
Geographic Location Determines Differentially Methylated Gene Expressions in Autoimmune Diseases

 , , ,
, , ,
Abstract
:1. Introduction
2. Patients and Methods
2.1. Patient Population
2.2. Methylation
2.3. RNAseq Analysis
3. Results
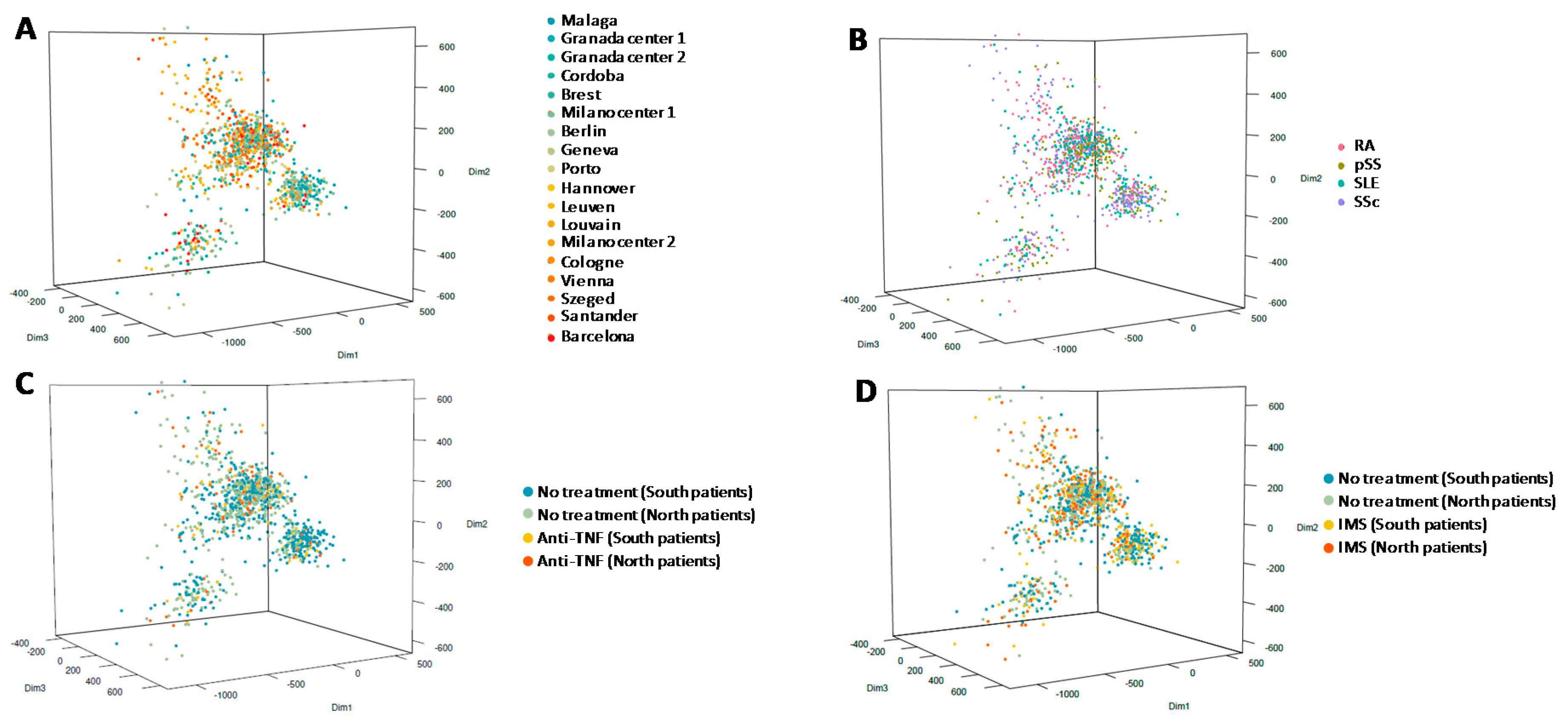
3.1. Characteristics of Selected Patients
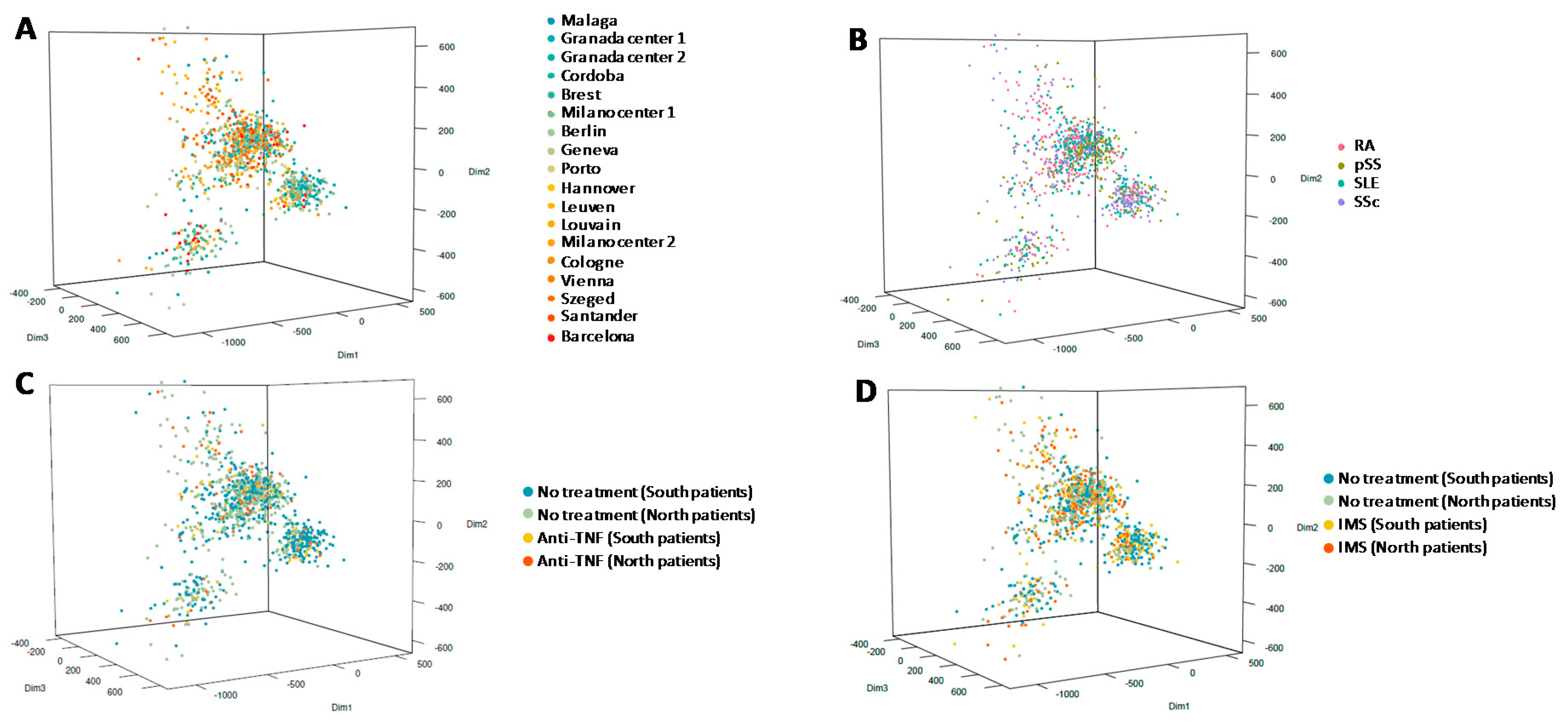
3.2. Methylation Analysis
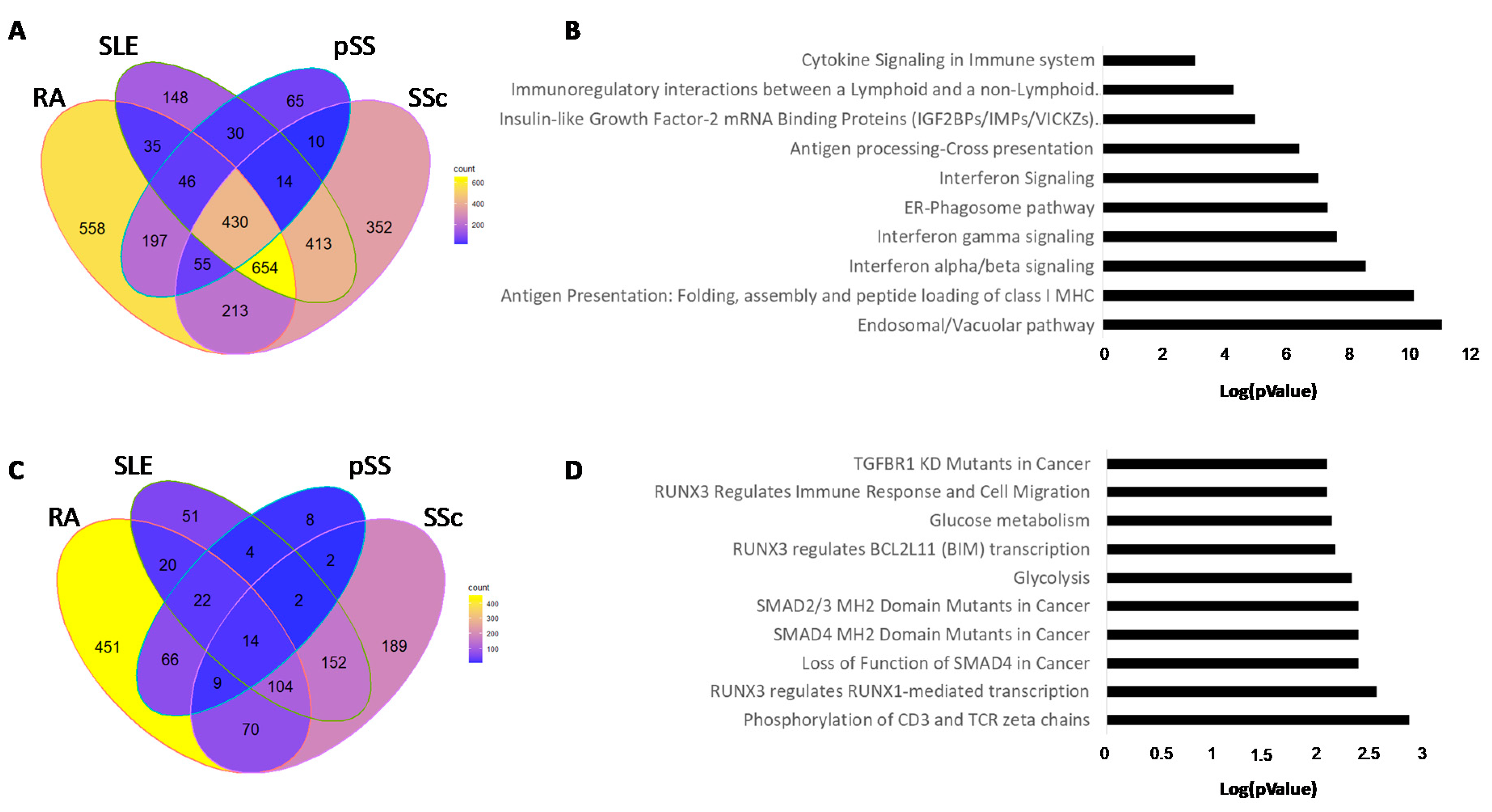
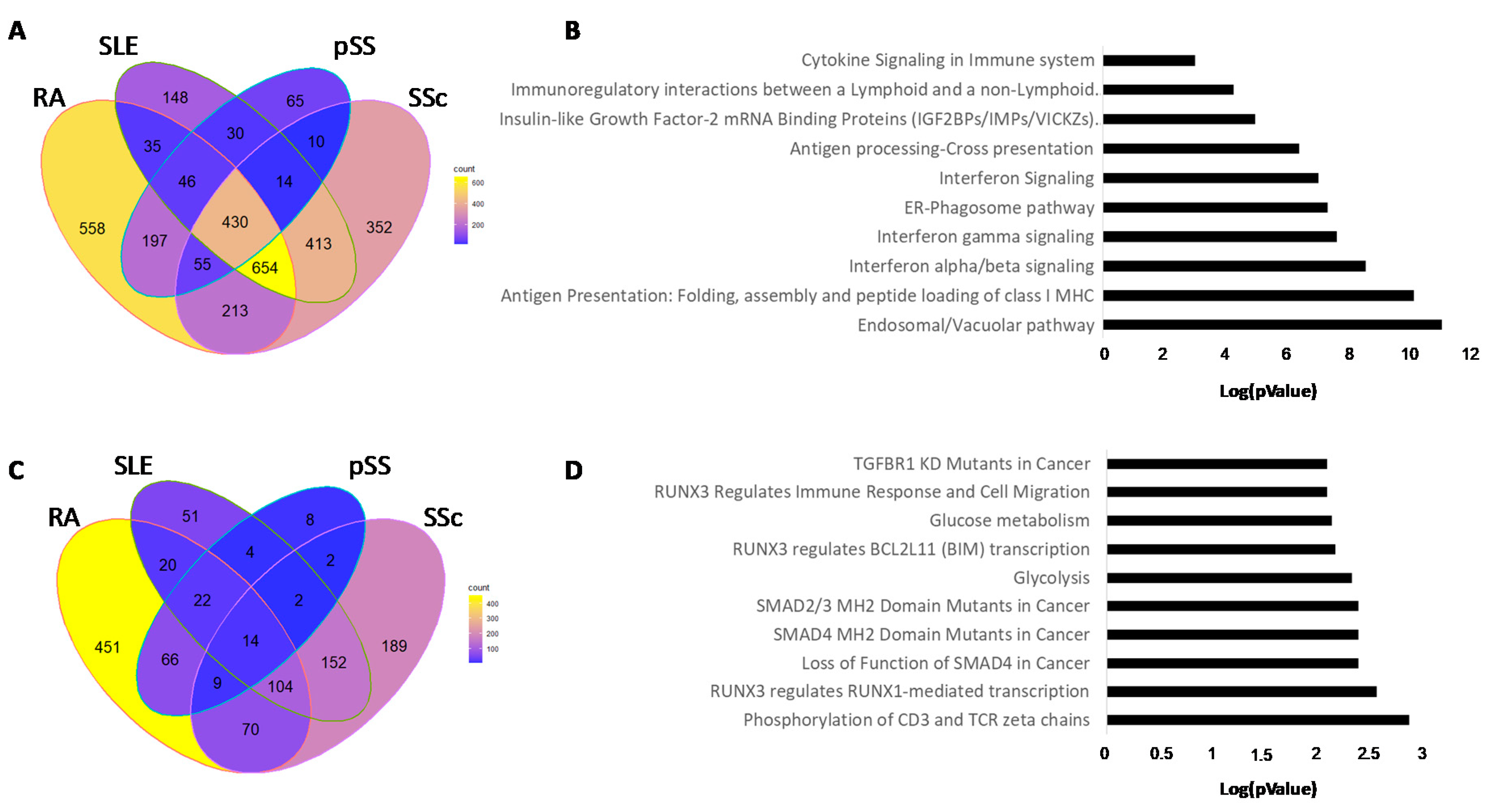
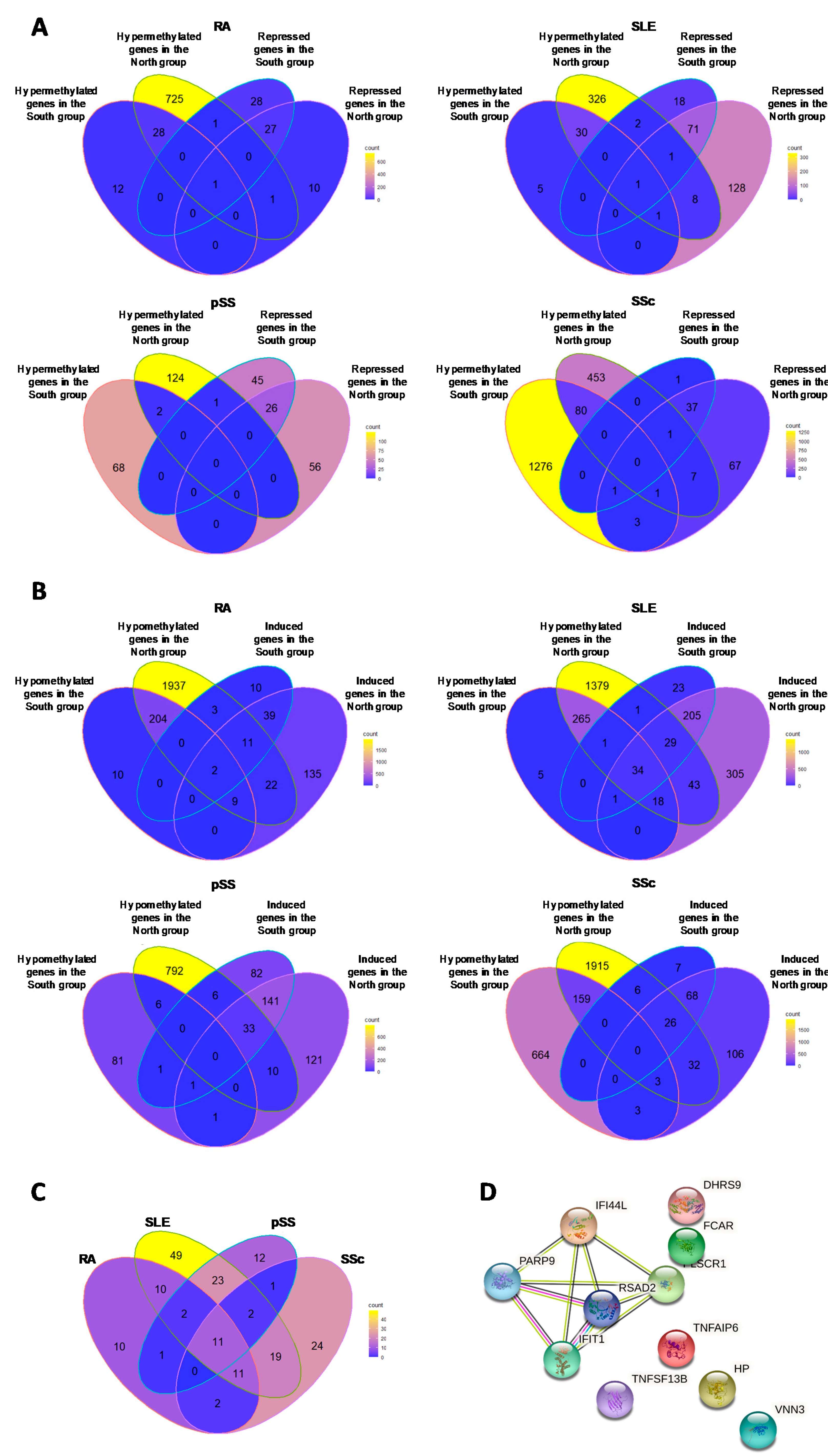
3.3. Hypomethylated Genes in Common in Each Autoimmune Disease
3.4. Hypermethylated Genes in Common in Each Autoimmune Disease
3.5. Comparison of Transcript Expression with Methylation Status
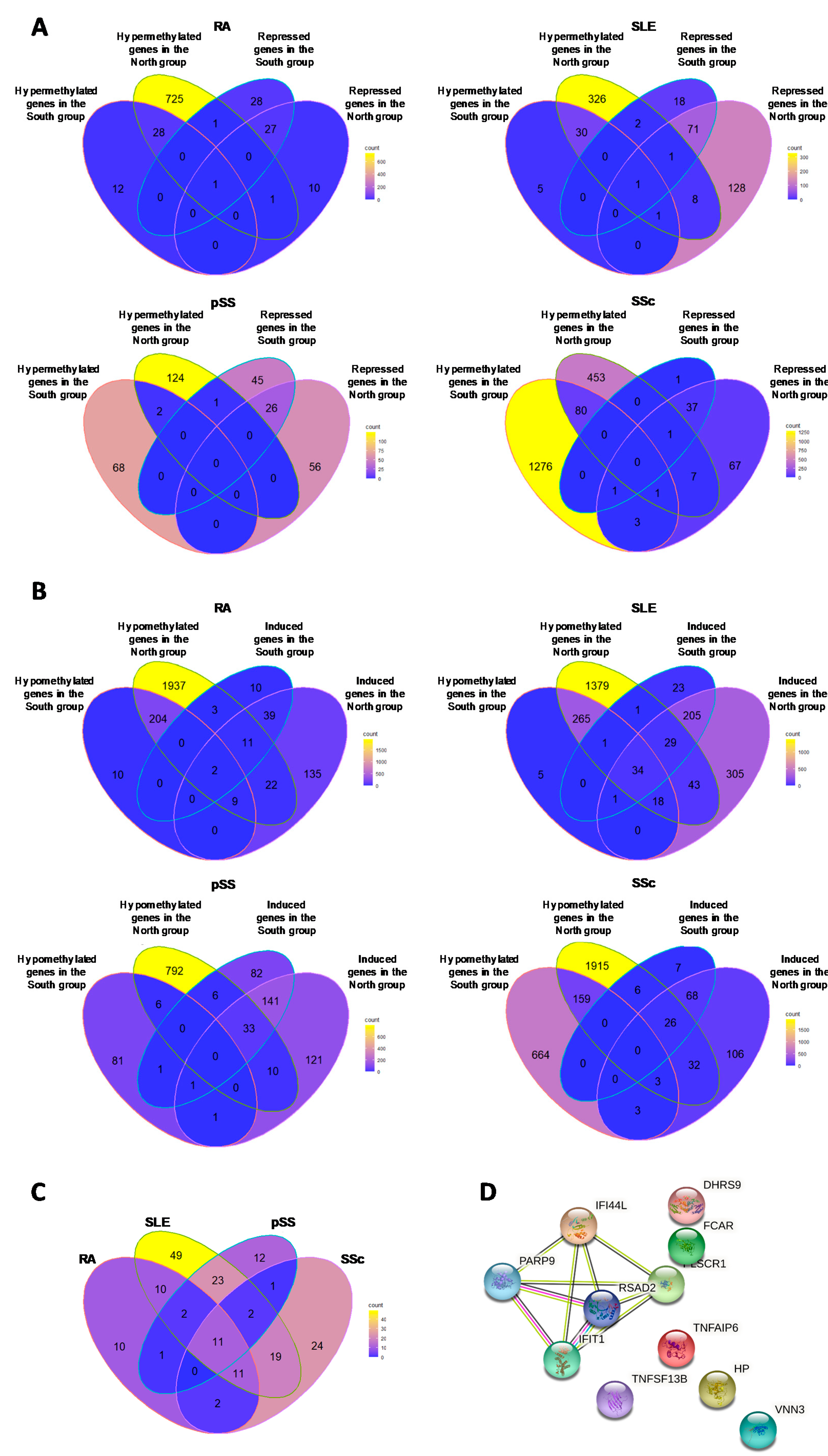
3.5.1. Hypermethylated (ΔBeta ≥ 0.05) Genes Associated with Repressed Transcript Expression (FC ≤ 1.3)
3.5.2. Hypomethylated Genes (ΔBeta ≤ 0.05) Associated with Induced Transcript Expression (FC ≥ 1.3)
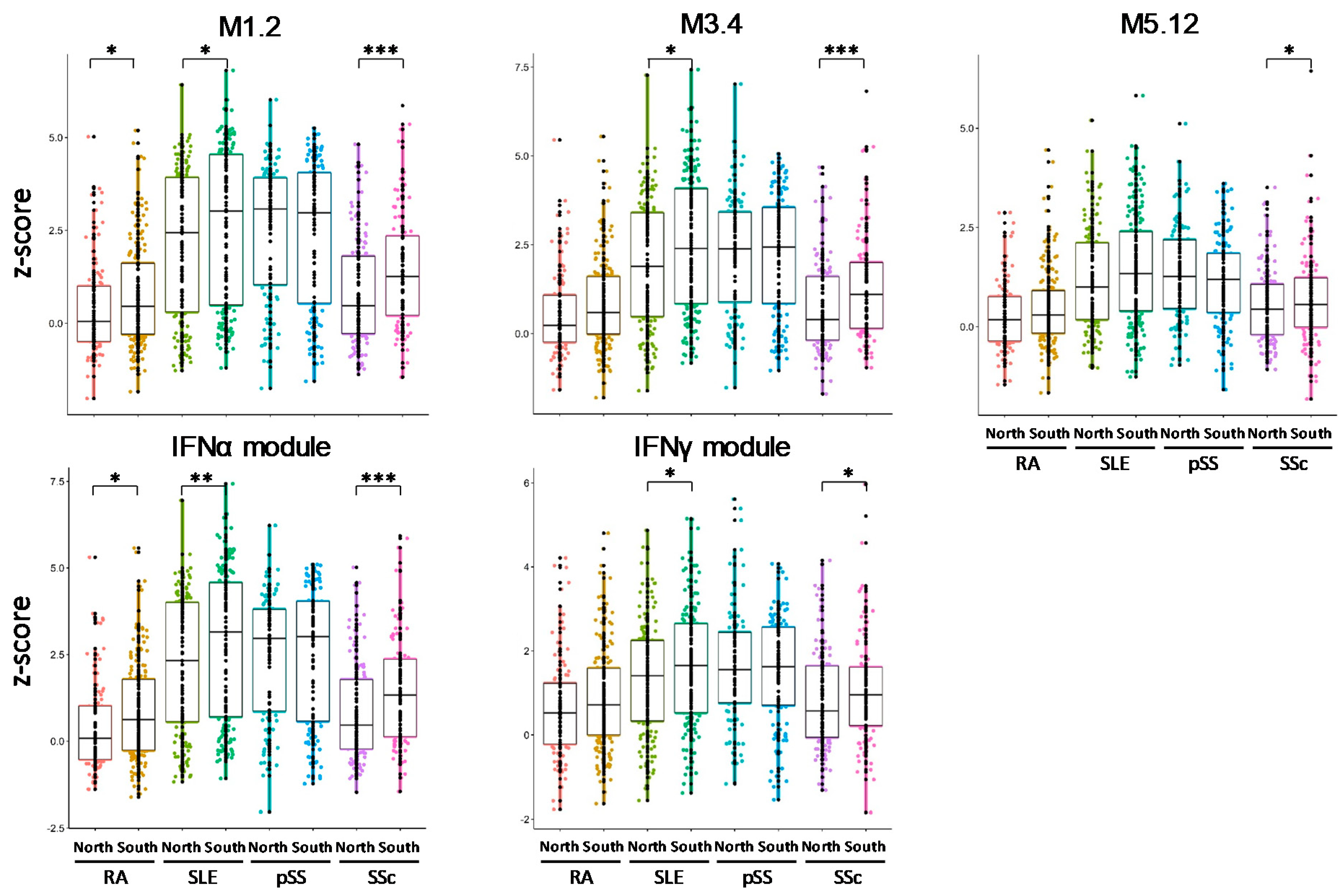
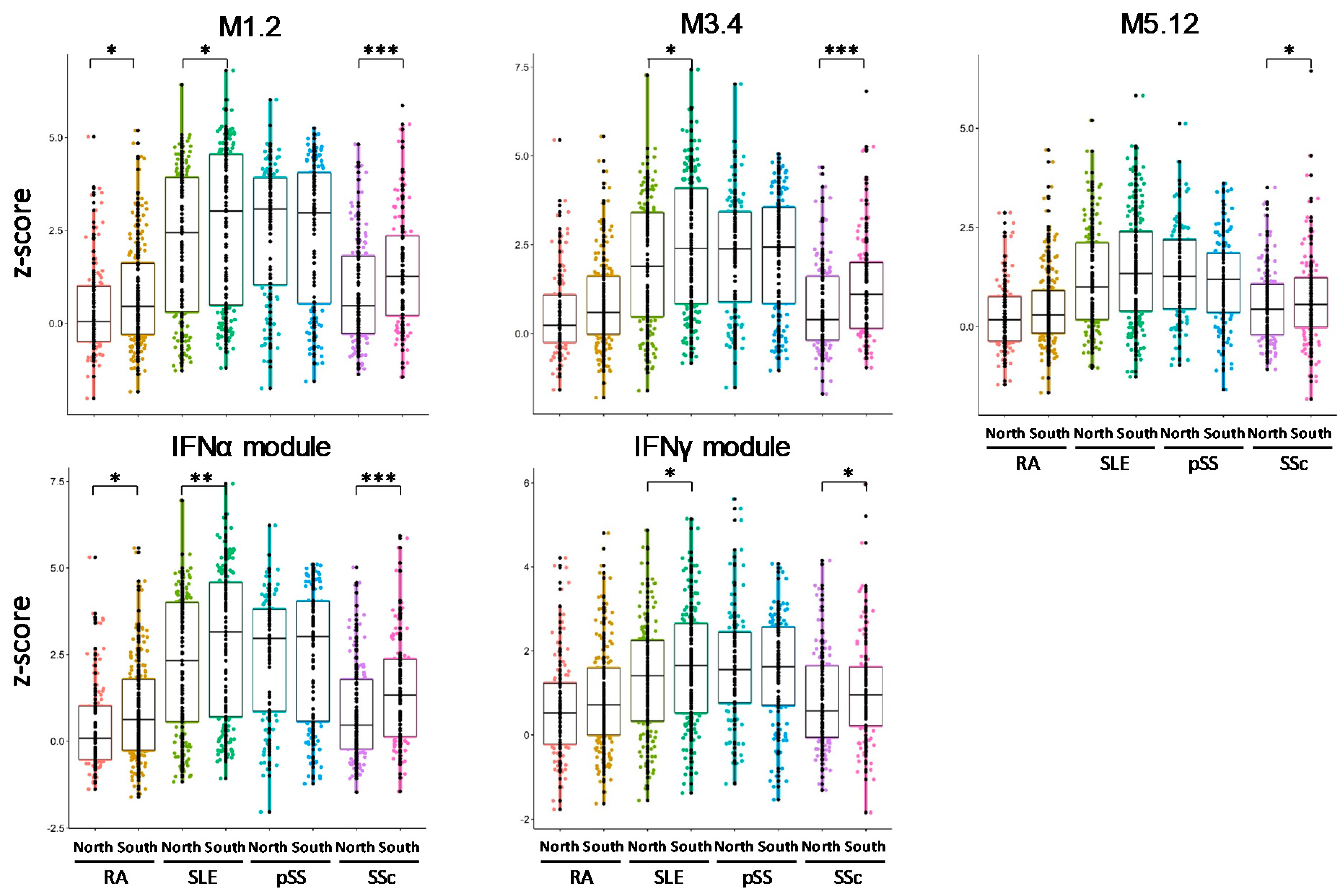
3.6. IFN Signatures Are Increased in Patients from the North of Europe
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blank, M.; Shoenfeld, Y.; Perl, A. Cross-talk of the environment with the host genome and the immune system through endogenous retroviruses in systemic lupus erythematosus. Lupus 2009, 18, 1136–1143. [Google Scholar] [CrossRef]
- Shapira, Y.; Agmon-Levin, N.; Shoenfeld, Y. Defining and analyzing geoepidemiology and human autoimmunity. J. Autoimmun. 2010, 34, J168–J177. [Google Scholar] [CrossRef] [PubMed]
