Abstract
Parkinson’s disease (PD) is a neurodegenerative movement disorder characterized by the loss of dopaminergic neurons, which results in motor impairment. The rationale and objective of the review article is to determine whether CCBs use contributes to a lower risk of developing a first-time diagnosis of PD. Ca2+ homeostasis disruption and mitochondrial dysfunction play a vital role in PD aetiology. In addition, the L-type voltage-gated calcium channel is expressed at high levels amongst nigral neurons, and could play a role in the pathogenesis of PD. In the dopaminergic neurons, Ca2+ entry through plasma membrane Cav1 channels drives a sustained feed-forward stimulation of mitochondrial oxidative phosphorylation. This study investigates the therapeutic potential of R- and T-type Ca2+ channel inhibition in light of new preclinical and clinical data and the feasibility of available Ca2+ channel blockers to cure PD progression. The R-type calcium channel is a type of voltage-dependent calcium channel. Available findings suggest that calcium homeostasis in dopaminergic neurons might be a valuable target for developing new drugs for PD patients. The limitations of our study include reports of observational studies with different follow-up periods. The specific roles of individual drugs and doses were also not mentioned because of nonreporting in the studies.
1. Introduction
Parkinson’s disease (PD) is a neurodegenerative disorder, characterized by cardinal motor symptoms such as bradykinesia, rigidity, and tremor [1]. PD is strongly associated with ageing, increases exponentially in incidence above the age of 65 years, and has no cure [2]. The motor symptoms of PD first appear clinically caused by the degeneration and death of selective dopaminergic (DA) neurons within the substantia nigra pars compacta (SNpc) [3]. The neurological processes underpinning dementia and its accompanying intellectual abnormalities are referred to as PD. Recent neuroscientist findings have begun to untangle the diverse participation of numerous separate neural networks underpinning cognitive impairments in Parkinson’s Disease Dementia (PDD) and their regulation by both dopaminergic and non-dopaminergic transmitter systems in the brain [4]. Cognitive impairment is a typical symptom of PD, which has a high morbidity and fatality rate. The severity of these symptoms ranges from modest executive dysfunction to full-blown dementia affecting numerous areas [5].
The pathological hallmarks of PD are the presence of Lewy bodies (LBs) and the loss of DA neurons containing neuromelanin [6]. LBs are spherical eosinophilic cytoplasmic protein aggregates composed of proteins including α-synuclein, ubiquitin, parkin, and neurofilaments, and these proteins are found in the affected regions of the brain [7,8]. LBs are most commonly found in the brain regions with the greatest neuron loss in PD, such as the SN, locus coeruleus, the dorsal motor nucleus of the vagus, and the nucleus basalis of Meynert, but they have also been found in the neocortex, diencephalon, spinal cord, and even peripheral autonomic ganglia [9]. DJ-1, encoded by the PARK7 gene, causes early-onset autosomal recessive PD and is likely the most thoroughly investigated [10]. DJ-1 is an essential regulator of the pro-inflammatory response, and knocking it out in astrocytes reduces inflammatory-related damage. PARK2 and PINK1 are both expressed at comparable amounts in astrocytes and neurons [11]. Interestingly, astrocytes lacking Parkin, expressed by the PARK2 gene, showed a stress-induced increase in NOD2 expression, a receptor that integrates endoplasmic reticulum stress and inflammation, and these astrocytes displayed increased cytokine release and reduced neurotrophic factor production [12]. Parkin has also been involved in astrocyte responses to inflammatory signals; stimulation with TNF- leads in Parkin overexpression, whereas activation with IL-1 results in Parkin downregulation [13]. PINK1 expression, which encodes the protein PTEN-induced putative kinase 1 (PINK1), is a loss of function mutation related to early-onset PD. Furthermore, neurotoxic kynurenine metabolites in plasma and cerebrospinal fluid (CSF) are related to symptom severity and nigral pathology in PD [14]. The various mechanisms involved in PD are mitochondrial dysfunction, oxidative stress (OS), neuroinflammation, gene mutation, and some environmental toxins [15] (Figure 1).
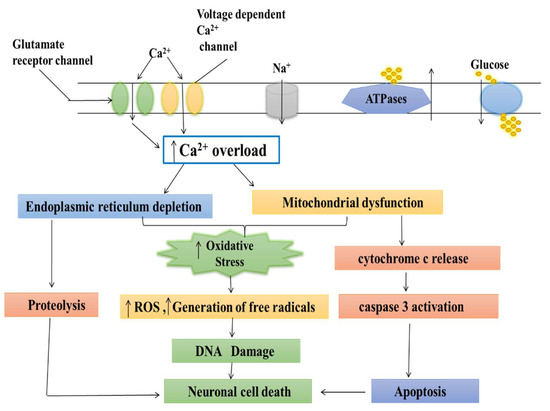
Figure 1.
Different mechanisms involved in Parkinson’s disease (mitochondrial dysfunctioning, oxidative stress (OS), neuroinflammation, gene mutation, and some environmental toxins).
Oxidative stress has long been thought to be one of the pathophysiological mechanisms implicated in PD, which led to the investigation of the antioxidant systems as a promising therapy more than two decades ago. A useful antioxidant must have certain characteristics: it must be capable of interacting with biologically relevant oxidants and free radicals; its reaction by-products must be harmless; and, finally, it must reach a sufficiently high concentration in tissue and cell compartments to ensure quantitatively relevant activity [16]. Patients with Parkinson’s disease (PD) frequently experienced gastrointestinal problems prior to the start of motor symptoms. Parkinson’s disease neuropathology has also been found in the enteric nervous system (ENS). Many studies have found substantial PD-related changes in gut microbiota. The microbiota–gut–brain axis is a dynamic bidirectional communication network that plays a role in the aetiology of PD. The aggregation of misfolded protein alpha-synuclein, the neuropathological characteristic of PD, is thought to start in the stomach and move to the CNS via the vagus nerve and olfactory bulb. Changes in the architecture of the gut microbiota raise the concentrations of short-chain fatty acids (SCFAs) and other metabolites, which act on the neuroendocrine system and modulate the concentrations of GABA, serotonin, and other neurotransmitters. Furthermore, it affects the vagus and intestinal nerve systems, impacting the brain and behavior through the activation of microglia and systemic cytokines. An increasing collection of experimental and clinical evidence suggests that gut dysbiosis and microbiota host interaction play a role in neurodegeneration [17]. Dopaminergic neurodegeneration is directly associated with metal accumulation or elevated inflammatory cytokines such as interleukin-1 (IL-1, IL-6), TNF-α, which causes neuroinflammation, and, ultimately, neuronal death. Metals are the primary natural elements of the earth’s crust and are spread throughout the biosphere by human activities. Metals are commonly found in mining, industrial waste, tailings, agricultural runoff, treated timber, paints, ageing water supply infrastructure, lead-acid batteries, vehicle emissions, fertilizers, and microplastics [18]. Metals’ significance in the aetiology of PD remains a key topic in neurotoxicology and medicinal chemistry. Heavy metals, such as Fe (III) and Mn (II), cause oxidative stress by boosting ROS generation via the Fenton and Haber–Weiss reaction and changing the antioxidant system in cells [19,20].
These metal toxins aggravate the oxidative stress process in the cell, resulting in cell death by producing an imbalance between free radical and antioxidant enzymes [21,22]. In addition, neuroinflammation aggravates oxidative stress pathways, which can lead to protein aggregation via changing the activity of the UPS. Protein aggregates can accumulate due to defective protein breakdown machinery, disrupting cellular activities and causing cell death [23]. In addition, heavy metals inhibit the action of mitochondrial complexes, resulting in a slowed metabolic process, increased ROS generation, and oxidative stress [24,25].
