
Signaling Pathways in Inflammation and Cardiovascular Diseases: An Update of Therapeutic Strategies


Abstract
1. Introduction
2. Inflammatory Mediators in Cardiovascular Diseases

{kind=link}
{kind=link}
{kind=link}
| Inflammatory Mediator | Anti-Inflammatory Agent | Experimental Study | Clinical Trial | Outcomes | References |
|---|---|---|---|---|---|
| CRP | Rosuvastatin | - | Aortic Stenosis Progression Observation: Measuring Effects of Rosuvastatin (ASTRONOMER) | ↓ CRP levels ↓ LDL cholesterol | [27] |
| - | GISSI-HF (Gruppo Italiano Per Lo Studio Della Sopravvivenza Nell’Insufficienza Cardiaca-Heart Failure) | [28] | |||
| - | Controlled Rosuvastatin Multinational Trial in Heart Failure (CORONA) | [29] | |||
| PTX3 | - | ✓ | - | ↑ PTX3 in patients with acute MI and infectious myocarditis | [33] |
| - | - | Lipid Assessment Trial Italian Network (LATIN) | PTX3 prognostic tool: 3 month mortality in patients with MI and ST elevation | [34] | |
| TNF-α | Etanercept | - | RENEWAL (Randomized Etanercept Worldwide Evaluation)—combined data from RECOVER and RENAISSANCE | No improvement on the rate of death or hospitalization in patients with NYHA class II to IV chronic HF | [43] |
| ✓ | - | ↓ TNF-α ↓ NF-κB in induced rheumatoid arthritis rats | [44] |
3. Reactive Oxygen Species (ROS), NADPH Oxidases (NOXs), and NF-κB: Putative Therapeutic Targets
4. Targeting Inflammatory Signaling Pathways
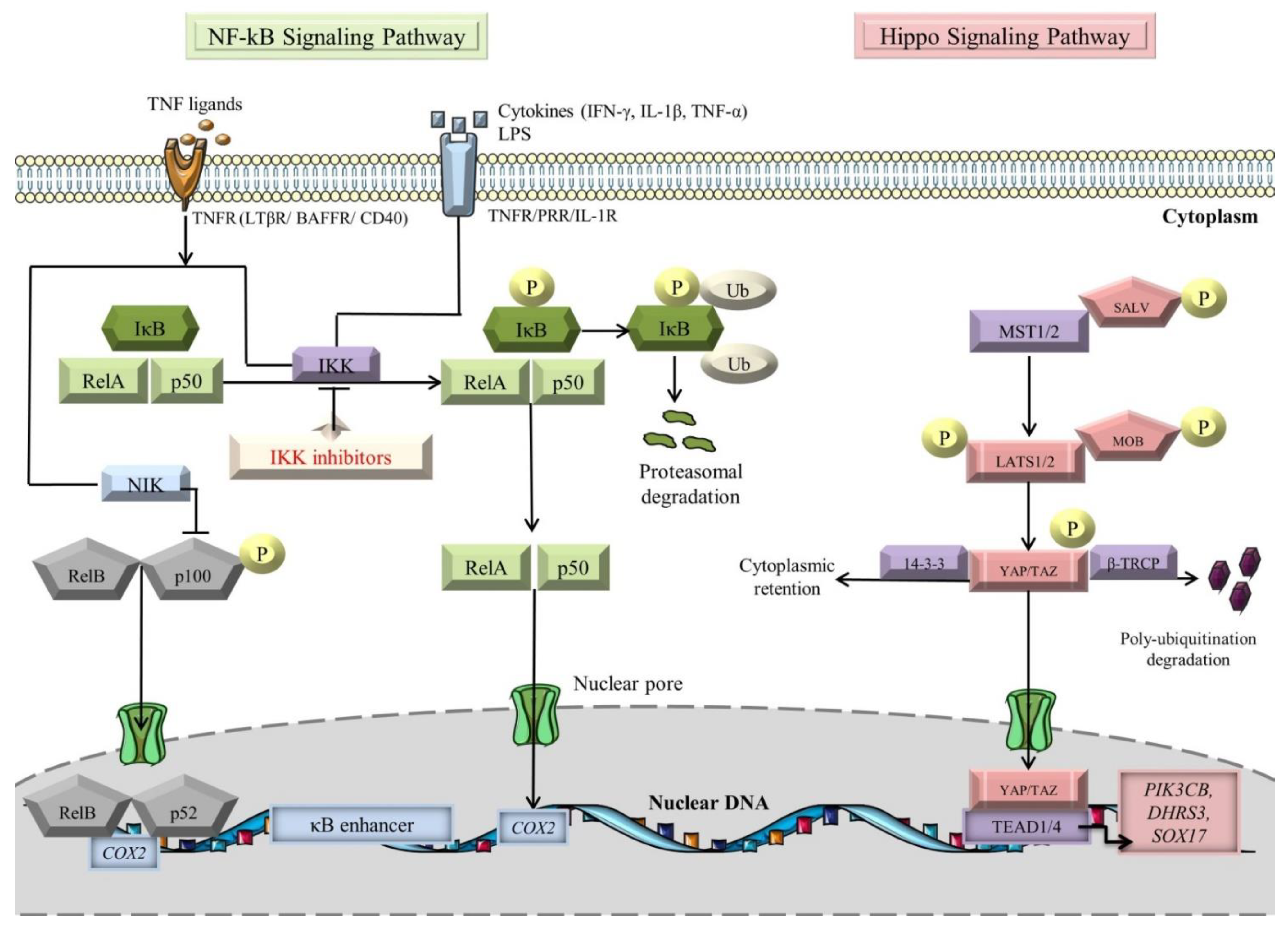
4.1. NF-κB Signaling Pathway and Its Role in Cardiovascular Biology
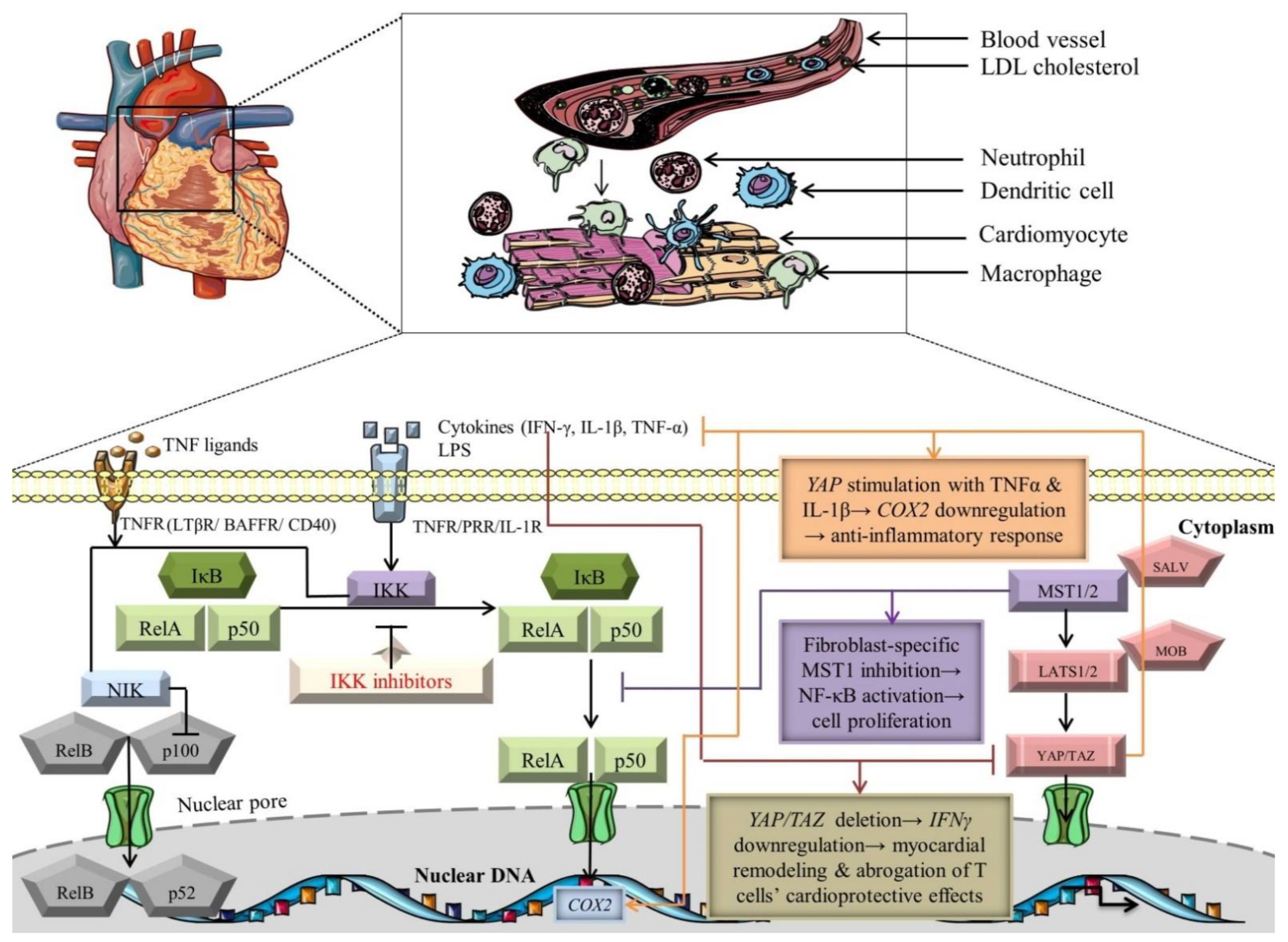
4.2. Crosstalk between Hippo and NF-κB Signaling Pathways
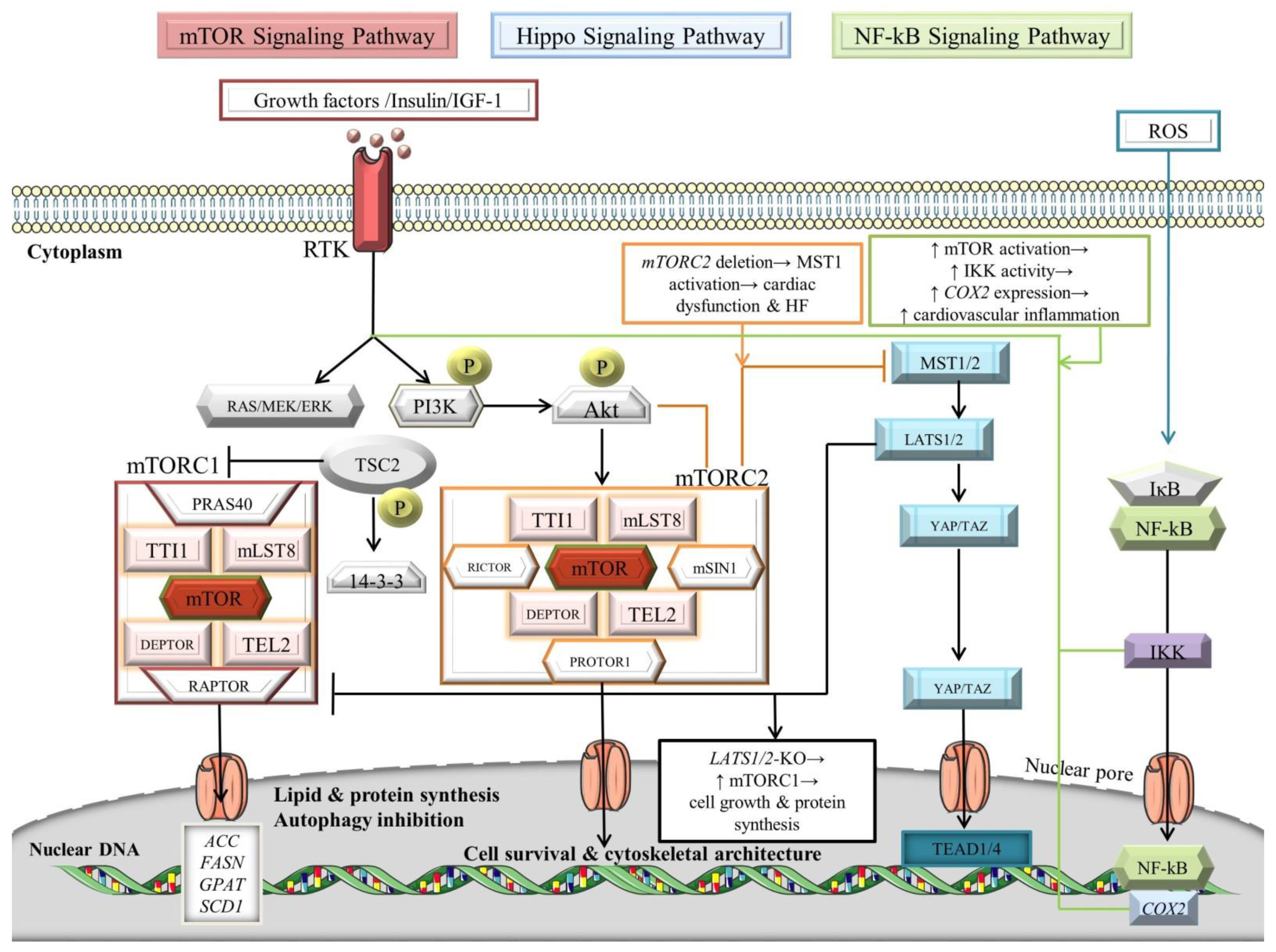
4.3. Crosstalk between Mechanistic/Mammalian Target of Rapamycin (mTOR), NF-κB, and Hippo Signaling Pathways
5. An Update of Targeted Therapy to Subdue Inflammatory Pathways
6. Conclusions and Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schwinger, R.H.G. Pathophysiology of heart failure. Cardiovasc. Diagn. Ther. 2021, 11, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Nakano, S. Cardiomyocyte and Heart Failure; Chapter 8; Muramatsu, T., Ed.; IntechOpen: Rijeka, Croatia, 2012. [Google Scholar]
- Chiong, M.; Wang, Z.V.; Pedrozo, Z.; Cao, D.J.; Troncoso, R.; Ibacache, M.; Criollo, A.; Nemchenko, A.; Hill, J.A.; Lavandero, S. Cardiomyocyte death: Mechanisms and translational implications. Cell Death Dis. 2011, 2, e244. [Google Scholar] [CrossRef] [PubMed]
- Tomasoni, D.; Vishram-Nielsen, J.K.K.; Pagnesi, M.; Adamo, M.; Lombardi, C.M.; Gustafsson, F.; Metra, M. Advanced heart failure: Guideline-directed medical therapy, diuretics, inotropes, and palliative care. ESC Heart Fail. 2022, 9, 1507–1523. [Google Scholar] [CrossRef] [PubMed]
- Ruparelia, N.; Chai, J.T.; Fisher, E.A.; Choudhury, R.P. Inflammatory processes in cardiovascular disease: A route to targeted therapies. Nat. Rev. Cardiol. 2017, 14, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Bäck, M.; Yurdagul, A.J.; Tabas, I.; Öörni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef]
- Willerson, J.T.; Ridker, P.M. Inflammation as a cardiovascular risk factor. Circulation 2004, 109, II2–II10. [Google Scholar] [CrossRef]
- Riehle, C.; Bauersachs, J. Key inflammatory mechanisms underlying heart failure. Herz 2019, 44, 96–106. [Google Scholar] [CrossRef]
- Vos, A.G.; Idris, N.S.; Barth, R.E.; Klipstein-Grobusch, K.; Grobbee, D.E. Pro-Inflammatory Markers in Relation to Cardiovascular Disease in HIV Infection. A Systematic Review. PLoS ONE 2016, 11, e0147484. [Google Scholar] [CrossRef]
- Tousoulis, D.; Antoniades, C.; Stefanadis, C. Assessing inflammatory status in cardiovascular disease. Heart 2007, 93, 1001–1007. [Google Scholar] [CrossRef]
- Zhao, Y.; Feng, G.; Wang, Y.; Yue, Y.; Zhao, W. A key mediator, PTX3, of IKK/IκB/NF-κB exacerbates human umbilical vein endothelial cell injury and dysfunction. Int. J. Clin. Exp. Pathol. 2014, 7, 7699–7707. [Google Scholar]
- Ristagno, G.; Fumagalli, F.; Bottazzi, B.; Mantovani, A.; Olivari, D.; Novelli, D.; Latini, R. Pentraxin 3 in cardiovascular disease. Front. Immunol. 2019, 10, 823. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.P.; Kakkar, R.; McCarthy, C.P.; Januzzi, J.L.J. Inflammation in Heart Failure: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2020, 75, 1324–1340. [Google Scholar] [CrossRef]
- Reina-Couto, M.; Vale, L.; Carvalho, J.; Bettencourt, P.; Albino-Teixeira, A.; Sousa, T. Resolving Inflammation in Heart Failure: Novel Protective Lipid Mediators. Curr. Drug Targets 2016, 17, 1206–1223. [Google Scholar] [CrossRef] [PubMed]
- Adamo, L.; Rocha-Resende, C.; Prabhu, S.D.; Mann, D.L. Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 2020, 17, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Hanna, A.; Frangogiannis, N.G. Inflammatory Cytokines and Chemokines as Therapeutic Targets in Heart Failure. Cardiovasc. Drugs Ther. 2020, 34, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Frostegård, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef]
- Reina-Couto, M.; Pereira-Terra, P.; Quelhas-Santos, J.; Silva-Pereira, C.; Albino-Teixeira, A.; Sousa, T. Inflammation in Human Heart Failure: Major Mediators and Therapeutic Targets. Front. Physiol. 2021, 12, 746494. [Google Scholar] [CrossRef]
- Golia, E.; Limongelli, G.; Natale, F.; Fimiani, F.; Maddaloni, V.; Pariggiano, I.; Bianchi, R.; Crisci, M.; D’Acierno, L.; Giordano, R.; et al. Inflammation and Cardiovascular Disease: From Pathogenesis to Therapeutic Target. Curr. Atheroscler. Rep. 2014, 16, 435. [Google Scholar] [CrossRef]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef]
