurons, its expression is constitutive and serves to persistently protect these tissues from inadvertent damage caused by activated T cells [19,20]. In other tissues throughout the body, PD-L1 expression has been found to be inducible in the presence of a number of pro-inflammatory cytokines, such as interferon gamma (IFN-γ), which are released by tissue-infiltrating effector T-cells [16,20,21,22]. The pro-inflammatory cytokines cause a localized induction of PD-L1 expression that, through binding PD-1, serves to protect host tissue that is experiencing active infection and inflammation by markedly affecting the effector activity of both CD8+ and CD4+ effector T-cells. Activation of the PD-1 receptor on effector CD8+ T-cells induces T-cell exhaustion, resulting in a significant inhibition of effector activity, a reduction in proliferation, and a decrease in cytokine production [8,23]. At the same time, PD-1 receptors on CD4+ T-cells play a critical role in the differentiation and establishment of Treg cell populations, which attenuate the immune state and prevent autoimmunity [24].
3. PD-L1 Overexpression in Tumors and PD-1/PD-L1 Immune Checkpoint Therapy
PD-L1 is overexpressed in several malignancies, allowing the tumor cells to evade elimination by the immune system through inducing localized immune tolerance within the tumor microenvironment [1,9,10,11,12]. Higher expression levels of PD-L1 correlate strongly with a poorer prognosis by suppressing the activation of T cells, leading to tumor progression [25,26,27,28,29,30,31]. Recognizing this role of the PD-1/PD-L1 axis, several anti-PD-1/PD-L1 monoclonal antibody therapies have been developed to bind either the receptor or ligand to prevent the transduction of the inhibitory signals to the effector T-cells, thereby allowing the immune system to mount a robust anti-tumor response unimpeded by the tumor’s PD-L1 expression levels [5,16,32,33,34,35]. The first of these ICIs were nivolumab (anti-PD-1) and pembrolizumab (anti-PD-1), both of which received FDA approval in 2014. Several additional agents targeting the PD-1/PD-L1 axis have since been approved, including atezolizumab (anti-PD-L1) in 2016, avelumab (anti-PD-L1) in 2017, durvalumab (anti-PD-L1) in 2017, cemiplimab (anti-PD-1) in 2018, and most recently dostarlimab (anti-PD-1) in 2021 [4].
PD-1/PD-L1 checkpoint inhibitor therapy has shown remarkable clinical efficacy in treating non-hematologic and hematologic malignancies. These treatments have become the standard of care for a select subset of neoplasms, most notably non-small-cell lung cancer (NSCLC) and metastatic melanoma [36]. Other clinical indications of PD-1/PD-L1 checkpoint inhibitors have continued to diversify in our ongoing pre-clinical and clinical trials [37]. Recently, a phase 2 clinical study by Cereck et al. has shown remarkable success with Dostarlimab, a PD-1/PD-L1 checkpoint inhibitor, in locally invasive, mismatch repair-deficient adenocarcinomas of the colon. After more than six months of treatment, all 12 patients enrolled in the study achieved a clinically complete response, as confirmed by 18F-fluorodeoxyglucose positron emission tomography (FDG PET), a digital rectal exam (DRE), T2-weighted magnetic resonance imaging (MRI), endoscopy, and/or biopsy at baseline, and at 6 weeks, 3 months, 6 months, and every 4 months following. No further surgery or radiation or chemotherapy was required in these patients, indicating that cancers with these molecular features are highly sensitive to PD-1/PD-L1 immunotherapy [38]. In light of the successes of PD-1/PD-L1 checkpoint inhibition, it is important to note the emerging recognition of the importance of PD-L2, which has an equivalent function to PD-L1 in immune regulation. Current research, such as that of Yearly et al., suggests that anti-PD-1 monoclonal antibodies such as Nivolumab derive benefit through the simultaneous blocking of PD-1/PD-L1 and PD-1/PD-L2 interactions, in addition to suggesting that the use of combination therapy targeting both PD-1 ligands in lieu of monotherapy targeting PD-L1 alone may provide greater clinical benefit, particularly in tumors with high PD-L2 expression [39,40].
4. Mechanisms of Resistance to PD-1/PD-L1 Blockade and Corresponding Therapeutic Strategies
Despite the clinical successes seen with PD-1/PD-L1 immune checkpoint therapy, patient response to therapy is heterogeneous and unpredictable, in terms of both the length and magnitude of therapeutic efficacy. An estimated 70–80% of patients experience cancer progression due to primary or secondary resistance mechanisms that prevent the immune system from mounting a robust and persistent anti-tumor response [14,15,41]. These resistance pathways have been an active area of research, and efforts are in place to discover innovative therapeutic strategies to overcome PD-1/PD-L1 resistance, of which the most notable are outlined in this section.
4.1. Immunosuppressive Cytokine Pathways
Regulatory T-cells (Treg) are a subpopulation of suppressor CD4+T-cells that are involved in the suppression of immune-mediated tissue destruction towards self-antigens, helping prevent the development of autoimmunity [42]. However, Treg cells support tumor progression by secreting inhibitory cytokines such as Transforming Growth Factor-β (TGFβ), Interleukin-10 (IL-10), and IL-35 inhibiting the anti-tumor immune response of effector T cells, natural killer (NK) cells and dendritic cells (DCs) resulting in tumor progression [43]. Targeting these immunosuppressive cytokines overcomes ICI resistance by increasing effector T cells and NK cell infiltration and by decreasing the myeloid-derived suppressor cell (MDSC) population within the TME [42]. It is therefore hypothesized that the modulation of certain cytokines, such as chemokine ligand 12 (CXCL12), TGFβ, and IL-17A, using monoclonal antibodies could be an avenue to potentiate the efficacy of PD-1/PD-L1 blockers by surpassing primary or acquired resistance in these therapy-refractory cancers [44,45,46,47].
CXCL12 is a cytokine that is localized in tumor cells and binds to chemokine receptor 4 (CXCR4) to activate multiple signaling pathways, including mitogen-activated protein kinase (MAPK), wingless-related integration site (wnt), and epithelial growth factor/receptor (EGF/EGFR) for cell cycle progression and migration [48,49]. Through utilizing an antagonist to inhibit CXCL12 binding to CXCR4 in pancreatic cancer murine models, one study showed a marked increase in effector T-cell accumulation in the tumors and a reduction of tumor cell volume despite the continued presence of Treg cells. These results suggest that by neutralizing CXCL12 signaling, Treg-induced inhibitory pathways in tumors can be bypassed, allowing the recruitment of T-cells and thereby re-sensitizing immune cells and tumor cells to PD-1/PD-L1 checkpoint inhibition, as illustrated in Figure 1 [48]. Similar results were seen in pre-clinical studies where CXCL12 antagonism in metastatic breast cancer led to increased T-cell infiltration and improved immunotherapeutic efficacy in murine models [50]. With the intent of clinical translation, clinical trials have combined CXCL12 antagonist and anti-PD-1/PD-L1 therapies and have shown these treatments to be safe and efficacious in metastatic pancreatic cancer and colorectal cancer [51,52]. In these studies, combination therapy consisting of CXCL12 antagonists and ICIs showed synergism and enriched TME with effector T-cells to generate a robust anti-tumor response.
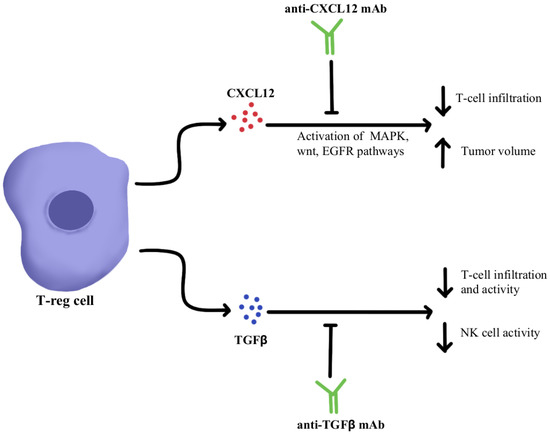
Figure 1.
Diagram of CXCL12 and TGFβ cytokine pathway modulation [48,49,50,51,52,53,54,55,56,57,58,59]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
TGFβ is a cytokine that drives numerous cell processes, ranging from proliferation, motility, apoptosis, differentiation, and immune regulation [53]. TGFβ signaling in the immune TME, as illustrated in Figure 1, represses the anti-tumor activity of the immune cell population, including T cells and NK cells, resulting in immunosuppression that further limits the efficacy of ICIs and, as a result, induces therapeutic resistance to ICIs [54]. Studies using TGFβ inhibitors have been shown to increase T cell infiltration, Thelper1 (Th1)-type immune response, upregulation of Interferon-γ (IFN-γ), and promoted the lytic function of tumor antigen-specific CD8+T cells [55]. Further studies using a dual approach by simultaneously blocking the PD-1/PD-L1 checkpoints as well as the TGFβ pathway were successful in mounting a significant anti-tumor response by overcoming the ICI resistance as well as enhancing the recruitment of T cells [55,56,57]. The clinical data of TGFβ pathway inhibitors alone and in combination with ICIs have been studied in multiple advanced treatment-refractory cancers including pancreatic, cervix, colorectal carcinoma, biliary tract cancer, urothelial carcinoma, and NSCLC, and have shown to be safe, well-tolerated and provide better clinical outcomes (NCT03631706, NCT03732274) [58,59].