- Shapira, Y.; Poratkatz, B.-S.; Gilburd, B.; Barzilai, O.; Ram, M.; Blank, M.; Lindeberg, S.; Frostegård, J.; Anaya, J.-M.; Bizzaro, N.; et al. Geographical differences in autoantibodies and anti-infectious agents antibodies among healthy adults. Clin. Rev. Allergy Immunol. 2012, 42, 154–163. [Google Scholar] [CrossRef]
- Waddington, C.H. The epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grolleau-Julius, A.; Ray, D.; Yung, R.L. The role of epigenetics in aging and autoimmunity. Clin. Rev. Allergy Immunol. 2010, 39, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Youinou, P.; Pers, J.-O.; Gershwin, M.E.; Shoenfeld, Y. Geo-epidemiology and autoimmunity. J. Autoimmun. 2010, 34, J163–J167. [Google Scholar] [CrossRef]
- Barturen, G.; Babaei, S.; Català-Moll, F.; Martínez-Bueno, M.; Makowska, Z.; Martorell-Marugán, J.; Carmona-Sáez, P.; Toro-Domínguez, D.; Carnero-Montoro, E.; Teruel, M.; et al. Integrative analysis reveals a molecular stratification of systemic autoimmune diseases. Arthritis Rheumatol. 2021, 73, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Busche, S.; Shao, X.; Caron, M.; Kwan, T.; Allum, F.; Cheung, W.A.; Ge, B.; Westfall, S.; Simon, M.-M.; Multiple Tissue Human Expression Resource; et al. Population whole-genome bisulfite sequencing across two tissues highlights the environment as the principal source of human methylome variation. Genome Biol. 2015, 16, 290. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. ChAMP: 450k chip analysis methylation pipeline. Bioinformatics 2014, 30, 428–430. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Franceschini, A.; Szklarczyk, D.; Frankild, S.; Kuhn, M.; Simonovic, M.; Roth, A.; Lin, J.; Minguez, P.; Bork, P.; von Mering, C.; et al. STRING v9.1: Protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013, 41, D808–D815. [Google Scholar] [CrossRef] [Green Version]
- Chiche, L.; Jourde-Chiche, N.; Whalen, E.; Presnell, S.; Gersuk, V.; Dang, K.; Anguiano, E.; Quinn, C.; Burtey, S.; Berland, Y.; et al. Modular transcriptional repertoire analyses of adults with systemic lupus erythematosus reveal distinct type I and type II interferon signatures. Arthritis Rheumatol. 2014, 66, 1583–1595. [Google Scholar] [CrossRef] [PubMed]
- Kirou, K.A.; Lee, C.; George, S.; Louca, K.; Papagiannis, I.G.; Peterson, M.G.E.; Ly, N.; Woodward, R.N.; Fry, K.E.; Lau, A.Y.-H.; et al. Coordinate overexpression of interferon-alpha-induced genes in systemic lupus erythematosus. Arthritis Rheumatol. 2004, 50, 3958–3967. [Google Scholar] [CrossRef]
- Johnson, A.A.; Akman, K.; Calimport, S.R.G.; Wuttke, D.; Stolzing, A.; de Magalhães, J.P. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012, 15, 483–494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Morgan, M.; Hutchison, K.; Calhoun, V.D. A study of the influence of sex on genome wide methylation. PLoS ONE 2010, 5, e10028. [Google Scholar] [CrossRef] [Green Version]