- Chatzizisis, Y.S.; Coskun, A.U.; Jonas, M.; Edelman, E.R.; Feldman, C.L.; Stone, P.H. Role of Endothelial Shear Stress in the Natural History of Coronary Atherosclerosis and Vascular Remodeling: Molecular, Cellular, and Vascular Behavior. J. Am. Coll. Cardiol. 2007, 49, 2379–2393. [Google Scholar] [CrossRef]
- Tabas, I.; Williams, K.J.; Borén, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef] [PubMed]
- Karadag, F.; Kirdar, S.; Karul, A.B.; Ceylan, E. The value of C-reactive protein as a marker of systemic inflammation in stable chronic obstructive pulmonary disease. Eur. J. Intern. Med. 2008, 19, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Wu, Y.; Liu, E. C-reactive protein and cardiovascular disease: From animal studies to the clinic (Review). Exp. Med. 2020, 20, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Avan, A.; Tavakoly Sany, S.B.; Ghayour-Mobarhan, M.; Rahimi, H.R.; Tajfard, M.; Ferns, G. Serum C-reactive protein in the prediction of cardiovascular diseases: Overview of the latest clinical studies and public health practice. J. Cell. Physiol. 2018, 233, 8508–8525. [Google Scholar] [CrossRef] [PubMed]
- Anand, I.S.; Latini, R.; Florea, V.G.; Kuskowski, M.A.; Rector, T.; Masson, S.; Signorini, S.; Mocarelli, P.; Hester, A.; Glazer, R.; et al. C-reactive protein in heart failure: Prognostic value and the effect of valsartan. Circulation 2005, 112, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.L.; Dumesnil, J.G.; Tam, J.; Ni, A.; Teo, K. Effect of rosuvastatin on C-reactive protein and progression of aortic stenosis. Am. Heart J. 2011, 161, 1133–1139. [Google Scholar] [CrossRef]
- Tavazzi, L.; Maggioni, A.P.; Marchioli, R.; Barlera, S.; Franzosi, M.G.; Latini, R.; Lucci, D.; Nicolosi, G.L.; Porcu, M.; Tognoni, G. Effect of rosuvastatin in patients with chronic heart failure (the GISSI-HF trial): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Kjekshus, J.; Apetrei, E.; Barrios, V.; Böhm, M.; Cleland, J.G.F.; Cornel, J.H.; Dunselman, P.; Fonseca, C.; Goudev, A.; Grande, P.; et al. Rosuvastatin in older patients with systolic heart failure. N. Engl. J. Med. 2007, 357, 2248–2261. [Google Scholar] [CrossRef] [PubMed]
- Garlanda, C.; Bottazzi, B.; Magrini, E.; Inforzato, A.; Mantovani, A. PTX3, a Humoral Pattern Recognition Molecule, in Innate Immunity, Tissue Repair, and Cancer. Physiol. Rev. 2018, 98, 623–639. [Google Scholar] [CrossRef]
- Fornai, F.; Carrizzo, A.; Forte, M.; Ambrosio, M.; Damato, A.; Ferrucci, M.; Biagioni, F.; Busceti, C.; Puca, A.A.; Vecchione, C. The inflammatory protein Pentraxin 3 in cardiovascular disease. Immun. Ageing 2016, 13, 25. [Google Scholar] [CrossRef] [PubMed]
- Norata, G.D.; Marchesi, P.; Pirillo, A.; Uboldi, P.; Chiesa, G.; Maina, V.; Garlanda, C.; Mantovani, A.; Catapano, A.L. Long pentraxin 3, a key component of innate immunity, is modulated by high-density lipoproteins in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 925–931. [Google Scholar] [CrossRef]
- Nebuloni, M.; Pasqualini, F.; Zerbi, P.; Lauri, E.; Mantovani, A.; Vago, L.; Garlanda, C. PTX3 expression in the heart tissues of patients with myocardial infarction and infectious myocarditis. Cardiovasc. Pathol. Off. J. Soc. Cardiovasc. Pathol. 2011, 20, e27–e35. [Google Scholar] [CrossRef]
- Latini, R.; Maggioni, A.P.; Peri, G.; Gonzini, L.; Lucci, D.; Mocarelli, P.; Vago, L.; Pasqualini, F.; Signorini, S.; Soldateschi, D.; et al. Prognostic Significance of the Long Pentraxin PTX3 in Acute Myocardial Infarction. Circulation 2004, 110, 2349–2354. [Google Scholar] [CrossRef]
- Schumacher, S.M.; Naga Prasad, S.V. Tumor Necrosis Factor-α in Heart Failure: An Updated Review. Curr. Cardiol. Rep. 2018, 20, 117. [Google Scholar] [CrossRef]
- Gullestad, L.; Ueland, T.; Vinge, L.E.; Finsen, A.; Yndestad, A.; Aukrust, P. Inflammatory Cytokines in Heart Failure: Mediators and Markers. Cardiology 2012, 122, 23–35. [Google Scholar] [CrossRef]
- Idriss, H.T.; Naismith, J.H. TNF alpha and the TNF receptor superfamily: Structure-function relationship(s). Microsc. Res. Tech. 2000, 50, 184–195. [Google Scholar] [CrossRef]
- Bartekova, M.; Radosinska, J.; Jelemensky, M.; Dhalla, N.S. Role of cytokines and inflammation in heart function during health and disease. Heart Fail. Rev. 2018, 23, 733–758. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front. Cell Dev. Biol. 2019, 7, 91. [Google Scholar] [CrossRef]
- Urschel, K.; Cicha, I. TNF-α in the cardiovascular system: From physiology to therapy. Int. J. Interf. Cytokine Mediat. Res. 2015, 7, 9–25. [Google Scholar] [CrossRef]
- Bozkurt, B.; Kribbs, S.B.; Clubb, F.J.; Michael, L.H.; Didenko, V.V.; Hornsby, P.J.; Seta, Y.; Oral, H.; Spinale, F.G.; Mann, D.L. Pathophysiologically Relevant Concentrations of Tumor Necrosis Factor-α Promote Progressive Left Ventricular Dysfunction and Remodeling in Rats. Circulation 1998, 97, 1382–1391. [Google Scholar] [CrossRef]
- Hussain, A.; Tarahomi, T.; Singh, L.; Bollampally, M.; Heydari-Kamjani, M.; Kesselman, M.M. Cardiovascular Risk Associated With TNF Alpha Inhibitor Use in Patients With Rheumatoid Arthritis. Cureus 2021, 13, e17938. [Google Scholar] [CrossRef]
- Mann, D.L.; McMurray, J.J.V.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef]
- Liu, C.-L.; Wang, Y.-Y. Effects of TNF-alpha/NF-kappa B signaling pathway on etanercept alleviating rheumatoid arthritis. Zhongguo Ying Yong Sheng Li Xue Za Zhi = Zhongguo Yingyong Shenglixue Zazhi = Chin. J. Appl. Physiol. 2017, 33, 373–376. [Google Scholar] [CrossRef]
- Panth, N.; Paudel, K.R.; Parajuli, K. Reactive Oxygen Species: A Key Hallmark of Cardiovascular Disease. Adv. Med. 2016, 2016, 9152732. [Google Scholar] [CrossRef]
- Moris, D.; Spartalis, M.; Tzatzaki, E.; Spartalis, E.; Karachaliou, G.-S.; Triantafyllis, A.S.; Karaolanis, G.I.; Tsilimigras, D.I.; Theocharis, S. The role of reactive oxygen species in myocardial redox signaling and regulation. Ann. Transl. Med. 2017, 5, 324. [Google Scholar] [CrossRef]