IL-17A is a proinflammatory cytokine that is produced by various cells, including Th17 cells, CD8+T-cells, γδT-cells, and NK cells within the TME [60]. IL-17A is expressed across different tumor types; however, the impact of IL-17A appears to be different based on the type of cancer. For instance, IL-17A serves as a tumor suppressor in melanoma and ovarian cancer, while in other cancers such as colorectal cancer and non-small cell lung cancer, IL-17A supports tumor growth [45,61,62,63]. Looking specifically at the impact of IL-17A and its relationship to checkpoint inhibitors, IL-17A may potentiate the recruitment of MDSC and increase the immunosuppressive activity of Tregs within the TME that ultimately leads to resistance to anti-PD-1/PD-L1 therapy, as illustrated in Figure 2 [64,65,66]. Pre-clinical evidence suggests that IL-17A can promote migration and invasion of cancer cells by upregulating matrix metalloproteinase, and inhibition of a nuclear factor-kb (NF-kb) abolishes the tumor-promoting effects of IL-17A. These laboratory observations have not yet been translated into clinical applications. Due to the differential effects of IL-17A seen in different cancer types, the exact role of IL-17A in protective and aberrant processes has yet to be fully elucidated; however, this cytokine remains a possible modulation target to immunopotentiate the TME in ICI-refractory tumors [67].
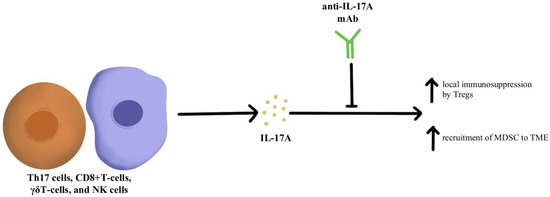
Figure 2.
Diagram of IL-17A cytokine pathway modulation [45,60,61,62,63,64,65,66,67]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
4.2. T-Cell Exhaustion and Depletion
T-cell exhaustion within the TME is a significant mechanism of resistance to anti-PD-1/PD-L1 therapy. Exhausted T cells have been found to upregulate multiple inhibitory receptors such as cytotoxic T-lymphocyte-associated protein 4 (CTLA4), T-cell immunoglobulin, mucin-3 (TIM-3), lymphocyte activation gene 3 (LAG-3), and the T-cell tyrosine-based inhibitory motif (ITIM) domain [68,69,70,71,72]. Mutation of the ITIM domain affects PD-1 signaling and T-cell functional activity [73]. Overexpression of the T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT) and increased infiltration of CD8+TIGIT+T-cells have been noted as correlating with worse prognosis in several cancers. Studies have shown that dual blockade of TIGIT/PD-1 blockers can overcome the T-cell dysfunction/exhaustion pathway of resistance [74]. CTLA4, discovered by 2018 Nobel Prize co-recipient James Allison, is an immune checkpoint protein expressed on activated T-cells that binds either CD80 and/or CD86 found on antigen-presenting cells (APCs) and regulates T-cell activity and effector functions [75]. CTLA4 and PD-1/PD-L1 pathways interact with each other in cancer in a complex manner, and studies have clearly shown the benefit of combining anti-CTLA4/anti-PD-1 treatments, both in preclinical and clinical settings, to reverse T-cell exhaustion [76]. LAG-3 interacts with major histocompatibility-II (MHC-II) to prohibit the binding of MHC molecules to T-cell receptor (TCR) and CD4+T cells, therefore, directly hindering TCR signaling in immune response [77]. A combination of anti-LAG-3 antibodies with nivolumab in a clinical study has shown it to be a safe and efficacious regimen in immunotherapy-resistant melanoma through its modulation of this pathway, as can be seen in Figure 3 [78].
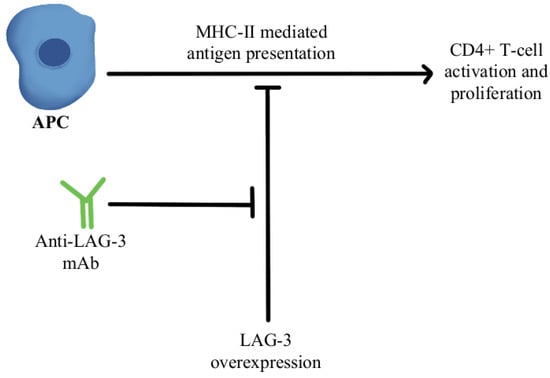
Figure 3.
Diagram of LAG-3 pathway modulation [77,78]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
Another significant mechanism of resistance is the upregulation of TIM-3 on dysfunctional tumor-infiltrating lymphocytes (TILs), leading to an exhaustion phenotype [79]. It has been shown that over-expression of TIM-3 on Treg cells is associated with the development of resistance to anti-PD-1/PD-L1 therapy in melanoma [80]. The use of TIM-3 antibody in combination with chemotherapy indirectly enhanced CD8+T-cell responses as well as increased chemokine ligand 9 (CXCL9) expression by DCs [81]. TIM-3 mediates the exhaustion of T-cells and escape of PD-1 inhibition by activating phospho-inositol-3 kinase (PI3K)/protein kinase B(Akt) signaling [82]. In phosphatase and tensin homolog (PTEN) deleted tumors, PI3K/AKT pathway enhances the expression of PD-L1 and inactivates T-cells. Further, a selective PI3Kβ inhibitor in murine cancer models has revealed improvements in the efficacy of both anti-PD-1 and anti-CTLA-4 antibodies [83]. Additionally, a clinical study combining anti-TIM-3 antibody with anti-PD-1 immunotherapy in 219 patients was deemed safe and efficacious in solid malignancies [84]. There is also an ongoing clinical trial, slated to complete in December of 2022, that is investigating the modulation of TIM-3′s ligand, galectin-9, in potentiating the efficacy of anti-PD1 therapy in the treatment of solid tumors (NCT04666688). Figure 4 contains a diagram of this pathway and its modulation targets.
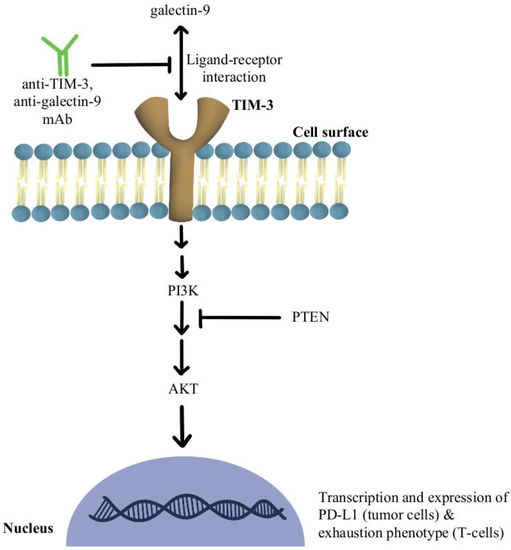
Figure 4.
Diagram of TIM-3 pathway modulation (NCT04666688) [79,80,81,82,83,84]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
Another reported mechanism of resistance to PD-1/PD-L1 checkpoint inhibitors is increased levels of collagen in the tumor extracellular matrix that activates leukocyte-associated immunoglobulin-like receptor (LAIR-1), a membrane-bound receptor expressed by CD8+T-cells following CD18 interaction with collagen, causing a signaling cascade that induces T-cell exhaustion through SHP-1 signaling [85]. Preliminary in vivo studies have shown that LAIR-2 protein, a soluble receptor with greater affinity for collagen than LAIR-1, regulates the immune system by preventing LAIR-1 cross-linking to collagen via competitive binding [86]. Experiments are currently underway using LAIR-1 fusion proteins to probe for potential ligands for this inhibitory receptor for pre-clinical and clinical use, as diagrammed in Figure 5 [87].
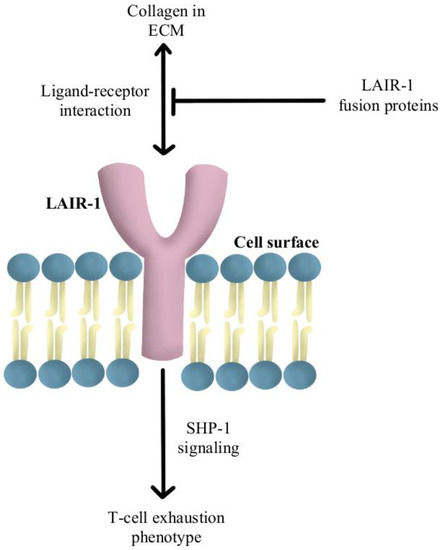
Figure 5.