- Bind, M.-A.; Zanobetti, A.; Gasparrini, A.; Peters, A.; Coull, B.; Baccarelli, A.; Tarantini, L.; Koutrakis, P.; Vokonas, P.; Schwartz, J. Effects of temperature and relative humidity on DNA methylation. Epidemiology 2014, 25, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Bind, M.-A.C.; Coull, B.A.; Baccarelli, A.; Tarantini, L.; Cantone, L.; Vokonas, P.; Schwartz, J. Distributional changes in gene-specific methylation associated with temperature. Environ. Res. 2016, 150, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricceri, F.; Trevisan, M.; Fiano, V.; Grasso, C.; Fasanelli, F.; Scoccianti, C.; Marco, L.D.; Tos, A.G.; Vineis, P.; Sacerdote, C. Seasonality modifies methylation profiles in healthy people. PLoS ONE 2014, 9, e106846. [Google Scholar] [CrossRef]
- Prunicki, M.; Cauwenberghs, N.; Lee, J.; Zhou, X.; Movassagh, H.; Noth, E.; Lurmann, F.; Hammond, S.K.; Balmes, J.R.; Desai, M.; et al. Air pollution exposure is linked with methylation of immunoregulatory genes, altered immune cell profiles, and increased blood pressure in children. Sci. Rep. 2021, 11, 4067. [Google Scholar] [CrossRef]
- Miller, C.N.; Dye, J.A.; Schladweiler, M.C.; Richards, J.H.; Ledbetter, A.D.; Stewart, E.J.; Kodavanti, U.P. Acute inhalation of ozone induces DNA methylation of apelin in lungs of long-evans rats. Inhal. Toxicol. 2018, 30, 178–186. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, N.F.P.; de Souza, B.F.; de Castro Coêlho, M. UV radiation and its relation to DNA methylation in epidermal cells: A review. Epigenomes 2020, 4, 23. [Google Scholar] [CrossRef]