Among these, mitochondrial dysfunction and an increase in oxidative stress play a significant role in the pathogenesis of PD [26]. Although the mechanisms are unclear, the mitochondrial dysfunction in dopaminergic neurons of idiopathic and familial PD is well known. Langston et al. and Burns et al. reported that the accidental administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) selectively inhibits complex I of the mitochondrial electron transport chain (ETC) and because mitochondria play a significant role in the pathogenesis of PD [27,28]. Rotenone, trichloroethylene, pyridaben are the other complex I inhibitors that induce dopaminergic neurodegeneration in PD [29]. MPTP, a strong inhibitor of mitochondrial complex-1 of the electron transport chain, induces parkinsonian symptoms in rats and mimics dopaminergic degenerations via the nigrostriatal pathway [30,31]. As a result, in animal PD models, MPTP is widely utilized to investigate the molecular mechanisms underlying dopaminergic neuronal degeneration and evaluate the efficiency of various neuroprotective agents [32]. Biomolecules such as lipids, proteins, and DNA are destroyed by reactive oxygen and nitrogen species (ROS and RNS), by-products found in the SN and striatum of human PD post-mortem brains [33,34]. Thus, lipid and protein oxidation can result in membrane integrity loss, enzyme deactivation, and cell death in neurodegenerative diseases [35]. Various natural products are used for testing their neuroprotective effects against MPTP-induced PD. Mucuna pruriens (Mp) exhibits various pharmacological properties like analgesic, anti-inflammatory, anti-neoplastic, anti-epileptic, and anti-microbial activities [36]. Mp has been found to be rich in bioactive compounds such as tannins, alkaloids, phenolic compounds, and flavonoids [37]. In addition, Mp extract was reported to significantly improve neuroinflammatory processes and restore biochemical and behavioral parameters and immunoreactivity. Mp shows anti-inflammatory action and its high antioxidant capabilities, which can be utilized to treat inflammatory conditions in the case of PD [38].
An increase in PD risk comes with increasing intake of foods that contain animal fat and foods containing vitamin D. Intake of fruits, vegetables, meats, bread and cereals, or foods containing vitamins A, C, E, or iron was not significantly related to PD risk. Vitamin use, in general, was also not found to be related to PD risk, although a significant trend of increasing risk of PD was noted for intake of vitamin A supplements. The tryptophan (TRP)-kynurenine (KYN) metabolic pathway is the primary catabolic route of TRP metabolism, converting over 95% of TRP into a variety of bioactive metabolites such as anti-inflammatory, antioxidative, proinflammatory, neurotoxic, neuroprotective, and immunologic compounds. Furthermore, kynurenine pathway (KP) enzymes influence inflammation and the immune system. Alterations in the KP enzymes’ activity and the levels of the KP metabolites have been linked to neurological disorders, cancer, autoimmune conditions, and inflammation. However, the functions of KP enzymes and metabolites in the development and progression of many diseases constitute a field of medicine that has received comparatively little attention. An illustration of this is the relationship between kynurenines (KYNs) and the KP enzymes, which have been linked to a variety of diseases, including cancer, autoimmune diseases, inflammatory diseases, neurologic diseases, and mental disorders. One of the key immune response regulators and a potential player in the inflammatory response in parkinsonism is the KP, the primary catabolic route for tryptophan. The KP produced various neuroactive compounds and has both neurotoxic and neuroprotective effects [39]. These disorders are related with amyloid-β (Aβ), alpha synuclein (α--Syn), and prion protein (PrP) depositions in the brain, which cause synaptic disconnection and eventual progressive neuronal death. Although continued progress has been made in understanding the aetiology of many neurological disorders, the precise mechanisms of their origins remain largely unclear. A growing body of evidence implies association between host microbiota, neuroinflammation, and dementia, either directly due to bacterial brain invasion via barrier leakage and the generation of toxins and inflammation, or indirectly by altering the immune response and causing PD-like symptoms [40].
Ursolic acid is a naturally occurring pentacyclic triterpenoid carboxylic acid found in many plants, including apples, basil, bilberries, cranberries, peppermint, rosemary, and oregano. Several biochemical and pharmacological actions of ursolic acid have been described in various experimental systems, including anti-inflammatory, antioxidative, anti-proliferative, anti-cancer, anti-mutagenic, antiatherosclerotic, anti-hypertensive, anti-leukemic, and antiviral characteristics [41,42]. Ursolic acid inhibits MPTP-induced dopaminergic neurotoxicity through the NF-B pathway. Ursolic acid’s anti-inflammatory action has been attributed mostly to its neuroprotective potential. Although the chemical mechanism behind ursolic acid’s neuroprotective impact remains unknown, our findings suggest that ursolic acid might be employed as a viable medication in the treatment of Parkinson’s disease symptoms. As a result, the ability of ursolic acid to rescue dopaminergic neurons from neurodegeneration may imply a role for therapeutic intervention in PD [43].
Chlorogenic acid (CGA), a polyphenolic molecule present in many plants, is particularly prevalent in green coffee beans, which contain roughly 5–12% CGA by weight [44]. CGA is a trans-cinnamic acid ester (which includes caffeic acid, ferulic acid, and p-coumaric acid) and quinic acid. It is widely consumed by individuals and may be found in various drinks and food items [45]. It is mainly found in fruits and vegetables such as apples, apricots, cherries, plums, and tomatoes. Wine, coffee, and tea are the most prevalent CGA-rich drinks [46]. They have anticancer efficacy, cardioprotective properties, and may have neuroprotective activities [47]. Evidence suggests that CGA has a variety of biological effects, including antioxidant, neuroprotective, and neurotrophic properties [48]. Therefore, CGA can potentially be a powerful anti-inflammatory drug in preventing neurodegeneration in PD. It exerts its effects primarily by suppressing the production of iNOS, TNF- α, and NF-κB in activated glial cells, ultimately decreasing neuroinflammation via increased anti-inflammatory and antioxidant activity [49]. Thus, CGA’s anti-inflammatory effect and its high antioxidant characteristics can be utilized to treat the inflammatory state associated with PD [50].
The symptoms of PD are motor symptoms such as bradykinesia, tremor, rigidity, and asymmetric manifestations. Patients with PD also exhibit various non-motor symptoms, which frequently precede motor symptoms by several years and have a negative impact on quality of life, increased caregiver burden, and annual economic costs. Given the variety of symptoms and the risk of dementia, therapy for cognitive impairment is a developing therapeutic concern. Given that up to 80% of Parkinson’s disease patients develop dementia after 15–20 years of disease, there is an urgent need to find early biomarkers in order to develop effective therapies and monitor cognitive impairment [51]. Generally, mitochondrial dysfunction is characterized by increased reactive oxygen species (ROS), decreased mitochondrial complex I enzyme activity, the release of cytochrome c, ATP depletion, and caspase 3 activations [52]. The impaired mitochondrial function leads to increased OS, causing degeneration of dopaminergic neurons. The ROS and OS not only cause cellular death but also lead to cell death by activating signaling pathways [52]. Cases of PD that occur primarily from excessive mitochondrial oxidant stress are more likely to react to calcium channel blocker medication, whereas those in which this mechanism of toxicity is less prominent may show less clinical improvement [53]. It is now simpler to regulate intracellular calcium levels pharmacologically with calcium channel blocker (CCB) medicines than it is to minimize α-synuclein aggregation. This is because calcium channel blocking therapies are accessible, have been approved for use in humans, and have a favorable safety profile. This investigation aimed to determine the neuroprotective effects of CCB in PD [54,55].
Although the pathway for Ca2+ buildup into mitochondria has long been described, its functional relevance in cell physiology and disease is just now becoming understood. Fundamental cell processes, such as proliferation and death, are regulated by mitochondrial metabolism’s adaptability and its subtle modifications to particular physiological or pathological situations. Particularly, Ca2+ signaling has become a crucial role that mitochondria use to adjust their activity in response to cell demand. The proper mitochondrial Ca2+ signal is greatly influenced by the functional interaction between mitochondria and endoplasmic reticulum (ER), which in turn modifies the bioenergetics and functioning of the cell. In fact, the mitochondria take up the Ca2+ that is produced by the ER and use it to control the activity of proteins, enzymes, and transporters that are involved in the metabolism of organelles both in the matrix and in the intermembrane space [56]. It has been suggested that calcium channel blockers (CCBs) be used to lower the risk of PD. This study aimed to evaluate the association between CCBs and its dose effect and the risk of PD in patients.
2. Normal Physiology and Pathology of Mitochondria
Mitochondria is the powerhouse of the cell, including the production of energy through the mitochondrial respiratory chain, cell death regulation, calcium metabolism, and production of ROS [57,58]. Mitochondria is the primary source of free radical generaation in the cell resulting in OS. The mitochondrial ETC involves five complexes I-V embedded in the inner mitochondrial membrane, which involve the transfer of reducing equivalents from high-energy compounds to oxygen through Kreb’s cycle [59,60]. The mutations in specific genes such as Parkin, alpha-synuclein, DJ-1, LRRK2, PTEN-induced kinase 1 (PINK 1), and vacuolar protein sorting 35 (VSP35) support the mitochondrial dysfunction in PD [61,62]. Also, the toxins such as rotenone, MPTP, and paraquat alter mitochondrial respiration in PD. These toxins cause the deficiency in mitochondrial complexes’ activity [63], reduce the movement of mitochondria, and mitigate generation of reactive oxygen species (ROS) [64], thereby leading to PD-like symptoms (Figure 2).