Diagram of LAIR-1 pathway modulation [85,86,87]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
T-cells recognize antigens presented by APCs. However, mutations in beta-2-microglobulin (B2M) can disrupt antigen presentation that ultimately leads to immune checkpoint blockade therapy resistance [88,89]. Whole exome sequencing of immunotherapy-resistant tumors has shown that truncating mutations in B2M and mutations in the genes encoding interferon-receptor-associated Janus kinase 1/2 (JAK1/2) result in a lack of response to IFN-γ with loss of surface expression of major histocompatibility complex class I [90]. With this loss of MHC class I, tumors with the B2M mutation respond to anti-PD-1 therapies through the activation of MHC class I-independent mechanisms mediated by CD4+T-cells or NK cells [91]. Bempegaldesleukin, a CD122-preferential IL-2 pathway agonist, has been shown to activate CD4+T-cells and NK cells regardless of the PD-1/PD-L1 status of tumors [92]. Additionally, when Bempegaldesleukin was used in combination with immunotherapeutics, including anti-PD-1 and anti-CTLA4, it resulted in intratumoral depletion of Treg cells and increased proliferation of CD8+T-cells [93]. As the mutations in JAK1/2 led to resistance to PD-1 blockade therapy, a preclinical study has shown that administration of Toll-like receptor 9 (TLR9) agonists can overcome JAK1/2 knockout-induced resistance mediated by NK and CD8+T-cells [94]. Following these observations, CMP-001 (a TLR9 agonist) has been developed for study in a phase Ib clinical study in combination with Pembrolizumab in advanced melanoma (NCT02680184). Another phase II study evaluated the combination of IMO (another TLR9 agonist) in combination with ipilimumab (anti-CTLA4) in immunotherapy-refractory melanoma and has shown a revival of immune responses in 15 out of 24 enrolled patients [95]. These drug combinations and their respective effects on the MHC-I-independent and MHC-I-dependent pathways are illustrated in Figure 6.
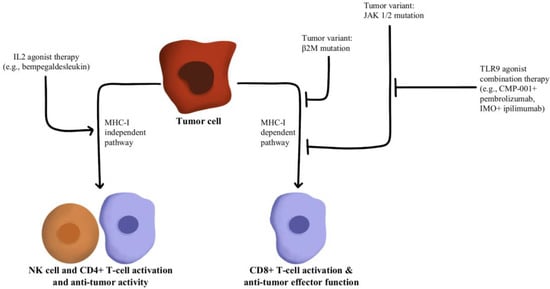
Figure 6.
Diagram of MHC-I-dependent and MHC-I-independent pathway modulation [88,89,90,91,92,93,94,95]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
Depletion of T-cells within the TME is facilitated by chronic, persistent type II interferon signaling, enabling signal transducer and activator of transcription (STAT) tumor-related epigenetic changes, resulting in increased expression of interferon-stimulated genes and inhibitory receptors (TCIRs) on multiple T-cells, including Galectin-9, MHC-II ligands, and immune inhibitory checkpoints, including TIM-3 and LAG-3. Increased co-expression of multiple TCIRs aggravates T-cell depletion, while blocking interferons can reverse resistance caused by T-cell depletion [96,97]. Konen and others have found that neurotrophic tyrosine receptor kinase (NTRK) is upregulated by anti-PD-1 therapy. NTRK also activates the JAK-STAT signaling pathway and by upregulating the expression of multiple inhibitory receptors on T-cell surfaces, promoting T-cell exhaustion, as illustrated in Figure 7 [98]. There are currently two FDA-approved drugs indicated for the treatment of solid tumors with NTRK gene fusion, entrectinib, and larotrectinib, which act through selective inhibition of the tyrosine kinase domain of the NTRK [4]. Patients experiencing resistance to ICI therapy may see therapeutic benefits with concurrent administration of one of these small molecule inhibitors due to its attenuation of that JAK-STAT signaling pathway and prevention of T-cell exhaustion, however, the repurposing of these drugs for treating ICI resistance remains an open avenue of investigation.
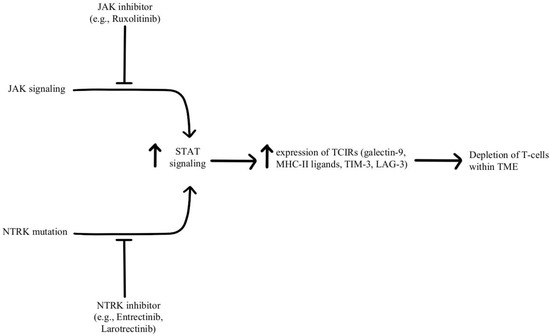
Figure 7.
Diagram of STAT pathway modulation [4,96,97,98]. Illustrated by Kaitlyn Mi of The Dartmouth Institute.
4.3. Adoptive Cellular Therapy
Chimeric antigen receptor T-cell (CAR-T) therapy has been shown to achieve durable remissions in cancers and is currently FDA-approved for the treatment of certain hematological malignancies, and its combination with checkpoint inhibitors targeting PD-1 has provided successful outcomes in several cancers such as B-cell non-Hodgkin lymphoma, hepatocellular carcinoma, and metastatic melanoma [99,100,101,102,103].
In a safety and efficacy clinical trial, the use of anti-CD19 CAR-T cells in combination with nivolumab in 11 patients with relapsed/refractory non-Hodgkin lymphoma has shown that this combined treatment regimen is safe and mediates potent anti-lymphoma activity [104]. Another study utilized HER-2+ transgenic mice models to study the effects of CAR-T cells with and without PD-1 inhibition, and it was observed that the combination therapy improved the therapeutic efficacy of CAR-T-cells against solid cancers [105]. The synergy seen in the combination therapy is likely multifactorial, being a product of increased T-cell effector activity and other effects, such as enhanced T-cell survival [106].
Investigations into the gene editing of CAR-T cells have revealed that PD1-knockout CAR-T cells are a clinically efficacious alternative to combination therapy. Specifically, it was observed in murine models that editing the PD1 gene on tumor-specific T-cells to reduce the expression of PD-1 resulted in a significant delay in tumor growth following administration to PD-L1 overexpressing tumors [107,108]. Further data supports these findings, in that PD-1 deficient CAR-T cells showed significantly better antitumor efficacy than wild-type CAR-T cells [109,110,111].
In pursuit of more effective treatments, the engineering of CAR-T cells has been explored, such as the modification of CAR-T cells to secrete PD-1 blockers–a modification that was found to control tumor growth in lung cancer models significantly better than standard CAR-T cell therapy [112]. In a similar study looking at humanized mice models of renal cell carcinoma, carbonic anhydrase-targeting CAR-T-cells engineered to secrete PD-L1 antibodies were shown to combat T-cell exhaustion and increase recruitment of NK cells to the tumor tissue, while in another study CAR-T cells were engineered to express an anti-PD-1 single chain variable fragment, which was shown to attenuate the PD-1-dependent inhibition of CAR-T cells as well as tumor-specific non-CAR-T cells, resulting in significant control of tumor growth when compared to wild type CAR-T-cells [113,114].
In addition, there are several ongoing clinical trials using CAR-T-cell therapy in combination with checkpoint blockers in the treatment of B-cell lymphoma, multiple myeloma, and Hodgkin lymphoma (NCT04134325, NCT04162119, NCT04163302, and NCT04213469). CAR-T-cell therapy and PD-1 inhibitors have both individually revolutionized the field of oncology, and their combination has been shown to be both synergistic and efficacious in overcoming resistance to checkpoint inhibition.
4.4. Tumor Neoantigen Vaccines
Heterogeneous response to PD-1/PD-L1 immunotherapy has prompted the investigation into strategies to overcome primary and acquired resistance. One such polypharmaceutical approach is the use of tumor neoantigen vaccines in combination with ICIs to potentiate the tumor-specific immune responses [115,116].
Tumor antigens, or neoantigens, are proteins displayed by tumor cells due to mutations in host DNA. Neoantigens stimulate CD4/8-MHC cell-mediated immune reactions, but clonal evolution promotes evasion of the normal response [117]. Studies support that tumor neoantigen vaccination is effective in individuals with an absence of pre-existing antitumor immunity. However, the TME expresses several immunosuppressive factors through prolonged exposure to cancer antigens, and/or poor infiltration of T-cells [118]. Anti-PD-1/PD-L1 immunotherapy has shown promising results in many solid tumors, with pancreatic cancer and glioblastomas being a few exceptions, possibly due to the lack of permeability of T-cells in tumor tissue [119]. A study outlined the following 3 distinct phenotypes describing the permeability of tumor cells based on epigenetic RNA N6-methyladenosine (m6A) modification in 1938 gastric cancer samples: immune-excluded, immune-inflamed, and immune-desert phenotypes. Based on their study, a low m6A score (indicating low levels of methylation and greater T-cell tissue infiltration) predicted an immune-inflamed phenotype, a better response to PD-1/PD-L1 immunotherapy, and a 5-year survival rate of 69.4% [120]. Cancer vaccines targeting neoantigens have been proposed as a tool to circumvent immunotherapy-induced resistance, increase T-cell permeability, and decrease T-cell exhaustion, potentially increasing the effectiveness of ICIs [118].