- Zhang, Y.; Murugesan, P.; Huang, K.; Cai, H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: Novel therapeutic targets. Nat. Rev. Cardiol. 2020, 17, 170–194. [Google Scholar] [CrossRef]
- Sirker, A.; Zhang, M.; Shah, A.M. NADPH oxidases in cardiovascular disease: Insights from in vivo models and clinical studies. Basic Res. Cardiol. 2011, 106, 735–747. [Google Scholar] [CrossRef]
- Augsburger, F.; Filippova, A.; Rasti, D.; Seredenina, T.; Lam, M.; Maghzal, G.; Mahiout, Z.; Jansen-Dürr, P.; Knaus, U.G.; Doroshow, J.; et al. Pharmacological characterization of the seven human NOX isoforms and their inhibitors. Redox Biol. 2019, 26, 101272. [Google Scholar] [CrossRef]
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef]
- Förstermann, U.; Li, H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br. J. Pharmacol. 2011, 164, 213–223. [Google Scholar] [CrossRef]
- Alp, N.J.; McAteer, M.A.; Khoo, J.; Choudhury, R.P.; Channon, K.M. Increased endothelial tetrahydrobiopterin synthesis by targeted transgenic GTP-cyclohydrolase I overexpression reduces endothelial dysfunction and atherosclerosis in ApoE-knockout mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 445–450. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Liang, T.; Zhou, Y.; Ju, H.; Song, D.; Fang, H. Telocytes reduce oxidative stress by downregulating DUOX2 expression in inflamed lungs of mice. Acta Biochim. Biophys. Sin. 2022, 54, 574–582. [Google Scholar] [CrossRef] [PubMed]
- Fuller, B.M.; Mohr, N.M.; Graetz, T.J.; Lynch, I.P.; Dettmer, M.; Cullison, K.; Coney, T.; Gogineni, S.; Gregory, R. The impact of cardiac dysfunction on acute respiratory distress syndrome and mortality in mechanically ventilated patients with severe sepsis and septic shock: An observational study. J. Crit. Care 2015, 30, 65–70. [Google Scholar] [CrossRef]
- Dimai, S.; Semmler, L.; Prabhu, A.; Stachelscheid, H.; Huettemeister, J.; Klaucke, S.C.; Lacour, P.; Blaschke, F.; Kruse, J.; Parwani, A.; et al. COVID19-associated cardiomyocyte dysfunction, arrhythmias and the effect of Canakinumab. PLoS ONE 2021, 16, e0255976. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Yu, M.-H.; Li, X.; Li, Q.; Mo, S.-J.; Ni, Y.; Han, F.; Wang, Y.-B.; Tu, Y.-X. SAA1 increases NOX4/ROS production to promote LPS-induced inflammation in vascular smooth muscle cells through activating p38MAPK/NF-κB pathway. BMC Mol. Cell Biol. 2019, 20, 15. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-κB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Dorrington, M.G.; Fraser, I.D.C. NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Misra, A.; Haudek, S.B.; Knuefermann, P.; Vallejo, J.G.; Chen, Z.J.; Michael, L.H.; Sivasubramanian, N.; Olson, E.N.; Entman, M.L.; Mann, D.L. Nuclear Factor-κB Protects the Adult Cardiac Myocyte Against Ischemia-Induced Apoptosis in a Murine Model of Acute Myocardial Infarction. Circulation 2003, 108, 3075–3078. [Google Scholar] [CrossRef]
- Santos, D.G.B.; Resende, M.F.; Mill, J.G.; Mansur, A.J.; Krieger, J.E.; Pereira, A.C. Nuclear Factor (NF) κB polymorphism is associated with heart function in patients with heart failure. BMC Med. Genet. 2010, 11, 89. [Google Scholar] [CrossRef]
- Chen, F.; Castranova, V.; Shi, X.; Demers, L.M. New insights into the role of nuclear factor-kappaB, a ubiquitous transcription factor in the initiation of diseases. Clin. Chem. 1999, 45, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell type specific roles of nf-kb linking inflamation and thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Saccani, S.; Pantano, S.; Natoli, G. Modulation of NF-kappaB activity by exchange of dimers. Mol. Cell 2003, 11, 1563–1574. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef]
- Marienfeld, R.; May, M.J.; Berberich, I.; Serfling, E.; Ghosh, S.; Neumann, M. RelB forms transcriptionally inactive complexes with RelA/p65. J. Biol. Chem. 2003, 278, 19852–19860. [Google Scholar] [CrossRef]
- Cartwright, T.; Perkins, N.D.; Wilson, C.L. NFKB1: A suppressor of inflammation, ageing and cancer. FEBS J. 2016, 283, 1812–1822. [Google Scholar] [CrossRef]
- Gordon, J.W.; Shaw, J.A.; Kirshenbaum, L.A. Multiple facets of NF-κB in the heart: To be or not to NF-κB. Circ. Res. 2011, 108, 1122–1132. [Google Scholar] [CrossRef]
- Huguet, C.; Crepieux, P.; Laudet, V. Rel/NF-κB transcription factors and IκB inhibitors: Evolution from a unique common ancestor. Oncogene 1997, 15, 2965–2974. [Google Scholar] [CrossRef][Green Version]
- Sun, S.-C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and Function of NF-κB Transcription Factors in the Immune System. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Greten, F.R.; Arkan, M.C.; Bollrath, J.; Hsu, L.C.; Goode, J.; Miething, C.; Göktuna, S.I.; Neuenhahn, M.; Fierer, J.; Paxian, S.; et al. NF-κB Is a Negative Regulator of IL-1β Secretion as Revealed by Genetic and Pharmacological Inhibition of IKKβ. Cell 2007, 130, 918–931. [Google Scholar] [CrossRef]
- Rex, J.; Lutz, A.; Faletti, L.E.; Albrecht, U.; Thomas, M.; Bode, J.G.; Borner, C.; Sawodny, O.; Merfort, I. IL-1β and TNFα Differentially Influence NF-κB Activity and FasL-Induced Apoptosis in Primary Murine Hepatocytes During LPS-Induced Inflammation. Front. Physiol. 2019, 10, 117. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Regulation of NF-κB by TNF family cytokines. Semin. Immunol. 2014, 26, 253–266. [Google Scholar] [CrossRef]
- Chen, L.-F.; Greene, W.C. Shaping the nuclear action of NF-kappaB. Nat. Rev. Mol. Cell Biol. 2004, 5, 392–401. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Caamaño, J.; Hunter, C.A. NF-kappaB family of transcription factors: Central regulators of innate and adaptive immune functions. Clin. Microbiol. Rev. 2002, 15, 414–429. [Google Scholar] [CrossRef]