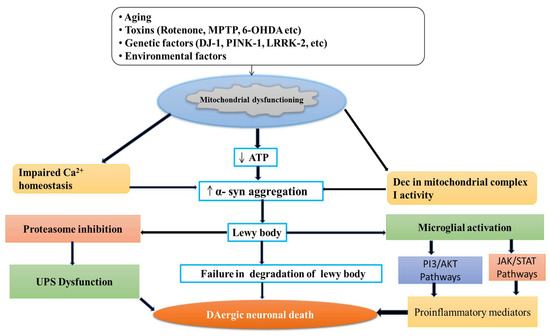
Figure 2.
Degeneration of DAergic neuronal death by different neurotoxins.
Paraquat (PQ) is a herbicide known to cause neurotoxicity by producing free radicals, resulting in oxidative stress [65]. The specific neurotoxicity produced by PQ in the SN area is attributable to the fact that it enters the CNS via the blood-brain barrier via neutral amino acid transporter. Maneb (MB), a fungicide, exhibits similar effects and has been reported to disrupt mitochondrial activity, resulting in oxidative stress [66,67,68]. When combined, MB and PQ are known to work synergistically, increasing neuronal toxicity and causing dopaminergic neurodegeneration and severe oxidative stress [69,70].
Withania somnifera (Ws) is a Solanaceae-family herbal medicinal plant and plays an essential role in Ayurveda. The biological activity of Ws extract demonstrated antioxidant and free radical scavenging capabilities [71]. Various parts of this medicinal plant have been used to cure various diseases since ancient times. It is known as “Indian Ginseng” due to its importance in traditional Indian medicine. Ws root extract contains withanolides, which are steroidal alkaloids and lactones of dopaminergic neurons in the SN area of the brain [72]. The anti-degeneration properties of the Ws root extract against MB–PQ-induced dopaminergic neurodegeneration in the PD animal model has been reported. Ws extract can increase the numbers of TH-positive cells in the SN region of the MB–PQ-induced PD animal brain while concurrently decreasing the oxidative stress occurring in nigrostriatal tissues [66,73]. Therefore, it appears that the up-regulation of TH expression in the SN area of the brain is the leading cause of the improvement in the walking pattern seen in the Ws-treated PD mouse. It is clear from our work that Ws has substantial antioxidant capability and that through preventing neurodegeneration, its ROS scavenging property plays a significant role in preventing PD. Taken as a whole, Ws extract looks to be a promising therapeutic candidate for Parkinson’s neuroprotection [66].
The mitochondrial function and calcium signaling are interlinked; the calcium (Ca2+) is the second messenger to transmit depolarization and synapses to the other neurons [74]. The concentration of Ca2+ in the cytosol stimulates the mitochondria to produce more energy. Ca2+ is maintained via the accumulation of Ca2+ in the mitochondrial matrix [75], leads to the activation of oxidative phosphorylation, and increases the production of ATP [74]. Environmental toxins such as rotenone and MPP+ reduce the level of Ca2+. The mitochondrial complex I deficiency power is not present in all patients with PD, either in the brain, platelets, or other tissues. The severity of the defect is about a 35% reduction in activity when the patient group is compared with control populations [76].
3. Role of Calcium in Mitochondria
Calcium is an essential ion with multiple roles in cell activity. Calcium enters mitochondria through a pore and is utilized in their energy production process [77]. Calcium acts as the key regulator of energy production in mitochondria, but excess calcium can trigger cell death [78]. If the pore fails to close, then mitochondria retain the energy synthesized in the form of ATP. This results in the accumulation of oxidants and calcium overloading, leading to mitochondrial swelling and cell stress, and resulting in numerous diseases including cardiovascular diseases, such as stroke and heart attack, and neurodegenerative disorders, such as PD and Alzheimer’s disease (AD) [79,80].
Several researchers reported that both calcium and magnesium ions were involved in controlling the shuttle. When these ions bind to the inside part of the calcium channel, the pore closes [81]. This helps explain the role of calcium transport protein in controlling mitochondrial calcium uptake and is important for understanding diseases associated with mitochondrial dysfunction [82].
The calcium was accumulated in the mitochondria neuron, resulting in the mitochondrial calcium uptake, sequestration, and release of the calcium-dependent responses that resulted in gene transcription and cell death [83,84]. The stimuli were activated by initiating the entry of (Ca2+) through plasma membrane channels and responded by neurons. However, the increase in free cytosolic (Ca2+) is strongly modulated by the activity of intracellular calcium stores [85]. In particular, Ca2+ uptake, sequestration, and release by the endoplasmic reticulum and mitochondria are the two major Ca2+-regulating organelles that play essential roles in modulating and interpreting Ca2+ signals [86]. Mitochondria play a critical role in neuronal (Ca2+) signaling. Also, the overloading of mitochondrial calcium and dysfunction may be important for triggering the cell death which follows ischemic and traumatic brain injury, and neurodegenerative disorders such as AD, PD, Huntington’s disease (HD), and Amyotrophic lateral sclerosis (ALS) [87,88].
3.1. Calcium Regulates Mitochondrial Function
Normally, under physiological conditions, the intracellular (Ca2+) is tightly and highly regulated in the cytosol and within the organelles by the calcium channels [89,90]. The resting total calcium concentration in neurons is typically about 1 mM, and most cytosolic proteins are bound to Ca2+ in the endoplasmic reticulum (ER). Consequently, the free cytosolic Ca2+ is usually maintained at approximately 100 nM, with stimulation causing global increases to approximately 1 μM; local increases may be substantially higher [91,92].
The uptake of neuronal mitochondrial Ca2+ is through uniporter, a channel sensitive to Ca2+ and opened by cytosolic Ca2+, which allows the influx of Ca2+ into the matrix. The release of mitochondrial Ca2+ is regulated by Na+/Ca2+ exchanger [93,94,95]. Therefore, the maximal uptake rate is much higher than the maximal release rate due to the continuous mitochondrial Ca2+ accumulation observed when the cytosolic Ca2+ is high. So, in resting cells, the net effect of mitochondrial Ca2+ is low but suddenly accumulates in a large amount of Ca2+ during the stimulation of influx Ca2+, and during the recovery state this calcium load is released [96,97,98]. The intracellular mitochondrial Ca2+ has numerous and significant physiological effects including aerobic ATP production, modulating the effects of elevated cytosolic Ca2+ on neurotransmitter release, synaptic transmission and excitability, regulating organelle dynamics and trafficking, mediating signaling to the nucleus, regulating the generation of reactive oxygen species (ROS), and activating the release of death signals [99,100].
3.2. Calcium Causes Mitochondrial Dysfunction
It is well known that the major excitatory neurotransmitter in the brain is glutamate, but a continuous exposure of neurons to excessive glutamate leads to excitotoxicity [101]. The process of excitotoxicity is implicated in the pathophysiology of various neurological disorders such as PD, AD, TBI, HD, and ALS. In excitotoxic injury, the N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methylisoxazole-4-propionate acid (AMPA) subtype of the glutamate receptor (NMDAR) plays a central role [102,103]. The physiologic activation of these receptors allows the flow of cations such as Na+ and Ca2+ through the ion channel essential for normal synaptic transmission and a variety of Ca2+-dependent signaling pathways [104,105]. On the other hand, the elevated level of glutamate triggers NMDAR stimulation which leads to the loss of ion homeostasis, that is cell swelling, mitochondrial dysfunction, and activation of cell death pathways, ultimately leading to necrotic death [106,107], and the numerous cell organelles are involved in the disruption of neuronal Ca2+ homeostasis in neurodegenerative disorders (Figure 3).
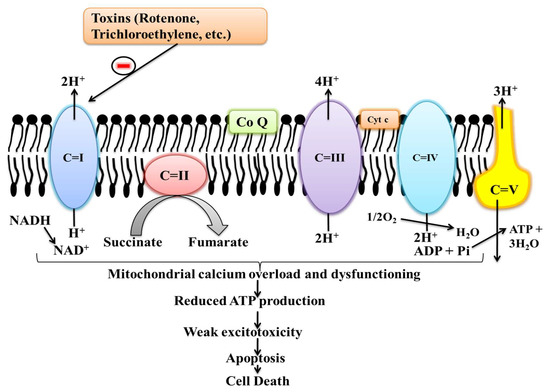
Figure 3.
Effects of different toxins in mitochondrial complexes activity.