A Phase 1b feasibility study used the DC neoantigen vaccine in combination with anti-PD-1 therapy in pancreatic ductal adenocarcinomas to reprogram the TME and showed positive outcomes, a step forward in using these advanced therapeutic platforms, especially for treatment-refractory cancers [118]. Another vaccine approach currently in studies is using PD-1/PD-L1 checkpoint inhibitors with the GVAX vaccine–an anti-cancer vaccine consisting of irradiated tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor (GM-CSF) [121]. GM-CSF is a hematopoietic cytokine responsible for inducing the production of white blood cells, including granulocytes, macrophages, and megakaryocytes. GM-CSF also functions as a major pro-inflammatory cytokine, coordinating communication between lymphocytes and myeloid cells [122]. Another study combined PD-1/CTLA-4 checkpoint blockade with the GVAX vaccine in preclinical models of colon cancer and ovarian cancer with the eradication of tumor cells [123]. In addition to the GVAX vaccine, GM-CSF is used in the similar yet distinct treatment modality of oncolytic virotherapy, appearing as one of the principal genetic modifications in the only current FDA-approved oncolytic virus, Talimogene laherparepvec (T-vec), wherein GM-CSF likewise serves a proinflammatory role, potentiating an anti-tumor immune response. T-vec has shown clinical efficacy in treating metastatic melanoma, for which it received its original FDA approval, while recent investigations into its combination with PD-1 immunotherapy have shown promising clinical benefits [4]. This success has further galvanized interest in the combination of oncolytic viruses with PD1 inhibitors, with the field seeing an explosion in the diversity of viruses, spanning both DNA and RNA viruses, used in combination with PD1 checkpoint therapy [124].
Several high-powered clinical trials are currently investigating the safety and efficacy of tumor neoantigen vaccines in combination with PD-1/PD-L1 immunotherapy, radiotherapy, and conventional chemotherapy in the treatment of tumors of both solid and hematogenous origin. A phase 2 trial concluding in 2019 assessed the safety and efficacy of the GVAX vaccine and cyclophosphamide combined with or without nivolumab. In total, 93 enrolled subjects with metastatic pancreatic adenocarcinoma were given the GVAX/cyclophosphamide with or without anti-PD-1 immunotherapy. After the conclusion of the trial, the overall survival was 5.88 months for the (+) nivolumab group (95% CI = 4.73–8.64), while it was 6.11 months for the (−) nivolumab group (95% CI = 3.52–7.00) (NCT02243371). More recently, another study used modified DC vaccines combined with nivolumab to treat six patients with malignant gliomas. Subjects receiving neoadjuvant therapy experienced a median progression-free survival of 4.3 months with a 95% confidence interval of 2.1–5.3 months, while those in the adjuvant therapy group had a median progression-free survival of 6.3 months with a 95% confidence interval of 4.7–10.7 months (NCT02529072). Radiotherapy with ICIs and/or vaccination may prove useful in maximizing the tumor microenvironment and treatment response. A study published by Zhang et al. found that combination E7 vaccination-radiotherapy proved efficacious in altering the ratio of CD8+ T cells/Tregs, increasing the number of CD45+CD8+ cells and effector CD8+ IFN-γ+ T cells, and decreasing overall tumor weight as compared to conventional monotherapy or other studied combinations [125]. Outside of these two trials, there are over 100 active and recruiting studies, indicating the available literature on this approach to overcoming PD-1/PD-L1 resistance will likely increase dramatically going forward.
4.5. Gut Microbiota
Another active field of study in overcoming ICI resistance is that of the gut microbiome and its manipulation. The gut microbiome is known to directly regulate the classically studied and well-understood gastrointestinal processes such as the absorption, metabolism, and excretion of nutrients and pharmaceutical agents, but it has further been found to regulate more complex processes such as the maintenance of the intestinal mucosal barrier, the synthesis of short-chain fatty acids, the modification of the regulation of key hormones and neurotransmitters such as CCK, GLP-1, and 5-HT, and host immunity [126,127,128,129]. When the homeostasis of the microbiome is disturbed due to antibiotics or some other destructive exposure (through environment, diet, lifestyle, smoking, alcohol, etc.), the sensitive balance between commensals, pathobionts, and pathogens can become unsettled and cause considerable pathology in the patient. If not self-resolved or remedied by medication, such disturbances in the microbiome can cause significant metabolic derangements in addition to initiating chronic inflammatory processes and inflicting permanent damage to the underlying mucosa [128,130]. The literature describes the relationship between the microbiome and drugs as “bidirectional,” due to their ability to both positively and negatively affect the functions and efficacy of one another [131]. Manipulation of the gut microbiome may therefore possess beneficial clinical applications and should continue to be assessed as a possible method of overcoming PD-1/PD-L1 blockade resistance.
Current evidence from murine models supports this hypothesis, showing that the gut microbiome plays a significant role in the clinical response to ICIs targeting the PD-1/PD-L1 axis. A study observed decreased efficacy of ICIs in germ-free mice and mice pretreated with antibiotics. After the manual inoculation of bacterial species in these mice, the use of ICIs led to significantly higher T-cell proliferation. Lastly, feces from ICI-responsive patients were transferred into mice, which showed increased efficacy of ICI immunotherapy, providing evidence to suggest fecal microbiota transplant (FMT) as a viable combination therapy to potentiate ICI efficacy [132]. Another study discovered that the use of Bifidobacterium species enhanced the efficacy of PD-L1 therapy [133]. Aside from murine models, numerous studies in the literature have retrospectively and prospectively assessed the content of microflora among human patients receiving PD-1/PD-L1 therapy for metastatic melanoma, hepatocellular carcinoma, gastrointestinal cancer, NSCLC, and several other diseases [131,134,135,136,137,138,139,140]. Heterogeneous response rates in ICI-induced resistance in cancers due to the diverse gut microbiome are an active area of investigation. Long-term data may serve to guide clinical decision-making regarding FMT and/or bacterial supplements concurrently with PD-1/PD-L1 therapy. Currently, 12 plus relevant trials are investigating the effect of microbiome alteration, either through FMT or probiotic supplementation, on the efficacy of anti-PD-1 therapy. A phase 1b study was designed to assess the safety and efficacy surrounding SER-401 FMT and nivolumab therapy in 14 patients between 2019 and 2022 who received pre-treatment with vancomycin or placebo followed by SER-401 or placebo and nivolumab; results are in progress (NCT03817125). Another ongoing clinical study aims to understand whether FMT can convert a patient’s ICI-refractory metastatic melanoma to ICI-responsive. These study patients will receive FMT from either ICI-responding or non-responding patients alongside their standard immunotherapy to investigate (NCT05251389). Meanwhile, another trial is assessing the feasibility of an FMT capsule with ICI therapy in treatment-refractory gastrointestinal cancers (NCT04130763). Other ongoing studies are assessing the use of FMT, prebiotics, and/or probiotics alongside ICI therapy for several cancer types, including NSCLC, hepatocellular carcinoma, and gastric cancer (NCT05008861, NCT03772899, NCT04101747, NCT04130763, NCT04924374, NCT04988841, NCT05001360, NCT05032014, NCT05094167, NCT05251389, and NCT05303493). More studies are required to modulate the gut microbiome in ICI-refractory cancers to maximize clinical efficacy.
5. Discussion
Monoclonal antibodies targeting PD-1/PD-L1 have demonstrated clinical benefits in several cancers, including melanoma, NSCLC, renal cell cancer, bladder cancer, head and neck cancers, and MSI-high colorectal cancer to name a few [141,142]. Long-term follow-up of patients in clinical trials utilizing ICIs has identified the following three broad groups: responders, non-responders, and responders that fail to respond over time [143]. Challenges remain in understanding the differences between responders and non-responders given intra- and inter-patient heterogeneity. Additionally, incomplete knowledge of clinical, molecular, and immunological mechanisms driving innate and acquired resistance poses a major problem. Our understanding about the PD-1/PD-L1 axis on immune cells and cancer cells is ever-evolving. Tumor-specific CD8+T-cells differentiate from effector T-cells, undergo clonal expansion, infiltrate TME, and aid in tumor killing by displaying tumor-associated antigens [115]. Lack of anti-tumor responses with ICI therapy can result from the suboptimal generation of anti-tumor T cells, the inadequate function of tumor-specific T-cells, and impaired memory T-cells [144,145]. Another set of mechanisms creating resistance to ICIs includes a lack of suitable neoantigen, dysfunctional neoantigen processing, and impaired presentation of neoantigen to form tumor-specific T-cells [115]. Depletion, exhaustion, and dysfunction of CD8+T-cells can arise through diverse immunosuppressive components of TME [146].
Immune evasion is a multifactorial process that includes genetic/epigenetic aberrations that lead to insufficient neoantigen presentation, malfunctional presentation, and further processing that disrupts the cytotoxic potential of CD8+T-cells [146]. The presence of non-cancerous cells or diverse host microbiota can also promote ICI resistance in tumors [147]. Anti-PD-1/PD-L1 blockers reactivate these tumor-specific T-cells. However, altered neoantigen production and processing can impair anti-tumor immune responses [148]. The presence of high levels of non-synonymous mutations in cancers leads to the highest response rates to ICIs. However, mutations in genes encoding antigen processing components such as B2M result in defective MHC-I presentation and hence suboptimal response to ICIs [149,150]. Ever changing mutational landscape and clonal expansion of neoantigens have been responsible for acquiring resistance to ICIs. A key strategy to improve immunogenic cell death is to stimulate innate immunity and DC functionality such as type 1 interferons, TLR ligands, oncolytic viruses, etc. that can promote neoantigen production in TME [146]. Specific oncogenic signaling such as loss of PTEN diminishes T-cell infiltration; alterations in wnt signaling lead to DC trafficking in TME; an oncogenic KRas mutation is associated with higher levels of T-cell exhaustion; the presence of immunosuppressive gene signature, they all lead to multigenic reversible pathways of resistance [83,151,152]. It is plausible to overcome ICI resistance by altering these intrinsic and extrinsic signals causing PD-1/PD-L1 resistance.