- Kim, H.-Y.; Lee, D.H.; Shin, M.H.; Shin, H.S.; Kim, M.-K.; Chung, J.H. UV-induced DNA methyltransferase 1 promotes hypermethylation of tissue inhibitor of metalloproteinase 2 in the human skin. J. Dermatol. Sci. 2018, 91, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snegarova, V.; Naydenova, D. Vitamin D: A review of its effects on epigenetics and gene regulation. Folia Med. 2020, 62, 662–667. [Google Scholar] [CrossRef]
- Barragan, M.; Good, M.; Kolls, J.K. Regulation of dendritic cell function by vitamin D. Nutrients 2015, 7, 8127–8151. [Google Scholar] [CrossRef]
- Almerighi, C.; Sinistro, A.; Cavazza, A.; Ciaprini, C.; Rocchi, G.; Bergamini, A. 1Alpha,25-Dihydroxyvitamin D3 Inhibits CD40L-Induced pro-Inflammatory and immunomodulatory activity in human monocytes. Cytokine 2009, 45, 190–197. [Google Scholar] [CrossRef]
- Harrison, S.R.; Li, D.; Jeffery, L.E.; Raza, K.; Hewison, M. Vitamin D, autoimmune disease and rheumatoid arthritis. Calcif. Tissue Int. 2020, 106, 58–75. [Google Scholar] [CrossRef] [Green Version]
- Dall’Ara, F.; Cutolo, M.; Andreoli, L.; Tincani, A.; Paolino, S. Vitamin D and systemic lupus erythematous: A review of immunological and clinical aspects. Clin. Exp. Rheumatol. 2018, 36, 153–162. [Google Scholar]
- Maugeri, A.; Barchitta, M. How dietary factors affect DNA methylation: Lesson from epidemiological studies. Medicina 2020, 56, 374. [Google Scholar] [CrossRef]