- Sen, R.; Baltimore, D. Multiple nuclear factors interact with the immunoglobulin enhancer sequences. Cell 1986, 46, 705–716. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Heissmeyer, V.; Krappmann, D.; Hatada, E.N.; Scheidereit, C. Shared pathways of IkappaB kinase-induced SCF(betaTrCP)-mediated ubiquitination and degradation for the NF-kappaB precursor p105 and IkappaBalpha. Mol. Cell. Biol. 2001, 21, 1024–1035. [Google Scholar] [CrossRef]
- Orian, A.; Gonen, H.; Bercovich, B.; Fajerman, I.; Eytan, E.; Israël, A.; Mercurio, F.; Iwai, K.; Schwartz, A.L.; Ciechanover, A. SCF(beta)(-TrCP) ubiquitin ligase-mediated processing of NF-kappaB p105 requires phosphorylation of its C-terminus by IkappaB kinase. EMBO J. 2000, 19, 2580–2591. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Gohda, J.; Matsumura, T.; Inoue, J. Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not toll/IL-1 receptor domain-containing adaptor-inducing IFN-beta (TRIF)-dependent pathway in TLR signaling. J. Immunol. 2004, 173, 2913–2917. [Google Scholar] [CrossRef]
- Hu, H.; Sun, S.-C. Ubiquitin signaling in immune responses. Cell Res. 2016, 26, 457–483. [Google Scholar] [CrossRef]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Sun, S.-C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Senftleben, U.; Cao, Y.; Xiao, G.; Greten, F.R.; Krähn, G.; Bonizzi, G.; Chen, Y.; Hu, Y.; Fong, A.; Sun, S.C.; et al. Activation by IKKalpha of a second, evolutionary conserved, NF-kappa B signaling pathway. Science 2001, 293, 1495–1499. [Google Scholar] [CrossRef]
- Zhang, H.; Sun, S.-C. NF-κB in inflammation and renal diseases. Cell Biosci. 2015, 5, 63. [Google Scholar] [CrossRef]
- Sun, S.-C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef]
- Tian, B.; Brasier, A.R. Identification of a nuclear factor kappa B-dependent gene network. Recent Prog. Horm. Res. 2003, 58, 95–130. [Google Scholar] [CrossRef]
- Maier, H.J.; Schips, T.G.; Wietelmann, A.; Krüger, M.; Brunner, C.; Sauter, M.; Klingel, K.; Böttger, T.; Braun, T.; Wirth, T. Cardiomyocyte-specific IκB kinase (IKK)/NF-κB activation induces reversible inflammatory cardiomyopathy and heart failure. Proc. Natl. Acad. Sci. USA 2012, 109, 11794–11799. [Google Scholar] [CrossRef]
- Bonomini, F. NF-Κb—A Key Factor in Atherogenesis and Atheroprogression; Chapter 2; Favero, G., Ed.; IntechOpen: Rijeka, Croatia, 2015. [Google Scholar]
- Gaspar-Pereira, S.; Fullard, N.; Townsend, P.A.; Banks, P.S.; Ellis, E.L.; Fox, C.; Maxwell, A.G.; Murphy, L.B.; Kirk, A.; Bauer, R.; et al. The NF-κB subunit c-Rel stimulates cardiac hypertrophy and fibrosis. Am. J. Pathol. 2012, 180, 929–939. [Google Scholar] [CrossRef]
- Dai, X.; Thiagarajan, D.; Fang, J.; Shen, J.; Annam, N.P.; Yang, Z.; Jiang, H.; Ju, D.; Xie, Y.; Zhang, K.; et al. SM22α suppresses cytokine-induced inflammation and the transcription of NF-κB inducing kinase (Nik) by modulating SRF transcriptional activity in vascular smooth muscle cells. PLoS ONE 2017, 12, e0190191. [Google Scholar] [CrossRef]
- Siednienko, J.; Jankowska, E.A.; Banasiak, W.; Gorczyca, W.A.; Ponikowski, P. Nuclear factor-kappaB activity in peripheral blood mononuclear cells in cachectic and non-cachectic patients with chronic heart failure. Int. J. Cardiol. 2007, 122, 111–116. [Google Scholar] [CrossRef]
- Tieri, P.; Termanini, A.; Bellavista, E.; Salvioli, S.; Capri, M.; Franceschi, C. Charting the NF-κB pathway interactome map. PLoS ONE 2012, 7, e32678. [Google Scholar] [CrossRef]
- Shaw, J.; Yurkova, N.; Zhang, T.; Gang, H.; Aguilar, F.; Weidman, D.; Scramstad, C.; Weisman, H.; Kirshenbaum, L.A. Antagonism of E2F-1 regulated Bnip3 transcription by NF-kappaB is essential for basal cell survival. Proc. Natl. Acad. Sci. USA 2008, 105, 20734–20739. [Google Scholar] [CrossRef]
- Hamid, T.; Guo, S.Z.; Kingery, J.R.; Xiang, X.; Dawn, B.; Prabhu, S.D. Cardiomyocyte NF-κB p65 promotes adverse remodelling, apoptosis, and endoplasmic reticulum stress in heart failure. Cardiovasc. Res. 2011, 89, 129–138. [Google Scholar] [CrossRef]
- Finsterwalder, R.; Ganesan, M.K.; Leb, H.; Habertheuer, A.; Basílio, J.; Lang, I.; Krunic, M.; Wiedemann, D.; Petzelbauer, P. Hypoxia/reperfusion predisposes to atherosclerosis. PLoS ONE 2018, 13, e0205067. [Google Scholar] [CrossRef] [PubMed]
- Cucu, I.; Nicolescu, M.I. A Synopsis of Signaling Crosstalk of Pericytes and Endothelial Cells in Salivary Gland. Dent. J. 2021, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.; Nguyen, M.-A.; Geoffrion, M.; Vreeken, D.; Lister, Z.; Cheng, H.S.; Otte, N.; Essebier, P.; Wyatt, H.; Kandiah, J.W.; et al. RIPK1 Expression Associates With Inflammation in Early Atherosclerosis in Humans and Can Be Therapeutically Silenced to Reduce NF-κB Activation and Atherogenesis in Mice. Circulation 2021, 143, 163–177. [Google Scholar] [CrossRef]
- Peri, G.; Introna, M.; Corradi, D.; Iacuitti, G.; Signorini, S.; Avanzini, F.; Pizzetti, F.; Maggioni, A.P.; Moccetti, T.; Metra, M.; et al. PTX3, A Prototypical Long Pentraxin, Is an Early Indicator of Acute Myocardial Infarction in Humans. Circulation 2000, 102, 636–641. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Wu, Y.; Gao, M.; Tian, Y.; Qi, P.; Shen, Y.; Huang, L.; Shi, L.; Wang, Y.; Liu, X. C-reactive protein promotes inflammation through TLR4/NF-κB/TGF-β pathway in HL-1 cells. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef]
- Ramadass, V.; Vaiyapuri, T.; Tergaonkar, V. Small Molecule NF-κB Pathway Inhibitors in Clinic. Int. J. Mol. Sci. 2020, 21, 5164. [Google Scholar] [CrossRef]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T. Randomized, Double-Blind, Placebo-Controlled, Pilot Trial of Infliximab, a Chimeric Monoclonal Antibody to Tumor Necrosis Factor-α, in Patients with Moderate-to-Severe Heart Failure. Circulation 2003, 107, 3133–3140. [Google Scholar] [CrossRef]