Increased TMB is associated with increased PD-L1 expression as seen in advanced melanoma, resulting in improved progression-free survival and overall survival when treated with combined or mono-immunotherapy versus patients with negative PD-L1 [153]. However, there are cancers with equally increased PD-L1 expression, yet no effective immune response from ICI, making PD-L1 not a very reliable biomarker for the desired responses from checkpoint blockade [154]. Moreover, inhospitable TME can threaten expanding the repertoire of anti-tumor T-cells because of high levels of alternate checkpoints of co-inhibitory receptors on T-cells (CTLA-4, TIM-3, and LAG-3), high levels of immune suppressive cytokines, and recruitment of MDSCs as well as Treg cells [115]. These cellular processes led to inadequate anti-tumor T-cell effector functions. Moreover, mutations in immune effector signaling pathways such as JAK1/JAK2 are associated with resistance to PD-1 blockade [150]. These mutations resulted in a loss of interferon responsiveness. Similarly, mutations in B2M, deletion of IFN-γ receptors, and mutations in components of the JAK/STAT pathway contribute to resistance to PD-/PD-L1 blockade [155]. Importantly, these signaling pathways can be inhibited and could overcome acquired resistance to ICI blockers.
The heterogeneity of PD-1+CD8+T-cells in TME responds differently to anti-PD-1 treatment [156]. Exhausted PD-1+CD8+T-cells display a unique genetic landscape compared to effector T cells [157]. These epigenetic regulators in exhausted and non-exhausted T cells drive variable responses to checkpoint blockade. One such TIL subpopulation, TCF1+PD-1+CD8+ T-cells with stem-like characteristics, has been identified as a primary mediator of the therapeutic response to PD1 checkpoint inhibition. This population proliferates in response to immunotherapy, forming terminally differentiated TCF1-PD1+CD8+ T-cells in addition to increasing the progenitor TCF1+PD-1+CD8+ population. Studies, such as that of Siddiqui, et al., have shown that ablation of this population restricts responses to PD1 checkpoint immunotherapy, lending further credence to the thought of modulating T-cell populations to potentiate checkpoint inhibitor efficacy [158,159,160]. In addition to checkpoint inhibitor monotherapy, co-inhibitory receptors can be blocked with combination checkpoint blockade using LAG-3+PD-1 and TIM-3+PD-1, which have shown better response rates in pre-clinical models as well as currently being studied in clinical trials [161]. PD-1/PD-L1 independent mechanisms of immune evasion such as Tregs, MDSCs, Th2 CD4+ T cells, and M2-polarized tumor-associated macrophages collectively influence the resistance to anti-PD-1/PD-L1 treatments. Removal of anti-PD-1 antibodies to inhibit its interaction with PD-1+CD8+T-cells via an FcγR-dependent manner has been reported [162]. Pre-clinical studies have been successful in eliminating this alternate population of immune cells that are driving the innate resistance to ICIs. In most tumors, the formation of memory T-cells as a response to either tumor formation or immune checkpoint therapies is impaired. These processes lead to a lack of durable responses. The transcriptional machinery in TME defines the fate of these T-cells in naïve, memory, and exhausted T-cell states [162]. Ongoing clinical studies are augmenting existing new T-cell responses or priming new populations of T-cells using adoptive cellular therapy like CAR-T cells.
Even if the T-cell activation process were not defective, there could be a disruption in T-cell trafficking and tumor infiltration because of the downregulation of chemokines such as CXCR3 required for T-cell recruitment [163]. Epigenetic regulation in silencing CXCL9 and CXCL10 in cancers leads to chemokine repression and tumor progression. T-cells traffic in lymph nodes by endothelin B receptors, and genetic changes in ETBR result in a potential immune escape mechanism [164]. Overexpression of VEGF downregulates T-cell adhesion, TILs, and shorter overall survival as a response to ICI. TME plays a crucial role by inducing the expression of PD-L1 and Tregs through several oncogenic signals, thereby decreasing cytotoxic T-cell functionality [165].
In summary, resistance to immune checkpoint inhibitors (PD-1/PD-L1) is driven by complex genetic/epigenetic interactions between immune cells and tumor cells. These innate and acquired resistance pathways overlap. Ongoing clinical trials are combining immunotherapeutic agents with targeted agents, cytotoxic chemotherapy, and/or radiation, all in an effort to provide a durable response to these revolutionary therapies. Alternative strategies, such as cancer neoantigen vaccines, CAR-T cellular therapies, genotype-targeted therapies, and modulation of the gut microbiome, are required to overcome resistance in non-responders. It is prudent to identify unique biomarkers, both predictive and prognostic, in responders vs. non-responders to ICIs. Checkpoint inhibitor immunotherapy has highlighted the possibility of curing cancers, yet a significant unmet clinical need is to be able to understand the landscape of resistance to ICI early on to customize therapies based on every patient’s unique tumor/immune genetic signature.
Author Contributions
Conceptualization, J.M. (John Moise) and S.A.; investigation, J.M. (John Moise), J.M. (Jeevan Murthy), D.D., S.Y., F.K., E.H. and S.A.; writing—original draft preparation, J.M. (John Moise), J.M. (Jeevan Murthy), S.Y. and S.A.; writing—review and editing, J.M. (John Moise); supervision, S.A.; project administration, J.M. (John Moise); funding acquisition, S.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The authors would like to extend their deepest gratitude to Kaitlyn Mi of The Dartmouth Institute for her illustrations of the biochemical pathways in Section 4.1 and Section 4.2.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, T.F.; Shachar, S.S.; Nyrop, K.A.; Muss, H.B. Safety and Tolerability of PD-1/PD-L1 Inhibitors Compared with Chemotherapy in Patients with Advanced Cancer: A Meta-Analysis. Oncologist 2017, 22, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhao, B. Efficacy of PD-1 or PD-L1 Inhibitors and PD-L1 Expression Status in Cancer: Meta-Analysis. BMJ 2018, 362, k3529. [Google Scholar] [CrossRef] [PubMed]
- US Food & Drug. Administration Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 19 April 2022).
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, upon Programmed Cell Death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of Lupus-like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a Third Member of the B7 Family, Co-Stimulates T-Cell Proliferation and Interleukin-10 Secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on Tumor Cells in the Escape from Host Immune System and Tumor Immunotherapy by PD-L1 Blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef]
- Lee, S.-J.; Jang, B.-C.; Lee, S.-W.; Yang, Y.-I.; Suh, S.-I.; Park, Y.-M.; Oh, S.; Shin, J.-G.; Yao, S.; Chen, L.; et al. Interferon Regulatory Factor-1 Is Prerequisite to the Constitutive Expression and IFN-Gamma-Induced Upregulation of B7-H1 (CD274). FEBS Lett. 2006, 580, 755–762. [Google Scholar] [CrossRef]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma Cells from Multiple Myeloma Patients Express B7-H1 (PD-L1) and Increase Expression after Stimulation with IFN-{gamma} and TLR Ligands via a MyD88-, TRAF6-, and MEK-Dependent Pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gajewski, T.F.; Kline, J. PD-1/PD-L1 Interactions Inhibit Antitumor Immune Responses in a Murine Acute Myeloid Leukemia Model. Blood 2009, 114, 1545–1552. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-Driven Biomarkers to Guide Immune Checkpoint Blockade in Cancer Therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, X.; Fu, J.; Wang, H. Progress and Challenges in Precise Treatment of Tumors With PD-1/PD-L1 Blockade. Front. Immunol. 2020, 11, 339. [Google Scholar] [CrossRef]
- Sun, J.-Y.; Zhang, D.; Wu, S.; Xu, M.; Zhou, X.; Lu, X.-J.; Ji, J. Resistance to PD-1/PD-L1 Blockade Cancer Immunotherapy: Mechanisms, Predictive Factors, and Future Perspectives. Biomark. Res. 2020, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD1:PD-L1/2 Pathway from Discovery to Clinical Implementation. Front. Immunol. 2016, 7, 550. [Google Scholar] [CrossRef]