- Vordenbäumen, S.; Sokolowski, A.; Rosenbaum, A.; Gebhard, C.; Raithel, J.; Düsing, C.; Chehab, G.; Richter, J.G.; Brinks, R.; Rehli, M.; et al. Methyl donor micronutrients, CD40-ligand methylation and disease activity in systemic lupus erythematosus: A cross-sectional association study. Lupus 2021, 30, 1773–1780. [Google Scholar] [CrossRef]
- Murdaca, G.; Tonacci, A.; Negrini, S.; Greco, M.; Borro, M.; Puppo, F.; Gangemi, S. Emerging role of vitamin D in autoimmune diseases: An update on evidence and therapeutic implications. Autoimmun. Rev. 2019, 18, 102350. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics of HCs | N | South, N = 196 1 | North, N = 122 1 | p-Value 2 |
| Age | 318 | 47 (13) | 43 (13) | 0.009 |
| MD | 0 | 0 | ||
| Gender | 318 | 0.091 | ||
| Female | 156/196 (80%) | 87/122 (71%) | ||
| Male | 40/196 (20%) | 35/122 (29%) | ||
| MD | 0 | 0 | ||
| Ethnic origin | 318 | 0.15 | ||
| Asian | 0/196 (0%) | 1/122 (0.8%) | ||
| Caucasian/White | 193/196 (98%) | 121/122 (99%) | ||
| Other | 3/196 (1.5%) | 0/122 (0%) | ||
| MD | 0 | 0 | ||
| Obesity | 316 | 12/194 (6.2%) | 12/122 (9.8%) | 0.2 |
| MD | 2 | 0 | ||
| Cigarette smoking | 303 | 27/185 (15%) | 17/118 (14%) | >0.9 |
| MD | 11 | 4 | ||
| Characteristics of patients | N | South, N = 523 1 | North, N = 554 1 | p-Value 2 |
| Age | 1077 | 55 (13) | 55 (15) | 0.9 |
| MD | 0 | 0 | ||
| Gender | 1077 | 0.057 | ||
| Female | 475/523 (91%) | 483/554 (87%) | ||
| Male | 48/523 (9.2%) | 71/554 (13%) | ||
| MD | 0 | 0 | ||
| Ethnic origin | 1077 | 0.2 | ||
| Asian | 1/523 (0.2%) | 6/554 (1.1%) | ||
| Black/African American | 1/523 (0.2%) | 4/554 (0.7%) | ||