- Weber, C.K.; Liptay, S.; Wirth, T.; Adler, G.; Schmid, R.M. Suppression of NF-kappaB activity by sulfasalazine is mediated by direct inhibition of IkappaB kinases alpha and beta. Gastroenterology 2000, 119, 1209–1218. [Google Scholar] [CrossRef]
- Tabit, C.E.; Holbrook, M.; Shenouda, S.M.; Dohadwala, M.M.; Widlansky, M.E.; Frame, A.A.; Kim, B.H.; Duess, M.-A.; Kluge, M.A.; Levit, A.; et al. Effect of sulfasalazine on inflammation and endothelial function in patients with established coronary artery disease. Vasc. Med. 2012, 17, 101–107. [Google Scholar] [CrossRef]
- Lv, Y.; Kim, K.; Sheng, Y.; Cho, J.; Qian, Z.; Zhao, Y.-Y.; Hu, G.; Pan, D.; Malik, A.B.; Hu, G. YAP Controls Endothelial Activation and Vascular Inflammation Through TRAF6. Circ. Res. 2018, 123, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Ramjee, V.; Li, D.; Manderfield, L.J.; Liu, F.; Engleka, K.A.; Aghajanian, H.; Rodell, C.B.; Lu, W.; Ho, V.; Wang, T.; et al. Epicardial YAP/TAZ orchestrate an immunosuppressive response following myocardial infarction. J. Clin. Investig. 2017, 127, 899–911. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hu, X.; Wang, J.; Xu, W.; Yi, C.; Ma, R.; Jiang, H. Inhibition of autophagy via activation of PI3K/Akt/mTOR pathway contributes to the protection of hesperidin against myocardial ischemia/reperfusion injury. Int. J. Mol. Med. 2018, 42, 1917–1924. [Google Scholar] [CrossRef]
- Klingenberg, R.; Stähli, B.E.; Heg, D.; Denegri, A.; Manka, R.; Kapos, I.; von Eckardstein, A.; Carballo, D.; Hamm, C.W.; Vietheer, J.; et al. Controlled-Level EVERolimus in Acute Coronary Syndrome (CLEVER-ACS)—A phase II, randomized, double-blind, multi-center, placebo-controlled trial. Am. Heart J. 2022, 247, 33–41. [Google Scholar] [CrossRef]
- Harvey, K.F.; Pfleger, C.M.; Hariharan, I.K. The Drosophila Mst ortholog, hippo, restricts growth and cell proliferation and promotes apoptosis. Cell 2003, 114, 457–467. [Google Scholar] [CrossRef]
- Wu, S.; Huang, J.; Dong, J.; Pan, D. hippo encodes a Ste-20 family protein kinase that restricts cell proliferation and promotes apoptosis in conjunction with salvador and warts. Cell 2003, 114, 445–456. [Google Scholar] [CrossRef]
- Halder, G.; Johnson, R.L. Hippo signaling: Growth control and beyond. Development 2011, 138, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Tumaneng, K.; Guan, K.-L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef]
- Flinn, M.A.; Link, B.A.; O’Meara, C.C. Upstream regulation of the Hippo-Yap pathway in cardiomyocyte regeneration. Semin. Cell Dev. Biol. 2020, 100, 11–19. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, L.; Zhao, B.; Guan, K.-L. The Hippo Pathway in Heart Development, Regeneration, and Diseases. Circ. Res. 2015, 116, 1431–1447. [Google Scholar] [CrossRef]
- Mo, J.-S.; Park, H.W.; Guan, K.-L. The Hippo signaling pathway in stem cell biology and cancer. EMBO Rep. 2014, 15, 642–656. [Google Scholar] [CrossRef] [PubMed]
- Ni, L.; Zheng, Y.; Hara, M.; Pan, D.; Luo, X. Structural basis for Mob1-dependent activation of the core Mst-Lats kinase cascade in Hippo signaling. Genes Dev. 2015, 29, 1416–1431. [Google Scholar] [CrossRef] [PubMed]
- Praskova, M.; Xia, F.; Avruch, J. MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr. Biol. 2008, 18, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.H.Y.; Nousiainen, M.; Chalamalasetty, R.B.; Schäfer, A.; Nigg, E.A.; Silljé, H.H.W. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene 2005, 24, 2076–2086. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Zha, Z.-Y.; Zhou, X.; Zhang, H.; Huang, W.; Zhao, D.; Li, T.; Chan, S.W.; Lim, C.J.; Hong, W.; et al. The hippo tumor pathway promotes TAZ degradation by phosphorylating a phosphodegron and recruiting the SCF{beta}-TrCP E3 ligase. J. Biol. Chem. 2010, 285, 37159–37169. [Google Scholar] [CrossRef]
- Freeman, A.K.; Morrison, D.K. 14-3-3 Proteins: Diverse functions in cell proliferation and cancer progression. Semin. Cell Dev. Biol. 2011, 22, 681–687. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Luo, J.; Hou, N. Molecular Mechanism of Hippo-YAP1/TAZ Pathway in Heart Development, Disease, and Regeneration. Front. Physiol. 2020, 11, 389. [Google Scholar] [CrossRef]
- Heallen, T.; Zhang, M.; Wang, J.; Bonilla-Claudio, M.; Klysik, E.; Johnson, R.L.; Martin, J.F. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 2011, 332, 458–461. [Google Scholar] [CrossRef]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Qi, X.; McAnally, J.; Schwartz, R.J.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. Regulation of insulin-like growth factor signaling by Yap governs cardiomyocyte proliferation and embryonic heart size. Sci. Signal. 2011, 4, ra70. [Google Scholar] [CrossRef]
- Cucu, I.; Nicolescu, M.I.; Busnatu, Ș.-S.; Manole, C.G. Dynamic Involvement of Telocytes in Modulating Multiple Signaling Pathways in Cardiac Cytoarchitecture. Int. J. Mol. Sci. 2022, 23, 5769. [Google Scholar] [CrossRef]
- Xin, M.; Kim, Y.; Sutherland, L.B.; Murakami, M.; Qi, X.; McAnally, J.; Porrello, E.R.; Mahmoud, A.I.; Tan, W.; Shelton, J.M.; et al. Hippo pathway effector Yap promotes cardiac regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13839–13844. [Google Scholar] [CrossRef] [PubMed]
- Del Re, D.P. Beyond the Cardiomyocyte: Consideration of HIPPO Pathway Cell-Type Specificity. Circ. Res. 2018, 123, 30–32. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Lu, J.; Li, W.; Wu, A.; Zhang, X.; Tong, W.; Ho, K.K.; Qin, L.; Song, H.; Mak, K.K. Reciprocal inhibition of YAP/TAZ and NF-κB regulates osteoarthritic cartilage degradation. Nat. Commun. 2018, 9, 4564. [Google Scholar] [CrossRef] [PubMed]