- Nurieva, R.; Thomas, S.; Nguyen, T.; Martin-Orozco, N.; Wang, Y.; Kaja, M.-K.; Yu, X.-Z.; Dong, C. T-Cell Tolerance or Function Is Determined by Combinatorial Costimulatory Signals. EMBO J. 2006, 25, 2623–2633. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Liang, X.-L.; Liu, X.-G.; Chen, N.-P. The Landscape of PD-L1 Expression and Somatic Mutations in Hepatocellular Carcinoma. J. Gastrointest. Oncol. 2021, 12, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue Expression of PD-L1 Mediates Peripheral T Cell Tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Eppihimer, M.J.; Gunn, J.; Freeman, G.J.; Greenfield, E.A.; Chernova, T.; Erickson, J.; Leonard, J.P. Expression and Regulation of the PD-L1 Immunoinhibitory Molecule on Microvascular Endothelial Cells. Microcirculation 2002, 9, 133–145. [Google Scholar] [CrossRef]
- Ju, X.; Zhang, H.; Zhou, Z.; Wang, Q. Regulation of PD-L1 Expression in Cancer and Clinical Implications in Immunotherapy. Am. J. Cancer Res. 2020, 10, 1–11. [Google Scholar] [PubMed]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed Death-1 Ligand 1 Interacts Specifically with the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 Pathway in Tolerance and Autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.H.; Gillett, M.D.; Cheville, J.C.; Lohse, C.M.; Dong, H.; Webster, W.S.; Krejci, K.G.; Lobo, J.R.; Sengupta, S.; Chen, L.; et al. Costimulatory B7-H1 in Renal Cell Carcinoma Patients: Indicator of Tumor Aggressiveness and Potential Therapeutic Target. Proc. Natl. Acad. Sci. USA 2004, 101, 17174–17179. [Google Scholar] [CrossRef]
- Ohigashi, Y.; Sho, M.; Yamada, Y.; Tsurui, Y.; Hamada, K.; Ikeda, N.; Mizuno, T.; Yoriki, R.; Kashizuka, H.; Yane, K.; et al. Clinical Significance of Programmed Death-1 Ligand-1 and Programmed Death-1 Ligand-2 Expression in Human Esophageal Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 2947–2953. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhu, Y.; Jiang, J.; Zhao, J.; Zhang, X.-G.; Xu, N. Immunohistochemical Localization of Programmed Death-1 Ligand-1 (PD-L1) in Gastric Carcinoma and Its Clinical Significance. Acta Histochem. 2006, 108, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed Cell Death 1 Ligand 1 and Tumor-Infiltrating CD8+ T Lymphocytes Are Prognostic Factors of Human Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef]
- Nakanishi, J.; Wada, Y.; Matsumoto, K.; Azuma, M.; Kikuchi, K.; Ueda, S. Overexpression of B7-H1 (PD-L1) Significantly Associates with Tumor Grade and Postoperative Prognosis in Human Urothelial Cancers. Cancer Immunol. Immunother. CII 2007, 56, 1173–1182. [Google Scholar] [CrossRef]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical Significance and Therapeutic Potential of the Programmed Death-1 Ligand/Programmed Death-1 Pathway in Human Pancreatic Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor Cell Expression of Programmed Cell Death-1 Ligand 1 Is a Prognostic Factor for Malignant Melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef]
- Abril-Rodriguez, G.; Ribas, A. SnapShot: Immune Checkpoint Inhibitors. Cancer Cell 2017, 31, 848–848.e1. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 Have Opposing Effects on the Response of T Cells to Stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, Durable Tumor Remission, and Long-Term Safety in Patients with Advanced Melanoma Receiving Nivolumab. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the Anti-PD-1 and Anti-PD-L1 Immune Checkpoint Antibodies. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef]
- Ai, L.; Chen, J.; Yan, H.; He, Q.; Luo, P.; Xu, Z.; Yang, X. Research Status and Outlook of PD-1/PD-L1 Inhibitors for Cancer Therapy. Drug Des. Dev. Ther. 2020, 14, 3625–3649. [Google Scholar] [CrossRef] [PubMed]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef]
- Couillault, C.; Srinivasamani, A.; Hedge, S.; Liu, Q.; Jaiswal, A.; Zha, D.; Curran, M. 291 Dual-Specific Antibodies Blocking Both PD-L1 and PD-L2 Engagement of PD-1 Restore Anti-Tumor Immunity. J. Immunother. Cancer 2021, 9, A315. [Google Scholar] [CrossRef]
- Nowicki, T.S.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. Sudbury Mass. 2018, 24, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Gianchecchi, E.; Fierabracci, A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front. Immunol. 2018, 9, 2374. [Google Scholar] [CrossRef]
- Cai, J.; Wang, D.; Zhang, G.; Guo, X. The Role Of PD-1/PD-L1 Axis In Treg Development And Function: Implications For Cancer Immunotherapy. OncoTargets Ther. 2019, 12, 8437–8445. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A Symbiotic Bridge Linking Cancer Cells and Their Stromal Neighbors in Oncogenic Communication Networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef]
- Liu, C.; Liu, R.; Wang, B.; Lian, J.; Yao, Y.; Sun, H.; Zhang, C.; Fang, L.; Guan, X.; Shi, J.; et al. Blocking IL-17A Enhances Tumor Response to Anti-PD-1 Immunotherapy in Microsatellite Stable Colorectal Cancer. J. Immunother. Cancer 2021, 9, e001895. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Frömming, A.; Vater, A. Increasing Tumor-Infiltrating T Cells through Inhibition of CXCL12 with NOX-A12 Synergizes with PD-1 Blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Riese, D.J.; Shen, J. The Role of the CXCL12/CXCR4/CXCR7 Chemokine Axis in Cancer. Front. Pharmacol. 2020, 11, 574667. [Google Scholar] [CrossRef]
- Chen, I.X.; Chauhan, V.P.; Posada, J.; Ng, M.R.; Wu, M.W.; Adstamongkonkul, P.; Huang, P.; Lindeman, N.; Langer, R.; Jain, R.K. Blocking CXCR4 Alleviates Desmoplasia, Increases T-Lymphocyte Infiltration, and Improves Immunotherapy in Metastatic Breast Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 4558–4566. [Google Scholar] [CrossRef] [PubMed]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: The COMBAT Trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Williams, A.; Schreiber, J.; Hohmann, N.; Pruefer, U.; Krauss, J.; Jäger, D.; Frömming, A.; Beyer, D.; Eulberg, D.; et al. Combined Inhibition of CXCL12 and PD-1 in MSS Colorectal and Pancreatic Cancer: Modulation of the Microenvironment and Clinical Effects. J. Immunother. Cancer 2021, 9, e002505. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, E.; Mishra, L. Transforming Growth Factor-Beta Signaling and Ubiquitinators in Cancer. Endocr. Relat. Cancer 2008, 15, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Strainic, M.G.; Shevach, E.M.; An, F.; Lin, F.; Medof, M.E. Absence of Signaling into CD4+ Cells via C3aR and C5aR Enables Autoinductive TGF-Β1 Signaling and Induction of Foxp3+ Regulatory T Cells. Nat. Immunol. 2013, 14, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Lind, H.; Gameiro, S.R.; Jochems, C.; Donahue, R.N.; Strauss, J.; Gulley, J.L.; Palena, C.; Schlom, J. Dual Targeting of TGF-β and PD-L1 via a Bifunctional Anti-PD-L1/TGF-ΒRII Agent: Status of Preclinical and Clinical Advances. J. Immunother. Cancer 2020, 8, e000433. [Google Scholar] [CrossRef]
- Knudson, K.M.; Hicks, K.C.; Luo, X.; Chen, J.-Q.; Schlom, J.; Gameiro, S.R. M7824, a Novel Bifunctional Anti-PD-L1/TGFβ Trap Fusion Protein, Promotes Anti-Tumor Efficacy as Monotherapy and in Combination with Vaccine. Oncoimmunology 2018, 7, e1426519. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced Preclinical Antitumor Activity of M7824, a Bifunctional Fusion Protein Simultaneously Targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10, eaan5488. [Google Scholar] [CrossRef]
- Strauss, J.; Heery, C.R.; Schlom, J.; Madan, R.A.; Cao, L.; Kang, Z.; Lamping, E.; Marté, J.L.; Donahue, R.N.; Grenga, I.; et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFβ, in Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1287–1295. [Google Scholar] [CrossRef]
- Yoo, C.; Oh, D.-Y.; Choi, H.J.; Kudo, M.; Ueno, M.; Kondo, S.; Chen, L.-T.; Osada, M.; Helwig, C.; Dussault, I.; et al. Phase I Study of Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Patients with Pretreated Biliary Tract Cancer. J. Immunother. Cancer 2020, 8, e000564. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, Y.; Ishigame, H.; Saijo, S.; Nakae, S. Functional Specialization of Interleukin-17 Family Members. Immunity 2011, 34, 149–162. [Google Scholar] [CrossRef]