The ability of mitochondria to accumulate enormous amounts of calcium in situ plays an important role in excitotoxic injury [108,109]. The excessive influx of Ca2+ across the outer mitochondrial membrane (OMM) occurs through the voltage-dependent anion channels (VDAC) [110]. VDAC is a large voltage-gated channel, fully opened with high conductance and weak anion-selectivity at low transmembrane potentials (<20–30 mV), but switching to cation selectivity and lower conductance at higher possibilities [24]. The ligand-gated ion channels in the plasma membrane are also responsible for the influx of Ca2+ and the release of neurotransmitters from presynaptic neurons [111,112,113]. The mitochondrial permeability transition (MPT) pore is a voltage- and Ca2+-dependent high-conductance channel breaching the inner mitochondrial membrane [114]. The activation of MPT is characterized by loss of the mitochondrial membrane potential, swelling of the mitochondrial matrix, and outer membrane rupture, followed by the release of internalized Ca2+ and apoptogenic protein [115,116].
3.3. Calcium Channel in the Brain
Calcium channels are ionic channel, also present in the brain, the same as the voltage-gated ion channel [117]. Voltage-gated channels are voltage-dependent calcium channels found in glial cells, muscles, and neurons’ membranes. These channels have selective permeability to cations and are slightly permeable to sodium ions [118]. These channels are normally closed at resting membrane potential and get activated at depolarized membrane potential. Normally, the concentration of Ca2+ ions is seven thousand times higher outside than inside the cell and results in the activation of calcium-sensitive potassium channels, muscular contraction excitation of neurons, up-regulation of gene expression, and release of neurotransmitters [119].
Each calcium channel is made up of four or five protein subunits (α1, α2, β, γ, δ) which are encoded by different genes [33]. The α1 subunit is the larger subunit in the calcium channel, consisting of four homologous domains, and each domain contains six transmembrane segments, forming the conduction pore [120]. The segments of domains serve as the voltage sensor determining ion selectivity and channel conductance. The other subunits modulate the pharmacological properties of ion channels and provide an additional basis for the diversity of channel types [121]. The voltage-gated Ca2+ channels (VGCC) have numerous roles in cellular signal transduction and have ten members. VGCCs are the transducers of electrical excitability with different physiological, pharmacological, and regulatory properties, and convert the electrical signal of action potential in the cell surface membrane to an intracellular Ca2+ transient [122,123]. The families of VGCCs are Cav1, Cav2, and Cav3. The Cav1 subfamily has four L-type calcium channel members (LTCC) from Cav1.1 to Cav1.4. This subfamily initiates contraction, secretion, gene expression regulation, synaptic input integration in neurons, and synaptic transmission in specialized sensory cells. The members of the Cav1 subfamily are sensitive to the low nanomolar concentration of dihydropyridines (DHP) [124]. Cav1.2 comprises 90% of all LTCCs in the CNS. The second subfamily of VGCCs is Cav2 and has three members, from Cav2.1 to Cav2.3, mediating VGCCs’ currents and located presynaptically. These help in the fast release of neurotransmitters at synapses. The third subfamily of VGCCs is Cav3 and these are low voltage channels. This subfamily involves three members from Cav3.1 to Cav3.3, composing the family of T type calcium channels [125]. This subfamily is essential for continuous firing of action potential in cardiac myocytes and thalamic neurons. The T type calcium channels get activated and inactivated at negative potential than Cav1 and Cav2 subfamily of VGCCS [126,127].
In SN DAergic neurons, the L-type calcium channels involve the family Cav1.3 α1subunits and activate at 10–20 mV, having more negative potential than the Cav1.2 channel [38]. These subfamilies Cav1.2 and Cav1.3 are present in the same cells, such as in postsynaptic neurons at somatodendritic locations, sinoatrial nodes, atrial cardiomyocytes, and adrenal chromaffin cells. They are widely expressed in vascular smooth muscle and heart muscle, and calcium channel blockers are used in cardiovascular diseases. These families regulate neuronal excitability and raise intracellular free Ca2+ concentration for many Ca2+-dependent signaling pathways [128,129].
3.4. Role of Calcium Channels in Parkinson’s Disease
The calcium ion plays a vital role in the healthy and diseased state of the brain. Normally, calcium ions trigger the signaling pathways essential for memory, but in excess amounts, the calcium is thought to cause damage to the brain. Calcium is an intracellular messenger which activates cell functions. In the absence of calcium, the central nervous systems (CNSs) have no outputs [130]. The L-type calcium channels (LTCCs) are also responsible for the calcium influx. These channels are expressed in the neurons of SNPc that degenerate in PD due to excitotoxicity [131]. During autonomous pacemaking or bursting in these cells, they contribute to somatodendritic Ca2+ oscillations [132]. Currently, it is well known that the Ca2+ overload causes excitotoxicity that is responsible for the neurodegeneration of dopaminergic neurons in PD by enhancing mitochondrial OS and multiple system atrophy [133].
There is growing evidence that disturbance of intracellular calcium homeostasis plays a significant role in the aetiology of PD [134]. The calcium pathway is related to mitochondrial function and oxidative stress, both of which are important in the aetiology of PD. It has recently been discovered that calcium regulation also interacts with endoplasmic reticulum function and the unfolded protein response [135]. The lysosome is increasingly recognized as a crucial component in calcium homeostasis, and lysosomal dysfunction is thought to have a role in PD [136]. Dopamine metabolism will worsen the calcium-mediated increase in oxidative stress, making substantia nigra pars compacta neurons more prone to injury. There are currently no effective or selective CaV1.3 channel inhibitors; however, modified pyrimidine-2,4,6-triones have recently been found as possible candidates in high-throughput screens [137]. Another possible neuroprotective target in PD is the plasma membrane CAV-1 L-type calcium channel [138]. Significant increased intracellular Ca2+ in susceptible to damage dopaminergic neurons throughactivation of CAV-1 L-type Ca2+ channels during autonomous pacemaking [139]. Several epidemiologic studies have found that people on dihydropyridine calcium channel blockers had a lower incidence of PD when compared to other antihypertensive medications [140].
Several neurological disorders involve the mechanism of cell death caused by the continuous action of NMDA receptors resulting in the excessive influx of calcium. Excess calcium leads to cytotoxicity and cell death by activating calcium-dependent protease, then producing eicosanoids that result in inflammation and free radicals, which causes tissue damage [141]. At Northwestern University in Chicago, Jaime Guzman and colleagues compared two brain areas of mice (the pacemaking SN and a neighboring area having no pacemaking activity) and reported the effect of calcium activity. They reported that the influx of calcium in SN causes OS and generates free radicals that damage proteins, DNA. The OS is known to a common process involved in the pathogenesis of PD [142].
In PD, the most characterized symptoms are dyskinesia, rigidity, and tremor. Among these, dyskinesia is characterized by an excess of motor activity which involves involuntary movements like tremors and writhing. The deposition of excess calcium in the brain’s basal ganglia and cerebellum regions results in involuntary movements [143]. James Surmeier said that calcium channels normally participate in the pacemaking activity but that they are not essential since other ion channels can pick up the slack. It is also reported that the absence of DJ-1 gene causes early-onset PD, and has a high level of damage to dopaminergic neurons of SN. Therefore, it may be suggested that treating PD in mice with a calcium channel blocker (CCB) prevented cell damage without hindering pacemaking activity [144]. In addition, the treatment with CCBs prevents the damage to dopaminergic neurons resistant to OS.
Depolarization of the membrane and increase of cytosolic calcium ions (Ca2+) activate large-conductance calcium and voltage-activated potassium channels (BKCa). BKCa channel activation requires Ca2+ concentrations that are generally seen near Ca2+ sources under normal physiological circumstances [145]. KCa channels affinity-purified from rat brain are assembled into macromolecular complexes with the voltage-gated calcium channels L-type, P/Q-type, and N-type. Heterologously expressed BKCa-Cav complexes reassemble a functional Ca2+ nanodomain in which Ca2+ influx via the Cav channel activates BKCa in the physiological voltage range with sub millisecond kinetics [146]. The formation of complexes with different Cav channels allows for BKCa-mediated membrane hyperpolarization, which regulates neuronal firing patterns and the release of hormones and transmitters in the central nervous system [147].
3.5. Treatment of PD with the Help of Calcium Channel Blockers (CCBs)
Christoph Meier at University Hospital Basel in Switzerland showed that calcium channel blocker hypertensive drugs reduce the risk of PD [148,149]. The calcium channel blocker hypertensive drugs include diltiazem, verapamil, and nifedipine, which inhibit Ca2+ currents through voltage-gated Ca2+ channels in arterial smooth muscles and cardiac myocytes, which exert vasorelaxant and cardio depressant actions. Surmeier said that using CCBs such as isradipine prevents the loss of dopamine by blocking the process of excitotoxicity [46].