- Tang, K.; Chen, W.; Tang, Z.; Yu, X.; Zhu, W.; Zhang, S.; Qiu, J. Role of the Hippo-YAP/NF-κB signaling pathway crosstalk in regulating biological behaviors of macrophages under titanium ion exposure. J. Appl. Toxicol. 2021, 41, 561–571. [Google Scholar] [CrossRef]
- Wang, S.; Zhou, L.; Ling, L.; Meng, X.; Chu, F.; Zhang, S.; Zhou, F. The Crosstalk Between Hippo-YAP Pathway and Innate Immunity. Front. Immunol. 2020, 11, 323. [Google Scholar] [CrossRef]
- Zhang, Q.; Han, X.; Chen, J.; Xie, X.; Xu, J.; Zhao, Y.; Shen, J.; Hu, L.; Xu, P.; Song, H.; et al. Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) mediate cell density-dependent proinflammatory responses. J. Biol. Chem. 2018, 293, 18071–18085. [Google Scholar] [CrossRef] [PubMed]
- Barry, E.R.; Simov, V.; Valtingojer, I.; Venier, O. Recent Therapeutic Approaches to Modulate the Hippo Pathway in Oncology and Regenerative Medicine. Cells 2021, 10, 2715. [Google Scholar] [CrossRef]
- Tolcher, A.W.; Lakhani, N.J.; McKean, M.; Lingaraj, T.; Victor, L.; Sanchez-Martin, M.; Kacena, K.; Malek, K.S.; Santillana, S. A phase 1, first-in-human study of IK-930, an oral TEAD inhibitor targeting the Hippo pathway in subjects with advanced solid tumors. J. Clin. Oncol. 2022, 40, TPS3168. [Google Scholar] [CrossRef]
- Matsuda, T.; Zhai, P.; Sciarretta, S.; Zhang, Y.; Jeong, J.I.; Ikeda, S.; Park, J.; Hsu, C.-P.; Tian, B.; Pan, D.; et al. NF2 Activates Hippo Signaling and Promotes Ischemia/Reperfusion Injury in the Heart. Circ. Res. 2016, 119, 596–606. [Google Scholar] [CrossRef]
- Mia, M.M.; Chelakkot-Govindalayathil, A.L.; Singh, M.K. Targeting NF2-Hippo/Yap signaling pathway for cardioprotection after ischemia/reperfusion injury. Ann. Transl. Med. 2016, 4, 545. [Google Scholar] [CrossRef]
- Zheng, B.; Wang, J.; Tang, L.; Shi, J.; Zhu, D. mTORC1 and mTORC2 play different roles in regulating cardiomyocyte differentiation from embryonic stem cells. Int. J. Dev. Biol. 2017, 61, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Liu, L.; Wang, S.; Dai, Z. ILK regulates MSCs survival and angiogenesis partially through AKT and mTOR signaling pathways. Acta Histochem. 2017, 119, 400–406. [Google Scholar] [CrossRef]
- Aoyagi, T.; Kusakari, Y.; Xiao, C.-Y.; Inouye, B.T.; Takahashi, M.; Scherrer-Crosbie, M.; Rosenzweig, A.; Hara, K.; Matsui, T. Cardiac mTOR protects the heart against ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2012, 303, H75–H85. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.-L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Limon, J.J.; Fruman, D.A. Akt and mTOR in B Cell Activation and Differentiation. Front. Immunol. 2012, 3, 228. [Google Scholar] [CrossRef]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef]
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawłowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.M.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 2007, 405, 513–522. [Google Scholar] [CrossRef]
- Schultze, S.M.; Hemmings, B.A.; Niessen, M.; Tschopp, O. PI3K/AKT, MAPK and AMPK signalling: Protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 2012, 14, e1. [Google Scholar] [CrossRef]
- Yoon, M.-S. The Role of Mammalian Target of Rapamycin (mTOR) in Insulin Signaling. Nutrients 2017, 9, 1176. [Google Scholar] [CrossRef] [PubMed]
- Dilly, A.K.; Rajala, R.V.S. Insulin growth factor 1 receptor/PI3K/AKT survival pathway in outer segment membranes of rod photoreceptors. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4765–4773. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Moerkens, M.; Ramaiahgari, S.; de Bont, H.; Price, L.; Meerman, J.; van de Water, B. Elevated insulin-like growth factor 1 receptor signaling induces antiestrogen resistance through the MAPK/ERK and PI3K/Akt signaling routes. Breast Cancer Res. 2011, 13, R52. [Google Scholar] [CrossRef] [PubMed]
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121. [Google Scholar] [CrossRef]
- Baffi, T.R.; Lordén, G.; Wozniak, J.M.; Feichtner, A.; Yeung, W.; Kornev, A.P.; King, C.C.; Del Rio, J.C.; Limaye, A.J.; Bogomolovas, J.; et al. mTORC2 controls the activity of PKC and Akt by phosphorylating a conserved TOR interaction motif. Sci. Signal. 2021, 14, eabe4509. [Google Scholar] [CrossRef]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef]
- Li, Y.; Inoki, K.; Vacratsis, P.; Guan, K.-L. The p38 and MK2 kinase cascade phosphorylates tuberin, the tuberous sclerosis 2 gene product, and enhances its interaction with 14-3-3. J. Biol. Chem. 2003, 278, 13663–13671. [Google Scholar] [CrossRef]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.-L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. An emerging role of mTOR in lipid biosynthesis. Curr. Biol. 2009, 19, R1046–R1052. [Google Scholar] [CrossRef] [PubMed]
- Deleyto-Seldas, N.; Efeyan, A. The mTOR–Autophagy Axis and the Control of Metabolism. Front. Cell Dev. Biol. 2021, 9, 1519. [Google Scholar] [CrossRef] [PubMed]
- Ling, N.X.Y.; Kaczmarek, A.; Hoque, A.; Davie, E.; Ngoei, K.R.W.; Morrison, K.R.; Smiles, W.J.; Forte, G.M.; Wang, T.; Lie, S.; et al. mTORC1 directly inhibits AMPK to promote cell proliferation under nutrient stress. Nat. Metab. 2020, 2, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Ma, M.; Zheng, Y.; Pan, Y.; Lin, X. mTOR and Beclin1: Two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. J. Cell. Physiol. 2019, 234, 12562–12568. [Google Scholar] [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12, eaav3249. [Google Scholar] [CrossRef]
- Matsui, Y.; Takagi, H.; Qu, X.; Abdellatif, M.; Sakoda, H.; Asano, T.; Levine, B.; Sadoshima, J. Distinct roles of autophagy in the heart during ischemia and reperfusion: Roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ. Res. 2007, 100, 914–922. [Google Scholar] [CrossRef]