| Caucasian/White | 512/523 (98%) | 538/554 (97%) | ||
| Native Hawaiian/Other Pacific Islander | 1/523 (0.2%) | 0/554 (0%) | ||
| Other | 8/523 (1.5%) | 6/554 (1.1%) | ||
| MD | 0 | 0 | ||
| Obesity | 1039 | 62/490 (13%) | 70/549 (13%) | >0.9 |
| MD | 33 | 5 | ||
| Cigarette smoking | 999 | 74/479 (15%) | 91/520 (18%) | 0.4 |
| MD | 44 | 34 | ||
| Duration disease | 1077 | 13 (9) | 12 (10) | 0.2 |
| MD | 0 | 0 | ||
| Physician global assessment | 1048 | 22 (18) | 26 (21) | 0.007 |
| MD | 0 | 29 | ||
| Treatment | ||||
| Antimalaria | 1077 | 167/523 (32%) | 182/554 (33%) | 0.7 |
| MD | 0 | 0 | ||
| Immunosuppressor | 1077 | 165/523 (32%) | 241/554 (44%) | <0.001 |
| MD | 0 | 0 | ||
| Steroids | 1077 | 178/523 (34%) | 217/554 (39%) | 0.081 |
| MD | 0 | 0 | ||
| Anti TNF | 1077 | 47/523 (9.0%) | 92/554 (17%) | <0.001 |
| MD | 0 | 0 | ||
| Tocilizumab | 39 | 12/12 (100%) | 27/27 (100%) | |
| MD | 511 | 527 | ||
| Abatacept | 27 | 11/11 (100%) | 16/16 (100%) | |
| MD | 512 | 538 |
| South Group | North Group | |||||||
|---|---|---|---|---|---|---|---|---|
| DMPs | Hypomethylated DMPs (%) | Genes | Hypomethylated Genes (%) | DMPs | Hypomethylated DMPs (%) | Genes | Hypomethylated Genes (%) | |
| RA | 365 | 87.7 | 266 | 84.6 | 4982 | 80.7 | 2810 | 77.9 |
| SLE | 522 | 90.8 | 360 | 90 | 3966 | 84.8 | 2058 | 86 |
| pSS | 264 | 53 | 159 | 56.6 | 1548 | 89.4 | 970 | 87.3 |
| SSc | 3295 | 39.7 | 2087 | 39.7 | 4970 | 81 | 2546 | 84.1 |
| South Group | North Group | |||||
|---|---|---|---|---|---|---|