Isradipine is in phase II clinical trial, used for people with early-stage PD, and Surmeier is planning to examine more active and effective drugs [14]. Nimodipine, a drug of the dihydropyridine (DHP) class of CCBs, possesses a neuroprotective effect after subarachnoid hemorrhage by preventing vasospasms [150,151]. DHP is a voltage-dependent blocker and binds to inactivated channel state with higher affinity [152].
LTCCs have been identified as the primary voltage-gated Ca2+ channel subtype causing stressed Ca2+ oscillations and hence a significant cause of SN DA neuronal cell death in recent years [153,154]. Epidemiological research indicated that brain-permeable DHP LTCC inhibitors (antihypertensives) lowered the incidence of PD [155]. Dihydropyridine (DHP) has been thoroughly investigated in preclinical PD models and demonstrated potentially beneficial protective effects [156]. Recently, the divergent results of DHP therapy in toxin-based PD animal models have been extensively explored, but no definite explanation has been found. In summary, eight out of thirteen reports of DHPs dramatically decreased mitochondrial-targeting toxin-induced SN DA cell death in mice, rats, and primates. However, the experimental designs of the investigations differed, making it difficult to draw a broad conclusion [157]. The differences included the PD model utilized (6-OHDA and MPTP), the animals (species, strain, age), the treatment regimen (DHP, treatment initiation, route of administration, and dosage interval), the readout (approach and technique), and plasma concentrations. Several factors may have contributed to the protective benefits of DHP, which may need an earlier therapy initiation since the onset of PD symptoms [158].
4. Medicinal Plants as Calcium Channel Blockers
The side effects of synthetic anti-hypertensive drugs have made researchers search for safer therapies to resolve hypertension. The preference for herbal alternatives to traditional, synthetic alternatives arises because herbal medications are both safer and less expensive than synthetic ones [159]. Furthermore, therapeutic herbs are more compatible with the human body. Medicinal plants provide a multitude of phytoconstituents that act on the numerous pharmacological targets implicated in hypertension. These plants can be used in the form of infusions, decoctions, and fresh fruits, or can be eaten raw [160]. Several medicinal plants with phytoconstituents that function as calcium channel blockers have been described in this review for the treatment of hypertension and are mentioned in Table 1.

Table 1.
Medicinal plants and their botanical names, chemical constituents responsible for activity, and the type of extracts used.
5. Conclusions and Future Perspectives
PD is a well-known neurodegenerative disorder characterized by motor and non-motor symptoms. The degeneration of neurons is due to alpha-synuclein accumulation and aggregation. Levodopa is a well-known drug for the symptomatic relief of PD, but it cannot cure the disease. Additionally, it cannot control the influx of calcium ions, causing excitotoxicity. Therefore, there is the need to study calcium channel inhibitors, that inhibits the influx of calcium ions, and preventing the neuron from degenerating. This emphasizes the need for effective, cheap, safe, and widely available Ca2+ channel blockers, a potential mechanism-based therapeutic approach for PD that merits further investigation in clinical trials. We also found a robust inverse relationship between continuous dihydropyridine calcium channel blocker exposure and incident PD, particularly in older individuals. Dihydropyridine calcium channel blocker usage was strongly related to lower mortality and increased life expectancy in PD patients globally. This emphasizes the significance of effective, safe, inexpensive, and widely available pharmaceuticals. Dihydropyridine calcium channel blockers are a promising therapy strategy for PD that should be investigated further in randomized trials. In addition, hypotension is a prevalent symptom that appears preclinically in many PD patients; further research and a more thorough knowledge of their biological and possible neuroprotective significance are required before evaluating anti-hypertensive medicines for therapeutic usage in PD. According to this study, people with newly diagnosed hypertension may experience a lower risk of Parkinson’s disease (PD) when receiving therapy with CCBs. When evaluating the risk of PD, clinicians may think about using CCBs to treat hypertension if they are not contraindicated. Further clinical studies concentrating on CCB medication are required to confirm its proposed neuroprotective effect in PD.
Literature Sources
The literature survey includes both clinical as well as preclinical studies that were taken from various sources like Google Scholar, Pubmed, ResearchGate, Scopus, and Web of Science.
Author Contributions
S.S. and K.R. worked on the conception, design and framing of the review, and both supervised, analyzed, and finalized the manuscript; M.R.A. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Author Contributions
S.S. and K.R. worked on the conception, design and framing of the review, and both supervised, analyzed, and finalized the manuscript; M.R.A. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare that there is no conflict of interest.
Abbreviations
| PD | Parkinson’s disease |
| SNpc | Substantia nigra pars compacta |
| ROS | Reactive oxygen species |
| LBs | Lewy bodies |
| AD | Alzheimer’s disease |
| VGCC | Voltage-gated Ca2+ channels |
| DHP | Dihydropyridines |
| CNS | Central Nervous System |
| LTCC | L-type calcium channels |
References
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef]
- Nazir, M.; Al-Ansari, A.; Al-Khalifa, K.; Alhareky, M.; Gaffar, B.; Almas, K. Global prevalence of periodontal disease and lack of its surveillance. Sci. World J. 2020, 2020, 2146160. [Google Scholar] [CrossRef] [PubMed]
- DeMaagd, G.; Philip, A. Parkinson’s disease and its management: Part 3: Nondopaminergic and nonpharmacological treatment options. Pharm. Ther. 2015, 40, 668. [Google Scholar]
- Cammisuli, D.M.; Cammisuli, S.M.; Fusi, J.; Franzoni, F.; Pruneti, C. Parkinson’s disease–mild cognitive impairment (PD-MCI): A useful summary of update knowledge. Front. Aging Neurosci. 2019, 11, 303. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, N.S.; Rodnitzky, R.L.; Uc, E.Y. Prefrontal dopamine signaling and cognitive symptoms of Parkinson’s disease. Rev. Neurosci. 2013, 24, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Conn, K.J.; Gao, W.; McKee, A.; Lan, M.S.; Ullman, M.D.; Eisenhauer, P.B.; Fine, R.E.; Wells, J.M. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004, 1022, 164–172. [Google Scholar] [CrossRef]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of α-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Kuusisto, E.; Parkkinen, L.; Alafuzoff, I. Morphogenesis of Lewy bodies: Dissimilar incorporation of α-synuclein, ubiquitin, and p62. J. Neuropathol. Exp. Neurol. 2003, 62, 1241–1253. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K. Potential pathways of abnormal tau and α-synuclein dissemination in sporadic Alzheimer’s and Parkinson’s diseases. Cold Spring Harb. Perspect. Biol. 2016, 8, a023630. [Google Scholar] [CrossRef]
- Taipa, R.; Pereira, C.; Reis, I.; Alonso, I.; Bastos-Lima, A.; Melo-Pires, M.; Magalhães, M. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain 2016, 139, 1680–1687. [Google Scholar] [CrossRef]