- Martin-Orozco, N.; Muranski, P.; Chung, Y.; Yang, X.O.; Yamazaki, T.; Lu, S.; Hwu, P.; Restifo, N.P.; Overwijk, W.W.; Dong, C. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. Immunity 2009, 31, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Banerjee, M.; Cheng, P.; Vatan, L.; Szeliga, W.; Wei, S.; Huang, E.; Finlayson, E.; Simeone, D.; Welling, T.H.; et al. Phenotype, Distribution, Generation, and Functional and Clinical Relevance of Th17 Cells in the Human Tumor Environments. Blood 2009, 114, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Benchetrit, F.; Ciree, A.; Vives, V.; Warnier, G.; Gey, A.; Sautès-Fridman, C.; Fossiez, F.; Haicheur, N.; Fridman, W.H.; Tartour, E. Interleukin-17 Inhibits Tumor Cell Growth by Means of a T-Cell-Dependent Mechanism. Blood 2002, 99, 2114–2121. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. ΓδT17 Cells Promote the Accumulation and Expansion of Myeloid-Derived Suppressor Cells in Human Colorectal Cancer. Immunity 2014, 40, 785–800. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ye, X.; Pitmon, E.; Lu, M.; Wan, J.; Jellison, E.R.; Adler, A.J.; Vella, A.T.; Wang, K. IL-17 Inhibits CXCL9/10-Mediated Recruitment of CD8+ Cytotoxic T Cells and Regulatory T Cells to Colorectal Tumors. J. Immunother. Cancer 2019, 7, 324. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Luber, B.; Tam, A.J.; Smith, K.N.; Siegel, N.; Awan, A.H.; Fan, H.; Oke, T.; Zhang, J.; Domingue, J.; et al. Intratumoral Adaptive Immunosuppression and Type 17 Immunity in Mismatch Repair Proficient Colorectal Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5250–5259. [Google Scholar] [CrossRef] [PubMed]
- Brevi, A.; Cogrossi, L.L.; Grazia, G.; Masciovecchio, D.; Impellizzieri, D.; Lacanfora, L.; Grioni, M.; Bellone, M. Much More Than IL-17A: Cytokines of the IL-17 Family Between Microbiota and Cancer. Front. Immunol. 2020, 11, 565470. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 Impair Tumor Antigen-Specific CD8+ T Cells in Melanoma Patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 Expression on HIV-Specific T Cells Is Associated with T-Cell Exhaustion and Disease Progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8(+) T Cell Effector Function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-Cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 Associate with Immunoreceptor Tyrosine-Based Switch Motif of Programmed Death 1 upon Primary Human T Cell Stimulation, but Only Receptor Ligation Prevents T Cell Activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef]
- Hosseinkhani, N.; Shadbad, M.A.; Asghari Jafarabadi, M.; Karim Ahangar, N.; Asadzadeh, Z.; Mohammadi, S.M.; Lotfinejad, P.; Alizadeh, N.; Brunetti, O.; Fasano, R.; et al. A Systematic Review and Meta-Analysis on the Significance of TIGIT in Solid Cancers: Dual TIGIT/PD-1 Blockade to Overcome Immune-Resistance in Solid Cancers. Int. J. Mol. Sci. 2021, 22, 10389. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.S.K.; Sansom, D.M. The Emerging Role of CTLA4 as a Cell-Extrinsic Regulator of T Cell Responses. Nat. Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dai, Z.; Wu, W.; Wang, Z.; Zhang, N.; Zhang, L.; Zeng, W.-J.; Liu, Z.; Cheng, Q. Regulatory Mechanisms of Immune Checkpoints PD-L1 and CTLA-4 in Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 184. [Google Scholar] [CrossRef]
- Goldberg, M.V.; Drake, C.G. LAG-3 in Cancer Immunotherapy. Curr. Top. Microbiol. Immunol. 2011, 344, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, P.A.; Melero, I.; Bhatia, S.; Bono, P.; Sanborn, R.E.; Lipson, E.J.; Callahan, M.K.; Gajewski, T.; Gomez-Roca, C.A.; Hodi, F.S.; et al. Initial Efficacy of Anti-Lymphocyte Activation Gene-3 (Anti–LAG-3; BMS-986016) in Combination with Nivolumab (Nivo) in Pts with Melanoma (MEL) Previously Treated with Anti–PD-1/PD-L1 Therapy. J. Clin. Oncol. 2017, 35, 9520. [Google Scholar] [CrossRef]
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 Expression Is Associated with Tumor Antigen-Specific CD8+ T Cell Dysfunction in Melanoma Patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Xu, Y.; Fang, J.; Liu, W.; Chen, J.; Liu, Z.; Xu, Q. Targeting STAT3 Abrogates Tim-3 Upregulation of Adaptive Resistance to PD-1 Blockade on Regulatory T Cells of Melanoma. Front. Immunol. 2021, 12, 654749. [Google Scholar] [CrossRef]
- de Mingo Pulido, Á.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103+ Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74. [Google Scholar] [CrossRef]
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive Resistance to Anti-PD1 Therapy by Tim-3 Upregulation Is Mediated by the PI3K-Akt Pathway in Head and Neck Cancer. Oncoimmunology 2017, 6, e1261779. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.-C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 3620–3629. [Google Scholar] [CrossRef] [PubMed]
- Peng, D.H.; Rodriguez, B.L.; Diao, L.; Chen, L.; Wang, J.; Byers, L.A.; Wei, Y.; Chapman, H.A.; Yamauchi, M.; Behrens, C.; et al. Collagen Promotes Anti-PD-1/PD-L1 Resistance in Cancer through LAIR1-Dependent CD8+ T Cell Exhaustion. Nat. Commun. 2020, 11, 4520. [Google Scholar] [CrossRef]
- Lebbink, R.J.; van den Berg, M.C.W.; de Ruiter, T.; Raynal, N.; van Roon, J.A.G.; Lenting, P.J.; Jin, B.; Meyaard, L. The Soluble Leukocyte-Associated Ig-like Receptor (LAIR)-2 Antagonizes the Collagen/LAIR-1 Inhibitory Immune Interaction. J. Immunol. 2008, 180, 1662–1669. [Google Scholar] [CrossRef] [PubMed]
- Meyaard, L.; Adema, G.J.; Chang, C.; Woollatt, E.; Sutherland, G.R.; Lanier, L.L.; Phillips, J.H. LAIR-1, a Novel Inhibitory Receptor Expressed on Human Mononuclear Leukocytes. Immunity 1997, 7, 283–290. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to Checkpoint Blockade Therapy through Inactivation of Antigen Presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 2019, 178, 933–948.e14. [Google Scholar] [CrossRef]
- Hurwitz, M.E.; Cho, D.C.; Balar, A.V.; Curti, B.D.; Siefker-Radtke, A.O.; Sznol, M.; Kluger, H.M.; Bernatchez, C.; Fanton, C.; Iacucci, E.; et al. Baseline Tumor-Immune Signatures Associated with Response to Bempegaldesleukin (NKTR-214) and Nivolumab. J. Clin. Oncol. 2019, 37, 2623. [Google Scholar] [CrossRef]
- Sharma, M.; Khong, H.; Fa’ak, F.; Bentebibel, S.-E.; Janssen, L.M.E.; Chesson, B.C.; Creasy, C.A.; Forget, M.-A.; Kahn, L.M.S.; Pazdrak, B.; et al. Bempegaldesleukin Selectively Depletes Intratumoral Tregs and Potentiates T Cell-Mediated Cancer Therapy. Nat. Commun. 2020, 11, 661. [Google Scholar] [CrossRef] [PubMed]
- Torrejon, D.Y.; Abril-Rodriguez, G.; Champhekar, A.S.; Tsoi, J.; Campbell, K.M.; Kalbasi, A.; Parisi, G.; Zaretsky, J.M.; Garcia-Diaz, A.; Puig-Saus, C.; et al. Overcoming Genetically Based Resistance Mechanisms to PD-1 Blockade. Cancer Discov. 2020, 10, 1140–1157. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Rahimian, S.; Haymaker, C.L.; Bernatchez, C.; Andtbacka, R.H.I.; James, M.; Johnson, D.B.; Markowitz, J.; Murthy, R.; Puzanov, I.; et al. A Phase 2 Study to Evaluate the Safety and Efficacy of Intratumoral (IT) Injection of the TLR9 Agonist IMO-2125 (IMO) in Combination with Ipilimumab (Ipi) in PD-1 Inhibitor Refractory Melanoma. J. Clin. Oncol. 2018, 36, 9515. [Google Scholar] [CrossRef]
- Ren, D.; Hua, Y.; Yu, B.; Ye, X.; He, Z.; Li, C.; Wang, J.; Mo, Y.; Wei, X.; Chen, Y.; et al. Predictive Biomarkers and Mechanisms Underlying Resistance to PD1/PD-L1 Blockade Cancer Immunotherapy. Mol. Cancer 2020, 19, 19. [Google Scholar] [CrossRef]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef]
- Konen, J.M.; Rodriguez, B.L.; Fradette, J.J.; Gibson, L.; Davis, D.; Minelli, R.; Peoples, M.D.; Kovacs, J.; Carugo, A.; Bristow, C.; et al. Ntrk1 Promotes Resistance to PD-1 Checkpoint Blockade in Mesenchymal Kras/P53 Mutant Lung Cancer. Cancers 2019, 11, 462. [Google Scholar] [CrossRef]
- Mian, A.; Hill, B.T. Brexucabtagene Autoleucel for the Treatment of Relapsed/Refractory Mantle Cell Lymphoma. Expert Opin. Biol. Ther. 2021, 21, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer Regression in Patients after Transfer of Genetically Engineered Lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Deng, Q.; Jiang, Y.-Y.; Zhang, R.; Zhu, H.-B.; Meng, J.-X.; Li, Y.-M. CAR-T 19 Combined with Reduced-Dose PD-1 Blockade Therapy for Treatment of Refractory Follicular Lymphoma: A Case Report. Oncol. Lett. 2019, 18, 4415–4420. [Google Scholar] [CrossRef]
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination With Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma. Front. Oncol. 2019, 9, 767. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Kershaw, M.H.; Darcy, P.K. Blockade of PD-1 Immunosuppression Boosts CAR T-Cell Therapy. Oncoimmunology 2013, 2, e26286. [Google Scholar] [CrossRef]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-Specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but Can Be Protected From Activation-Induced Cell Death by PD-1 Blockade. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef]
- Marotte, L.; Simon, S.; Vignard, V.; Dupre, E.; Gantier, M.; Cruard, J.; Alberge, J.-B.; Hussong, M.; Deleine, C.; Heslan, J.-M.; et al. Increased Antitumor Efficacy of PD-1-Deficient Melanoma-Specific Human Lymphocytes. J. Immunother. Cancer 2020, 8, e000311. [Google Scholar] [CrossRef]