| Genes | Induced Genes (%) | Repressed Genes (%) | Genes | Induced Genes (%) | Repressed Genes (%) | |
| RA | 122 | 53.3 | 46.7 | 257 | 84.5 | 15.5 |
| SLE | 387 | 75.9 | 24.1 | 845 | 75.1 | 24.9 |
| pSS | 336 | 78.6 | 21.4 | 389 | 78.9 | 21.1 |
| SSc | 147 | 72.8 | 27.2 | 355 | 67 | 33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pers, J.-O.; Bahane, H.; Le Dantec, C.; Foulquier, N.; Alarcon-Riquelme, M.E.; Youinou, P.; PRECISESADS Clinical Consortium. Geographic Location Determines Differentially Methylated Gene Expressions in Autoimmune Diseases. Immuno 2021, 1, 529-544. https://doi.org/10.3390/immuno1040037
Pers J-O, Bahane H, Le Dantec C, Foulquier N, Alarcon-Riquelme ME, Youinou P, PRECISESADS Clinical Consortium. Geographic Location Determines Differentially Methylated Gene Expressions in Autoimmune Diseases. Immuno. 2021; 1(4):529-544. https://doi.org/10.3390/immuno1040037
Chicago/Turabian StylePers, Jacques-Olivier, Hajar Bahane, Christelle Le Dantec, Nathan Foulquier, Marta E. Alarcon-Riquelme, Pierre Youinou, and PRECISESADS Clinical Consortium. 2021. "Geographic Location Determines Differentially Methylated Gene Expressions in Autoimmune Diseases" Immuno 1, no. 4: 529-544. https://doi.org/10.3390/immuno1040037
APA StylePers, J.-O., Bahane, H., Le Dantec, C., Foulquier, N., Alarcon-Riquelme, M. E., Youinou, P., & PRECISESADS Clinical Consortium. (2021). Geographic Location Determines Differentially Methylated Gene Expressions in Autoimmune Diseases. Immuno, 1(4), 529-544. https://doi.org/10.3390/immuno1040037