- MacMahon Copas, A.N.; McComish, S.F.; Fletcher, J.M.; Caldwell, M.A. The pathogenesis of Parkinson’s disease: A complex interplay between astrocytes, microglia, and T lymphocytes? Front. Neurol. 2021, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, T.; Liang, M.; Xie, J.; Song, N. Astrocyte dysfunction in Parkinson’s disease: From the perspectives of transmitted α-synuclein and genetic modulation. Transl. Neurodegener. 2021, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Khasnavis, S.; Pahan, K. Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neuroimmune Pharmacol. 2014, 9, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Heilman, P.L.; Wang, E.W.; Lewis, M.M.; Krzyzanowski, S.; Capan, C.D.; Burmeister, A.R.; Du, G.; Escobar Galvis, M.L.; Brundin, P.; Huang, X.; et al. Tryptophan metabolites are associated with symptoms and nigral pathology in parkinson’s disease. Mov. Disord. 2020, 35, 2028–2037. [Google Scholar] [CrossRef]
- Mani, S.; Sevanan, M.; Krishnamoorthy, A.; Sekar, S. A systematic review of molecular approaches that link mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurol. Sci. 2021, 42, 4459–4469. [Google Scholar] [CrossRef] [PubMed]
- Duarte-Jurado, A.P.; Gopar-Cuevas, Y.; Saucedo-Cardenas, O.; Loera-Arias, M.D.; Montes-de-Oca-Luna, R.; Garcia-Garcia, A.; Rodriguez-Rocha, H. Antioxidant therapeutics in Parkinson’s disease: Current challenges and opportunities. Antioxidants 2021, 10, 453. [Google Scholar] [CrossRef]
- Harsanyiova, J.; Buday, T.; Kralova Trancikova, A. Parkinson’s disease and the gut: Future perspectives for early diagnosis. Front. Neurosci. 2020, 14, 626. [Google Scholar] [CrossRef]
- Raj, K.; Kaur, P.; Gupta, G.D.; Singh, S. Metals associated neurodegeneration in Parkinson’s disease: Insight to physiological, pathological mechanisms and management. Neurosci. Lett. 2021, 753, 135873. [Google Scholar] [CrossRef]
- Andrade, V.M.; Aschner, M.; Dos Santos, A.P.M. Neurotoxicity of metal mixtures. In Neurotoxicity of Metals; Springer: Berlin/Heidelberg, Germany, 2017; pp. 227–265. [Google Scholar]
- Engwa, G.A.; Ferdinand, P.U.; Nwalo, F.N.; Unachukwu, M.N. Mechanism and health effects of heavy metal toxicity in humans. Poisoning Mod. World New Tricks Old Dog 2019, 10, 70–90. [Google Scholar]
- Pinto, E.; Sigaud-kutner, T.C.; Leitao, M.A.; Okamoto, O.K.; Morse, D.; Colepicolo, P. Heavy metal–induced oxidative stress in algae 1. J. Phycol. 2003, 39, 1008–1018. [Google Scholar] [CrossRef]
- Srivastava, S.; Singh, D.; Patel, S.; Singh, M.R. Role of enzymatic free radical scavengers in management of oxidative stress in autoimmune disorders. Int. J. Biol. Macromol. 2017, 101, 502–517. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Basic mechanisms of neurodegeneration: A critical update. J. Cell. Mol. Med. 2010, 14, 457–487. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Xi, S. The effects of heavy metals on human metabolism. Toxicol. Mech. Methods 2020, 30, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Li, Y.; Shi, L.; Hussain, R.; Mehmood, K.; Tang, Z.; Zhang, H. Heavy metals induced mitochondrial dysfunction in animals: Molecular mechanism of toxicity. Toxicology 2022, 21, 153136. [Google Scholar] [CrossRef]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797. [Google Scholar] [CrossRef] [PubMed]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Burns, R.S.; LeWitt, P.A.; Ebert, M.H.; Pakkenberg, H.; Kopin, I.J. The clinical syndrome of striatal dopamine deficiency. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6tetrahydropyridine (MPTP). N. Engl. J. Med. 1985, 312, 1418–1421. [Google Scholar] [CrossRef]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139, 216–231. [Google Scholar] [CrossRef]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol. 2012, 2012, 845618. [Google Scholar] [CrossRef]
- Jiang, P.; Dickson, D.W. Parkinson’s disease: Experimental models and reality. Acta Neuropathol. 2018, 135, 13–32. [Google Scholar] [CrossRef]
- Rai, S.N.; Birla, H.; Singh, S.S.; Zahra, W.; Patil, R.R.; Jadhav, J.P.; Gedda, M.R.; Singh, S.P. Mucuna pruriens protects against MPTP intoxicated neuroinflammation in Parkinson’s disease through NF-κB/pAKT signaling pathways. Front. Aging Neurosci. 2017, 9, 421. [Google Scholar] [CrossRef] [PubMed]
- Fubini, B.; Hubbard, A. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) generation by silica in inflammation and fibrosis. Free. Radic. Biol. Med. 2003, 34, 1507–1516. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Taso, O.V.; Philippou, A.; Moustogiannis, A.; Zevolis, E.; Koutsilieris, M. Lipid peroxidation products and their role in neurodegenerative diseases. Ann. Res. Hosp. 2019, 3. [Google Scholar] [CrossRef]
- Rai, S.N.; Chaturvedi, V.K.; Singh, P.; Singh, B.K.; Singh, M.P. Mucuna pruriens in Parkinson’s and in some other diseases: Recent advancement and future prospective. 3 Biotech. 2020, 10, 522. [Google Scholar] [CrossRef] [PubMed]
- Benhammou, N.; Bekkara, F.A.; Panovska, T.K. Antioxidant activity of methanolic extracts and some bioactive compounds of Atriplex halimus. Comptes Rendus Chim. 2009, 12, 1259–1266. [Google Scholar] [CrossRef]
- Rachsee, A.; Chiranthanut, N.; Kunnaja, P.; Sireeratawong, S.; Khonsung, P.; Chansakaow, S.; Panthong, A. Mucuna pruriens (L.) DC. seed extract inhibits lipopolysaccharide-induced inflammatory responses in BV2 microglial cells. J. Ethnopharmacol. 2021, 267, 113518–113531. [Google Scholar] [CrossRef]
- Török, N.; Tanaka, M.; Vécsei, L. Searching for peripheral biomarkers in neurodegenerative diseases: The tryptophan-kynurenine metabolic pathway. Int. J. Mol. Sci. 2020, 21, 9338. [Google Scholar] [CrossRef]
- González-Sanmiguel, J.; Schuh, C.M.; Muñoz-Montesino, C.; Contreras-Kallens, P.; Aguayo, L.G.; Aguayo, S. Complex Interaction between resident microbiota and misfolded proteins: Role in neuroinflammation and neurodegeneration. Cells 2020, 9, 2476. [Google Scholar] [CrossRef]
- Rai, S.N.; Zahra, W.; Singh, S.S.; Birla, H.; Keswani, C.; Dilnashin, H.; Rathore, A.S.; Singh, R.; Singh, R.K.; Singh, S.P. Anti-inflammatory activity of ursolic acid in MPTP-induced parkinsonian mouse model. Neurotox. Res. 2019, 36, 452–462. [Google Scholar] [CrossRef]
- Ikeda, Y.; Murakami, A.; Ohigashi, H. Ursolic acid An anti-and pro-inflammatory triterpenoid. Mol. Nutr. Food Res. 2008, 52, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Checker, R.; Sandur, S.K.; Sharma, D.; Patwardhan, R.S.; Jayakumar, S.; Kohli, V.; Sethi, G.; Aggarwal, B.B.; Sainis, K.B. Potent anti-inflammatory activity of ursolic acid, a triterpenoid antioxidant, is mediated through suppression of NF-κB, AP-1 and NF-AT. PLoS ONE 2012, 7, 31318–31334. [Google Scholar] [CrossRef] [PubMed]
- Ayelign, A.; Sabally, K. Determination of chlorogenic acids (CGA) in coffee beans using HPLC. Am. J. Res. Commun. 2013, 1, 78–91. [Google Scholar]
- Clifford, M.N. Chlorogenic acids and other cinnamates–nature, occurrence, dietary burden, absorption and metabolism. J. Sci. Food Agric. 2000, 80, 1033–1043. [Google Scholar] [CrossRef]
- Kuhnert, N.; Karaköse, H.; Jaiswal, R. Analysis of chlorogenic acids and other hydroxycinnamates in food, plants and pharmacokinetic studies. In Handbook of Analysis of Active Compounds in Functional Foods; CRC Press: Boca Raton, FL, USA, 2012; pp. 1–52. [Google Scholar]
- Jantas, D.; Chwastek, J.; Malarz, J.; Stojakowska, A.; Lasoń, W. Neuroprotective effects of methyl caffeate against hydrogen peroxide-induced cell damage: Involvement of caspase 3 and cathepsin D inhibition. Biomolecules 2020, 10, 1530. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Kumar, G.; Gedda, M.R.; Tiwari, N.; Patnaik, R.; Singh, R.K.; Singh, S.P. Effect of chlorogenic acid supplementation in MPTP-intoxicated mouse. Front. Pharmacol. 2018, 9, 757. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Singh, S.P. NF-κB-mediated neuroinflammation in Parkinson’s disease and potential therapeutic effect of polyphenols. Neurotox. Res. 2020, 37, 491–507. [Google Scholar] [CrossRef]