- Marotte, L.; Capitao, M.; Deleine, C.; Beauvais, T.; Cadiou, G.; Perrin, J.; Chérel, M.; Scotet, E.; Guilloux, Y.; Bruchertseifer, F.; et al. Anti-Tumor Efficacy of a Combination Therapy with PD-L1 Targeted Alpha Therapy and Adoptive Cell Transfer of PD-1 Deficient Melanoma-Specific Human T-Lymphocytes. Oncoimmunology 2021, 10, 1940676. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-Tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef] [PubMed]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Human Mesothelin-Targeted CAR T Cell Effector Functions. Cancer Immunol. Immunother. CII 2019, 68, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef]
- Nakajima, M.; Sakoda, Y.; Adachi, K.; Nagano, H.; Tamada, K. Improved Survival of Chimeric Antigen Receptor-Engineered T (CAR-T) and Tumor-Specific T Cells Caused by Anti-Programmed Cell Death Protein 1 Single-Chain Variable Fragment-Producing CAR-T Cells. Cancer Sci. 2019, 110, 3079–3088. [Google Scholar] [CrossRef] [PubMed]
- Suarez, E.R.; Chang, D.K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric Antigen Receptor T Cells Secreting Anti-PD-L1 Antibodies More Effectively Regress Renal Cell Carcinoma in a Humanized Mouse Model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.L.; Smyth, M.J. Resistance to PD1/PDL1 Checkpoint Inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef]
- Pennock, G.K.; Chow, L.Q.M. The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. The Oncologist 2015, 20, 812–822. [Google Scholar] [CrossRef]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen Vaccine: An Emerging Tumor Immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef]
- Bassani-Sternberg, M.; Digklia, A.; Huber, F.; Wagner, D.; Sempoux, C.; Stevenson, B.J.; Thierry, A.-C.; Michaux, J.; Pak, H.; Racle, J.; et al. A Phase Ib Study of the Combination of Personalized Autologous Dendritic Cell Vaccine, Aspirin, and Standard of Care Adjuvant Chemotherapy Followed by Nivolumab for Resected Pancreatic Adenocarcinoma-A Proof of Antigen Discovery Feasibility in Three Patients. Front. Immunol. 2019, 10, 1832. [Google Scholar] [CrossRef]
- Nesselhut, J.; Marx, D.; Lange, H.; Regalo, G.; Cillien, N.; Chang, R.Y.; Nesselhut, T. Systemic Treatment with Anti-PD-1 Antibody Nivolumab in Combination with Vaccine Therapy in Advanced Pancreatic Cancer. J. Clin. Oncol. 2016, 34, 3092. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. M6A Regulator-Mediated Methylation Modification Patterns and Tumor Microenvironment Infiltration Characterization in Gastric Cancer. Mol. Cancer 2020, 19, 53. [Google Scholar] [CrossRef]
- Nemunaitis, J. Vaccines in Cancer: GVAX, a GM-CSF Gene Vaccine. Expert Rev. Vaccines 2005, 4, 259–274. [Google Scholar] [CrossRef]
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973. [Google Scholar] [CrossRef] [PubMed]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual Blockade of PD-1 and CTLA-4 Combined with Tumor Vaccine Effectively Restores T-Cell Rejection Function in Tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef]
- Chen, C.-Y.; Hutzen, B.; Wedekind, M.F.; Cripe, T.P. Oncolytic Virus and PD-1/PD-L1 Blockade Combination Therapy. Oncolytic Virotherapy 2018, 7, 65–77. [Google Scholar] [CrossRef]
- Zhang, F.; Zheng, Z.; Barman, A.K.; Wang, Z.; Wang, L.; Zeng, W.; Wang, L.; Qin, Y.; Pandey, A.; Zhang, C.; et al. Optimal Combination Treatment Regimens of Vaccine and Radiotherapy Augment Tumor-Bearing Host Immunity. Commun. Biol. 2021, 4, 78. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The Gut-Brain Axis: Interactions between Enteric Microbiota, Central and Enteric Nervous Systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar] [PubMed]
- Holmes, E.; Li, J.V.; Athanasiou, T.; Ashrafian, H.; Nicholson, J.K. Understanding the Role of Gut Microbiome-Host Metabolic Signal Disruption in Health and Disease. Trends Microbiol. 2011, 19, 349–359. [Google Scholar] [CrossRef]
- Martin, A.M.; Sun, E.W.; Rogers, G.B.; Keating, D.J. The Influence of the Gut Microbiome on Host Metabolism Through the Regulation of Gut Hormone Release. Front. Physiol. 2019, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The Gut Microbiome in Health and in Disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, X.; Zhang, Y.; Zheng, K.; Xiang, Q.; Chen, N.; Chen, Z.; Zhang, N.; Zhu, J.; He, Q. Antibiotic-Induced Disruption of Gut Microbiota Alters Local Metabolomes and Immune Responses. Front. Cell. Infect. Microbiol. 2019, 9, 99. [Google Scholar] [CrossRef]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between Drugs and the Gut Microbiome. Gut 2020, 69, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer Immunotherapy by CTLA-4 Blockade Relies on the Gut Microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium Promotes Antitumor Immunity and Facilitates Anti-PD-L1 Efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to Anti-PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef]
- Jin, Y.; Dong, H.; Xia, L.; Yang, Y.; Zhu, Y.; Shen, Y.; Zheng, H.; Yao, C.; Wang, Y.; Lu, S. The Diversity of Gut Microbiome Is Associated With Favorable Responses to Anti-Programmed Death 1 Immunotherapy in Chinese Patients With NSCLC. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, 1378–1389. [Google Scholar] [CrossRef]
- Mao, J.; Wang, D.; Long, J.; Yang, X.; Lin, J.; Song, Y.; Xie, F.; Xun, Z.; Wang, Y.; Wang, Y.; et al. Gut Microbiome Is Associated with the Clinical Response to Anti-PD-1 Based Immunotherapy in Hepatobiliary Cancers. J. Immunother. Cancer 2021, 9, e003334. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Cheng, S.; Kou, Y.; Wang, Z.; Jin, R.; Hu, H.; Zhang, X.; Gong, J.-F.; Li, J.; Lu, M.; et al. The Gut Microbiome Is Associated with Clinical Response to Anti-PD-1/PD-L1 Immunotherapy in Gastrointestinal Cancer. Cancer Immunol. Res. 2020, 8, 1251–1261. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut Microbiome Influences Efficacy of PD-1-Based Immunotherapy against Epithelial Tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut Microbiome Affects the Response to Anti-PD-1 Immunotherapy in Patients with Hepatocellular Carcinoma. J. Immunother. Cancer 2019, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Andrews, M.C.; Duong, C.P.M.; Gopalakrishnan, V.; Iebba, V.; Chen, W.-S.; Derosa, L.; Khan, M.A.W.; Cogdill, A.P.; White, M.G.; Wong, M.C.; et al. Gut Microbiota Signatures Are Associated with Toxicity to Combined CTLA-4 and PD-1 Blockade. Nat. Med. 2021, 27, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.-J.; Weber, J.S.; et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Kasic, T.; Gri, G.; Gallana, K.; Borsellino, G.; Marigo, I.; Battistini, L.; Iafrate, M.; Prayer-Galetti, T.; Pagano, F.; et al. Boosting Antitumor Responses of T Lymphocytes Infiltrating Human Prostate Cancers. J. Exp. Med. 2005, 201, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Zitvogel, L.; Pitt, J.M.; Daillère, R.; Smyth, M.J.; Kroemer, G. Mouse Models in Oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-Intrinsic β-Catenin Signalling Prevents Anti-Tumour Immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed]
- Havel, J.J.; Chowell, D.; Chan, T.A. The Evolving Landscape of Biomarkers for Checkpoint Inhibitor Immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The Epigenetic Landscape of T Cell Exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T Cells That Provide the Proliferative Burst after PD-1 Therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of Exhausted CD8+ T Cells Differentially Mediate Tumor Control and Respond to Checkpoint Blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211. [Google Scholar] [CrossRef]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 Pathways to Reverse T Cell Exhaustion and Restore Anti-Tumor Immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In Vivo Imaging Reveals a Tumor-Associated Macrophage-Mediated Resistance Pathway in Anti-PD-1 Therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [PubMed]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-Redundant Requirement for CXCR3 Signalling during Tumoricidal T-Cell Trafficking across Tumour Vascular Checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.-J. CXCL9, CXCL10, CXCL11/CXCR3 Axis for Immune Activation—A Target for Novel Cancer Therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-T.; Sun, Z.-J. Turning Cold Tumors into Hot Tumors by Improving T-Cell Infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).