- Valentim, L.; Laurence, K.M.; Townsend, P.A.; Carroll, C.J.; Soond, S.; Scarabelli, T.M.; Knight, R.A.; Latchman, D.S.; Stephanou, A. Urocortin inhibits Beclin1-mediated autophagic cell death in cardiac myocytes exposed to ischaemia/reperfusion injury. J. Mol. Cell. Cardiol. 2006, 40, 846–852. [Google Scholar] [CrossRef]
- Hariharan, N.; Zhai, P.; Sadoshima, J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid. Redox Signal. 2011, 14, 2179–2190. [Google Scholar] [CrossRef]
- Ma, X.; Liu, H.; Foyil, S.R.; Godar, R.J.; Weinheimer, C.J.; Hill, J.A.; Diwan, A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation 2012, 125, 3170–3181. [Google Scholar] [CrossRef]
- Haar, L.; Ren, X.; Liu, Y.; Koch, S.E.; Goines, J.; Tranter, M.; Engevik, M.A.; Nieman, M.; Rubinstein, J.; Jones, W.K. Acute consumption of a high-fat diet prior to ischemia-reperfusion results in cardioprotection through NF-κB-dependent regulation of autophagic pathways. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1705–H1713. [Google Scholar] [CrossRef]
- Zeng, M.; Wei, X.; Wu, Z.; Li, W.; Li, B.; Zhen, Y.; Chen, J.; Wang, P.; Fei, Y. NF-κB-mediated induction of autophagy in cardiac ischemia/reperfusion injury. Biochem. Biophys. Res. Commun. 2013, 436, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Sciarretta, S.; Zhai, P.; Maejima, Y.; Del Re, D.P.; Nagarajan, N.; Yee, D.; Liu, T.; Magnuson, M.A.; Volpe, M.; Frati, G.; et al. mTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep. 2015, 11, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Dai, X.; Dai, X.; Xie, J.; Yin, S.; Zhu, J.; Wang, C.; Liu, Y.; Guo, J.; Wang, M.; et al. LATS suppresses mTORC1 activity to directly coordinate Hippo and mTORC1 pathways in growth control. Nat. Cell Biol. 2020, 22, 246–256. [Google Scholar] [CrossRef] [PubMed]
- van Leent, M.M.T.; Beldman, T.J.; Toner, Y.C.; Lameijer, M.A.; Rother, N.; Bekkering, S.; Teunissen, A.J.P.; Zhou, X.; van der Meel, R.; Malkus, J.; et al. Prosaposin mediates inflammation in atherosclerosis. Sci. Transl. Med. 2021, 13, eabe1433. [Google Scholar] [CrossRef] [PubMed]
- Evers, B.M.; Rodriguez-Navas, C.; Tesla, R.J.; Prange-Kiel, J.; Wasser, C.R.; Yoo, K.S.; McDonald, J.; Cenik, B.; Ravenscroft, T.A.; Plattner, F.; et al. Lipidomic and Transcriptomic Basis of Lysosomal Dysfunction in Progranulin Deficiency. Cell Rep. 2017, 20, 2565–2574. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Q.; Li, W.; Zhang, Q.; Jiang, Y.; Guo, D.; Sun, X.; Lu, W.; Li, C.; Wang, Y. TFEB-NF-κB inflammatory signaling axis: A novel therapeutic pathway of Dihydrotanshinone I in doxorubicin-induced cardiotoxicity. J. Exp. Clin. Cancer Res. 2020, 39, 93. [Google Scholar] [CrossRef]
- Palmerini, T.; Kirtane, A.J.; Serruys, P.W.; Smits, P.C.; Kedhi, E.; Kereiakes, D.; Sangiorgi, D.; Reggiani, L.B.; Kaiser, C.; Kim, H.-S.; et al. Stent Thrombosis With Everolimus-Eluting Stents. Circ. Cardiovasc. Interv. 2012, 5, 357–364. [Google Scholar] [CrossRef][Green Version]
- Buss, S.J.; Muenz, S.; Riffel, J.H.; Malekar, P.; Hagenmueller, M.; Weiss, C.S.; Bea, F.; Bekeredjian, R.; Schinke-Braun, M.; Izumo, S.; et al. Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J. Am. Coll. Cardiol. 2009, 54, 2435–2446. [Google Scholar] [CrossRef]
- Stähli, B.E.; Roland, K.; Dik, H.; Mattia, B.; Robert, M.; Ioannis, K.; Oliver, M.; Andrea, D.; Rahel, K.; Florence, B.; et al. Mammalian Target of Rapamycin Inhibition in Patients With ST-Segment Elevation Myocardial Infarction. J. Am. Coll. Cardiol. 2022, 80, 1802–1814. [Google Scholar] [CrossRef]
- Kaldirim, M.; Lang, A.; Pfeiler, S.; Fiegenbaum, P.; Kelm, M.; Bönner, F.; Gerdes, N. Modulation of mTOR Signaling in Cardiovascular Disease to Target Acute and Chronic Inflammation. Front. Cardiovasc. Med. 2022, 9, 907348. [Google Scholar] [CrossRef]
- Lis, K.; Kuzawińska, O.; Bałkowiec-Iskra, E. Tumor necrosis factor inhibitors—State of knowledge. Arch. Med. Sci. 2014, 10, 1175–1185. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, J.; Wang, Y.; Jain, P.; Wang, M. Zanubrutinib in lymphoproliferative disorders: A comprehensive review. Ther. Adv. Hematol. 2022, 13, 20406207221093980. [Google Scholar] [CrossRef] [PubMed]
- Perel, G.; Bliss, J.; Thomas, C.M. Carfilzomib (Kyprolis): A Novel Proteasome Inhibitor for Relapsed And/or Refractory Multiple Myeloma. Pharm. Ther. 2016, 41, 303–307. [Google Scholar]
- Gupta, N.; Hanley, M.J.; Xia, C.; Labotka, R.; Harvey, R.D.; Venkatakrishnan, K. Clinical Pharmacology of Ixazomib: The First Oral Proteasome Inhibitor. Clin. Pharmacokinet. 2019, 58, 431–449. [Google Scholar] [CrossRef]
- Kuruvilla, J.; Savona, M.; Baz, R.; Mau-Sorensen, P.M.; Gabrail, N.; Garzon, R.; Stone, R.; Wang, M.; Savoie, L.; Martin, P.; et al. Selective inhibition of nuclear export with selinexor in patients with non-Hodgkin lymphoma. Blood 2017, 129, 3175–3183. [Google Scholar] [CrossRef]
- Seijkens, T.T.P.; van Tiel, C.M.; Kusters, P.J.H.; Atzler, D.; Soehnlein, O.; Zarzycka, B.; Aarts, S.A.B.M.; Lameijer, M.; Gijbels, M.J.; Beckers, L.; et al. Targeting CD40-Induced TRAF6 Signaling in Macrophages Reduces Atherosclerosis. J. Am. Coll. Cardiol. 2018, 71, 527–542. [Google Scholar] [CrossRef]
- Arya, P.; Nabi, S.; Bhandari, U. Modulatory role of atorvastatin against high-fat diet and zymosan-induced activation of TLR2/NF-κB signaling pathway in C57BL/6 mice. Iran. J. Basic Med. Sci. 2021, 24, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Neininger, A.C.; Dai, X.; Liu, Q.; Burnette, D.T. The Hippo pathway regulates density-dependent proliferation of iPSC-derived cardiac myocytes. Sci. Rep. 2021, 11, 17759. [Google Scholar] [CrossRef]