- Giordano, R.; Saii, Z.; Fredsgaard, M.; Hulkko, L.S.; Poulsen, T.B.; Thomsen, M.E.; Henneberg, N.; Zucolotto, S.M.; Arendt-Nielsen, L.; Papenbrock, J.; et al. Pharmacological insights into halophyte bioactive extract action on anti-inflammatory, pain relief and antibiotics-type mechanisms. Molecules 2021, 26, 3140. [Google Scholar] [CrossRef]
- Biundo, R.; Weis, L.; Antonini, A. Cognitive decline in Parkinson’s disease: The complex picture. NPJ Park. Dis. 2016, 2, 16018. [Google Scholar] [CrossRef]
- Engelhardt, J.F. Redox-mediated gene therapies for environmental injury: Approaches and concepts. Antioxid Redox Signal 1999, 1, 5–27. [Google Scholar] [CrossRef]
- Swart, T.; Hurley, M.J. Calcium channel antagonists as disease-modifying therapy for Parkinson’s disease: Therapeutic rationale and current status. CNS Drugs 2016, 30, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Cooper, G.; Dunne, S.F.; Dusel, B.; Luan, C.H.; Surmeier, D.J.; Silverman, R.B. CaV1. 3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson’s disease. Nat. Commun. 2012, 3, 1146. [Google Scholar] [CrossRef] [PubMed]
- Phillips, W.J.; Currier, B.L. Analgesic pharmacology: II. Specific analgesics. JAAOS J. Am. Acad. Orthop. Surg. 2004, 12, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Et Biophys. Acta Mol Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef]
- Van Aken, O.; Van Breusegem, F. Licensed to kill: Mitochondria, chloroplasts, and cell death. Trends Plant Sci. 2015, 20, 754–766. [Google Scholar] [CrossRef]
- Azzam, E.I.; Jay-Gerin, J.P.; Pain, D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012, 327, 48–60. [Google Scholar] [CrossRef]
- Valenti, D.; de Bari, L.; De Filippis, B.; Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef]
- Lesage, S.; Brice, A. Role of Mendelian genes in “sporadic” Parkinson’s disease. Park. Relat. Disord. 2012, 18, S66–S70. [Google Scholar] [CrossRef]
- Hernandez, D.G.; Reed, X.; Singleton, A.B. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J. Neurochem. 2016, 139, 59–74. [Google Scholar] [CrossRef]
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Taylor, G.; Sherer, T.; Greenamyre, J.T. Rotenone model of Parkinson disease: Multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J. Biol. Chem. 2005, 280, 42026–42035. [Google Scholar] [CrossRef] [PubMed]
- Borland, M.K.; Trimmer, P.A.; Rubinstein, J.D.; Keeney, P.M.; Mohanakumar, K.; Liu, L.; Bennett, J.P. Chronic, lowdose rotenone reproduces Lewy neurites found in early stages of Parkinson’s disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol. Neurodegener. 2008, 3, 21. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Li, S.; Rodriguez-Rocha, H.; Burns, M.; Panayiotidis, M.I. Molecular mechanisms of pesticide-induced neurotoxicity: Relevance to Parkinson’s disease. Chem. Biol. Interact. 2010, 188, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Prakash, J.; Yadav, S.K.; Chouhan, S.; Singh, S.P. Neuroprotective role of Withania somnifera root extract in Maneb–Paraquat induced mouse model of parkinsonism. Neurochem. Res. 2013, 38, 972–980. [Google Scholar] [CrossRef] [PubMed]
- Dinis-Oliveira, R.J.; Remiao, F.; Carmo, H.; Duarte, J.A.; Navarro, A.S.; Bastos, M.L.; Carvalho, F. Paraquat exposure as an etiological factor of Parkinson’s disease. Neurotoxicology 2006, 27, 1110–1122. [Google Scholar] [CrossRef] [PubMed]
- Colle, D.; Farina, M. Oxidative stress in paraquat-induced damage to nervous tissues. In Toxicology; Academic Press: Cambridge, MA, USA, 2021; pp. 69–78. [Google Scholar]
- Gupta, S.P.; Patel, S.; Yadav, S.; Singh, A.K.; Singh, S.; Singh, M.P. Involvement of nitric oxide in maneb-and paraquat-induced Parkinson’s disease phenotype in mouse: Is there any link with lipid peroxidation? Neurochem. Res. 2010, 35, 1206–1213. [Google Scholar] [CrossRef]
- Ahmad, I.; Kumar, A.; Shukla, S.; Prasad Pandey, H.; Singh, C. The involvement of nitric oxide in maneb-and paraquat-induced oxidative stress in rat polymorphonuclear leukocytes. Free. Radic. Res. 2008, 42, 849–862. [Google Scholar] [CrossRef]
- Sharma, V.; Sharma, S.; Pracheta, R.P. Withania somnifera: A rejuvenating ayurvedic medicinal herb for the treatment. Int. J. Pharm. Tech. Res. 2011, 3, 187–192. [Google Scholar]
- Singh, N.; Bhalla, M.; de Jager, P.; Gilca, M. An overview on ashwagandha: A Rasayana (rejuvenator) of Ayurveda. Afr. J. Tradit. Complement. Altern. Med. 2011, 8, 208–213. [Google Scholar] [CrossRef]
- Vegh, C.; Wear, D.; Okaj, I.; Huggard, R.; Culmone, L.; Eren, S.; Cohen, J.; Rishi, A.K.; Pandey, S. Combined Ubisol-Q10 and Ashwagandha Root Extract Target Multiple Biochemical Mechanisms and Reduces Neurodegeneration in a Paraquat-Induced Rat Model of Parkinson’s Disease. Antioxidants 2021, 10, 563. [Google Scholar] [CrossRef]
- Gleichmann, M.; Mattson, M.P. Neuronal calcium homeostasis and dysregulation. Antioxid. Redox Signal 2011, 14, 1261–1273. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol. 2008, 7, 97–109. [Google Scholar] [CrossRef]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [PubMed]
- Kristián, T. Metabolic stages, mitochondria and calcium in hypoxic/ischemic brain damage. Cell Calcium 2004, 36, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Contributions of mitochondria to animal physiology: From homeostatic sensor to calcium signalling and cell death. J. Physiol. 1999, 516, 1–7. [Google Scholar] [CrossRef]
- Halestrap, A.P.; Pasdois, P. The role of the mitochondrial permeability transition pore in heart disease. Biochim. Et Biophys. Acta Bioenerg. 2009, 1787, 1402–1415. [Google Scholar] [CrossRef] [PubMed]
- Duchen, M.R. Roles of mitochondria in health and disease. Diabetes 2004, 53, S96–S102. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Gamal El-Din, T.M.; Payandeh, J.; Martinez, G.Q.; Heard, T.M.; Scheuer, T.; Zheng, N.; Catterall, W.A. Structural basis for Ca2+ selectivity of a voltage-gated calcium channel. Nature 2014, 505, 56–61. [Google Scholar] [CrossRef]
- Vafai, S.B.; Mootha, V.K. Mitochondrial disorders as windows into an ancient organelle. Nature 2012, 491, 374–383. [Google Scholar] [CrossRef]
- Pivovarova, N.B.; Andrews, S.B. Calcium-dependent mitochondrial function and dysfunction in neurons. FEBS J. 2010, 277, 3622–3636. [Google Scholar] [CrossRef]
- Moon, H.E.; Paek, S.H. Mitochondrial dysfunction in Parkinson’s disease. Exp. Neurobiol. 2015, 24, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Cullen, P.J.; Lockyer, P.J. Integration of calcium and Ras signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Lizama, B.N.; Chu, C.T. Excitotoxicity, calcium and mitochondria: A triad in synaptic neurodegeneration. Transl. Neurodegener. 2022, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative stress: Major threat in traumatic brain injury. CNS Neurol. Disord. Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef]
- Meldolesi, J.; Pozzan, T. The endoplasmic reticulum Ca2+ store: A view from the lumen. Trends Biochem. Sci. 1998, 23, 10–14. [Google Scholar] [CrossRef]
- Greotti, E.; Capitanio, P.; Wong, A.; Pozzan, T.; Pizzo, P.; Pendin, D. Familial Alzheimer’s disease-linked presenilin mutants and intracellular Ca2+ handling: A single-organelle, FRET-based analysis. Cell Calcium 2019, 79, 44–56. [Google Scholar] [CrossRef]
- Babcock, D.F.; Herrington, J.; Goodwin, P.C.; Park, Y.B.; Hille, B. Mitochondrial participation in the intracellular Ca2+ network. J. Cell Biol. 1997, 136, 833–844. [Google Scholar] [CrossRef]
- Esteras, N.; Abramov, A.Y. Mitochondrial calcium deregulation in the mechanism of beta-amyloid and tau pathology. Cells 2020, 9, 2135. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Kim, S.Y.; Cecen, F.S.C.; Cho, Y.; Kwon, S.-K. Dysfunction of mitochondrial Ca2+ regulatory machineries in brain aging and neurodegenerative diseases. Front. Cell Dev. Biol. 2020, 8, 599792. [Google Scholar] [CrossRef] [PubMed]