- Triastuti, E.; Nugroho, A.B.; Zi, M.; Prehar, S.; Kohar, Y.S.; Bui, T.A.; Stafford, N.; Cartwright, E.J.; Abraham, S.; Oceandy, D. Pharmacological inhibition of Hippo pathway, with the novel kinase inhibitor XMU-MP-1, protects the heart against adverse effects during pressure overload. Br. J. Pharmacol. 2019, 176, 3956–3971. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, K.; Ji, K.; Zhang, C.; Jiang, Y.; Zhang, Q.; Tian, Z.; Wang, X.; Zhang, M.; Li, X. microRNA-365 inhibits YAP through TLR4-mediated IRF3 phosphorylation and thereby alleviates gastric precancerous lesions. Cancer Cell Int. 2020, 20, 549. [Google Scholar] [CrossRef]
- Jiao, S.; Guan, J.; Chen, M.; Wang, W.; Li, C.; Wang, Y.; Cheng, Y.; Zhou, Z. Targeting IRF3 as a YAP agonist therapy against gastric cancer. J. Exp. Med. 2018, 215, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Marin, T.M.; Keith, K.; Davies, B.; Conner, D.A.; Guha, P.; Kalaitzidis, D.; Wu, X.; Lauriol, J.; Wang, B.; Bauer, M.; et al. Rapamycin reverses hypertrophic cardiomyopathy in a mouse model of LEOPARD syndrome-associated PTPN11 mutation. J. Clin. Investig. 2011, 121, 1026–1043. [Google Scholar] [CrossRef] [PubMed]
- Kuzman, J.A.; O’Connell, T.D.; Gerdes, A.M. Rapamycin prevents thyroid hormone-induced cardiac hypertrophy. Endocrinology 2007, 148, 3477–3484. [Google Scholar] [CrossRef] [PubMed]
- Vitiello, D.; Neagoe, P.-E.; Sirois, M.G.; White, M. Effect of everolimus on the immunomodulation of the human neutrophil inflammatory response and activation. Cell. Mol. Immunol. 2015, 12, 40–52. [Google Scholar] [CrossRef]
- Arriola Apelo, S.I.; Neuman, J.C.; Baar, E.L.; Syed, F.A.; Cummings, N.E.; Brar, H.K.; Pumper, C.P.; Kimple, M.E.; Lamming, D.W. Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell 2016, 15, 28–38. [Google Scholar] [CrossRef]
- Shioi, T.; McMullen, J.R.; Tarnavski, O.; Converso, K.; Sherwood, M.C.; Manning, W.J.; Izumo, S. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation 2003, 107, 1664–1670. [Google Scholar] [CrossRef]
- Gao, G.; Chen, W.; Yan, M.; Liu, J.; Luo, H.; Wang, C.; Yang, P. Rapamycin regulates the balance between cardiomyocyte apoptosis and autophagy in chronic heart failure by inhibiting mTOR signaling. Int. J. Mol. Med. 2020, 45, 195–209. [Google Scholar] [CrossRef]
- Shi, G.; Chiramel, A.I.; Majdoul, S.; Lai, K.K.; Dempsey, T.; Kenney, A.; Zani, A.; Eddy, A.; Zhang, L.; Beare, P.A.; et al. Rapalogs downmodulate intrinsic immunity and promote cell entry of SARS-CoV-2. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hoda, M.A.; Mohamed, A.; Ghanim, B.; Filipits, M.; Hegedus, B.; Tamura, M.; Berta, J.; Kubista, B.; Dome, B.; Grusch, M.; et al. Temsirolimus inhibits malignant pleural mesothelioma growth in vitro and in vivo: Synergism with chemotherapy. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2011, 6, 852–863. [Google Scholar] [CrossRef]
- Ohara, T.; Takaoka, M.; Toyooka, S.; Tomono, Y.; Nishikawa, T.; Shirakawa, Y.; Yamatsuji, T.; Tanaka, N.; Fujiwara, T.; Naomoto, Y. Inhibition of mTOR by temsirolimus contributes to prolonged survival of mice with pleural dissemination of non-small-cell lung cancer cells. Cancer Sci. 2011, 102, 1344–1349. [Google Scholar] [CrossRef]
- Chiong, E.; Lee, I.-L.; Dadbin, A.; Sabichi, A.L.; Harris, L.; Urbauer, D.; McConkey, D.J.; Dickstein, R.J.; Cheng, T.; Grossman, H.B. Effects of mTOR inhibitor everolimus (RAD001) on bladder cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 2863–2873. [Google Scholar] [CrossRef] [PubMed]
- Mita, M.; Sankhala, K.; Abdel-Karim, I.; Mita, A.; Giles, F. Deforolimus (AP23573) a novel mTOR inhibitor in clinical development. Expert Opin. Investig. Drugs 2008, 17, 1947–1954. [Google Scholar] [CrossRef] [PubMed]



| Signaling Pathway | Drug/Genetic Study | Clinical Trial | Pre-Clinical Study | Cardiovascular Events | References |
|---|---|---|---|---|---|
| NF-κB pathway | Infliximab | ATTACH (Anti-TNF alpha Therapy Against Chronic Heart failure) | - | ↑ mortality and hospitalization No improvement in clinical status of III-IV NYHA HF patients | [110] |
| Sulfasalazine | Sulfasalazine and Endothelial Function (NCT00554203) | - | ↓ NF-κB activation ↓ inflammatory TNFα-induced genes No amelioration of endothelial dysfunction in patients with coronary artery disease No effects on systemic inflammatory biomarkers | [112] | |
| Hippo pathway | Endothelial-specific YAP deletion | - | ✓ | ↓ NF-κB activation ↓ TAK1 induction ↓ proinflammatory cytokines | [113] |
| YAP and TAZ deletion in the epicardium | - | ✓ | ↓ IFN-γ → defective recruitment of T-regulatory cells → inhibition of cardioprotective effects → myocardial injury | [114] | |
| mTOR pathway | Hesperidin | - | ✓ | ↓ Beclin1 ↑ mTOR and Akt PI3K ↓ myocardial I/R injury | [115] |
| Everolimus | Controlled Level EVERolimus in Acute Coronary Syndromes (CLEVER-ACS) | - | No reduction in MI size No improvement of microvascular obstruction | [116] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cucu, I. Signaling Pathways in Inflammation and Cardiovascular Diseases: An Update of Therapeutic Strategies. Immuno 2022, 2, 630-650. https://doi.org/10.3390/immuno2040039
Cucu I. Signaling Pathways in Inflammation and Cardiovascular Diseases: An Update of Therapeutic Strategies. Immuno. 2022; 2(4):630-650. https://doi.org/10.3390/immuno2040039
Chicago/Turabian StyleCucu, Ioana. 2022. "Signaling Pathways in Inflammation and Cardiovascular Diseases: An Update of Therapeutic Strategies" Immuno 2, no. 4: 630-650. https://doi.org/10.3390/immuno2040039
APA StyleCucu, I. (2022). Signaling Pathways in Inflammation and Cardiovascular Diseases: An Update of Therapeutic Strategies. Immuno, 2(4), 630-650. https://doi.org/10.3390/immuno2040039