- Schapira, A.H. Mitochondrial dysfunction in Parkinson’s disease. Cell Death Differ. 2007, 14, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- Colegrove, S.L.; Albrecht, M.A.; Friel, D.D. Quantitative analysis of mitochondrial Ca2+ uptake and release pathways in sympathetic neurons: Reconstruction of the recovery after depolarization-evoked [Ca2+] i elevations. J. Gen. Physiol. 2000, 115, 371–388. [Google Scholar] [CrossRef]
- Somlyo, A.P.; Himpens, B. Cell calcium and its regulation in smooth muscle. FASEB J. 1989, 3, 2266–2276. [Google Scholar] [CrossRef]
- Hu, Q.; Wang, G. Mitochondrial dysfunction in Parkinson’s disease. Transl. Neurodegener. 2016, 5, 1–8. [Google Scholar] [CrossRef]
- Vos, M.; Lauwers, E.; Verstreken, P. Synaptic mitochondria in synaptic transmission and organization of vesicle pools in health and disease. Front. Synaptic Neurosci. 2010, 2, 139. [Google Scholar] [CrossRef]
- Lovinger, D.M. Excitotoxicity and alcohol-related brain damage. Alcohol. Clin. Exp. Res. 1993, 17, 19–27. [Google Scholar] [CrossRef]
- Mattson, M.P. Calcium and neurodegeneration. Aging Cell 2007, 6, 337–350. [Google Scholar] [CrossRef]
- Shaikh, S.; Dubey, R.; Joshi, Y.M.; Kadam, V.J. Excitotoxicity and cell Damage-A Review. Int. J. Pharm. Sci. Res. 2013, 4, 2062. [Google Scholar]
- Rajani, V.; Sengar, A.S.; Salter, M.W. Tripartite signalling by NMDA receptors. Mol. Brain 2020, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Salkoff, L.; Butler, A.; Ferreira, G.; Santi, C.; Wei, A. High-conductance potassium channels of the SLO family. Nat. Rev. Neurosci. 2006, 7, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.X.; Wang, Y.; Qin, Z.H. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol. Sin. 2009, 30, 379–387. [Google Scholar] [CrossRef]
- Ludhiadch, A.; Sharma, R.; Muriki, A.; Munshi, A. Role of calcium homeostasis in ischemic stroke: A review. CNS Neurol. Disord. -Drug Targets 2022, 21, 52–61. [Google Scholar] [CrossRef]
- Zündorf, G.; Reiser, G. Calcium dysregulation and homeostasis of neural calcium in the molecular mechanisms of neurodegenerative diseases provide multiple targets for neuroprotection. Antioxid. Redox Signal. 2011, 14, 1275–1288. [Google Scholar] [CrossRef]
- Báthori, G.; Csordás, G.; Garcia-Perez, C.; Davies, E.; Hajnóczky, G. Ca2+-dependent control of the permeability properties of the mitochondrial outer membrane and voltage-dependent anion-selective channel (VDAC). J. Biol. Chem. 2006, 281, 17347–17358. [Google Scholar] [CrossRef]
- Lehmann-Horn, F.; Jurkat-Rott, K. Voltage-gated ion channels and hereditary disease. Physiol. Rev. 1999, 79, 1317–1372. [Google Scholar] [CrossRef]
- Ponnalagu, D.; Singh, H. Anion channels of mitochondria. In Pharmacology of Mitochondria; Springer: Berlin/Heidelberg, Germany, 2016; pp. 71–101. [Google Scholar]
- Pusch, M.; Jentsch, T.J. Molecular physiology of voltage-gated chloride channels. Physiol. Rev. 1994, 74, 813–827. [Google Scholar] [CrossRef]
- Pepe, S. Mitochondrial function in ischaemia and reperfusion of the ageing heart. Clin. Exp. Pharmacol. Physiol. 2000, 27, 745–750. [Google Scholar] [CrossRef]
- Pivovarova, N.B.; Nguyen, H.V.; Winters, C.A.; Brantner, C.A.; Smith, C.L.; Andrews, S.B. Excitotoxic calcium overload in a subpopulation of mitochondria triggers delayed death in hippocampal neurons. J. Neurosci. 2004, 24, 5611–5622. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, A.; Fiskum, G. Calcium induced release of mitochondrial cytochrome c by different mechanisms selective for brain versus liver. Cell Death Differ. 1999, 6, 825–832. [Google Scholar] [CrossRef]
- Halestrap, A.P. What is the mitochondrial permeability transition pore? J. Mol. Cell Cardiol. 2009, 46, 821–831. [Google Scholar] [CrossRef]
- Bernardi, P.; Krauskopf, A.; Basso, E.; Petronilli, V.; Blachly-Dyson, E.; Di Lisa, F.; Forte, M.A. The mitochondrial permeability transition from in vitro artifact to disease target. FEBS J. 2006, 273, 2077–2099. [Google Scholar] [CrossRef] [PubMed]
- Striggow, F.; Ehrlich, B.E. Ligand-gated calcium channels inside and out. Curr. Opin. Cell Biol. 1996, 8, 490–495. [Google Scholar] [CrossRef]
- Hall, J.E. Guyton and Hall Textbook of Medical Physiology with Student Consult Online Access, 12th ed.; Elsevier Saunders: Philadelphia, PA, USA, 2011; p. 64. ISBN 978-1-4160-4574-8. [Google Scholar]
- Wilson, D.P.; Susnjar, M.; Kiss, E.; Sutherland, C.; Walsh, M.P. Thromboxane A2-induced contraction of rat caudal arterial smooth muscle involves activation of Ca2+ entry and Ca2+ sensitization: Rho-associated kinase-mediated phosphorylation of MYPT1 at Thr-855, but not Thr-697. Biochem. J. 2005, 389, 763–774. [Google Scholar] [CrossRef]
- Perez-Reyes, E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol. Rev. 2003, 83, 117–161. [Google Scholar] [CrossRef]
- Yang, S.N.; Berggren, P.O. The role of voltage-gated calcium channels in pancreatic β-cell physiology and pathophysiology. Endocr. Rev. 2006, 27, 621–676. [Google Scholar] [CrossRef]
- Striessnig, J.; Pinggera, A.; Kaur, G.; Bock, G.; Tuluc, P. L-type Ca2+ channels in heart and brain. Wiley Interdisciplinary Reviews: Membr. Transp. Signal. 2014, 3, 15–38. [Google Scholar]
- Liss, B.; Striessnig, J. The potential of L-type calcium channels as a drug target for neuroprotective therapy in Parkinson’s disease. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 263–289. [Google Scholar] [CrossRef]
- Lee, S. Pharmacological inhibition of voltage-gated Ca2+ channels for chronic pain relief. Curr. Neuropharmacol. 2013, 11, 606–620. [Google Scholar] [PubMed]
- Striessnig, J.; Koschak, A. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene-knockout models. Channels 2008, 2, 233–251. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A. Physiology and pathophysiology of the calcium store in the endoplasmic reticulum of neurons. Physiol. Rev. 2005, 85, 201–279. [Google Scholar] [CrossRef] [PubMed]
- Locatelli, A.; Cosconati, S.; Micucci, M.; Leoni, A.; Marinelli, L.; Bedini, A.; Ioan, P.; Spampinato, S.M.; Novellino, E.; Chiarini, A.; et al. Ligand based approach to L-type calcium channel by imidazo [2, 1-b] thiazole-1, 4-dihydropyridines: From heart activity to brain affinity. J. Med. Chem. 2013, 56, 3866–3877. [Google Scholar] [CrossRef]
- Paul, S.; Mahanta, S. Association of heat-shock proteins in various neurodegenerative disorders: Is it a master key to open the therapeutic door? Mol. Cell. Biochem. 2014, 386, 45–61. [Google Scholar] [CrossRef]
- Baumgart, J.; Perez-Reyes, E. Voltage-gated calcium channels. In Ion Channels: From Structure to Function; Kew, J., Davies, C., Eds.; Oxford Press: Oxford, UK, 2010; pp. 104–122. [Google Scholar]
- Calì, T.; Ottolini, D.; Brini, M. Calcium signaling in Parkinson’s disease. Cell Tissue Res. 2014, 357, 439–454. [Google Scholar] [CrossRef]
- Jellinger, K.A. The relevance of metals in the pathophysiology of neurodegeneration, pathological considerations. In International Review of Neurobiology; Academic Press: Cambridge, MA, USA, 2013; Volume 110, pp. 1–47. [Google Scholar]
- Zaichick, S.V.; McGrath, K.M.; Caraveo, G. The role of Ca2+ signaling in Parkinson’s disease. Dis. Model. Mech. 2017, 10, 519–535. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.X.; Tanaka, L.Y.; Wosniak, J., Jr.; Laurindo, F.R. Mechanisms and implications of reactive oxygen species generation during the unfolded protein response: Roles of endoplasmic reticulum oxidoreductases, mitochondrial electron transport, and NADPH oxidase. Antioxid. Redox Signal. 2009, 11, 2409–2427. [Google Scholar] [CrossRef]
- Navarro-Romero, A.; Montpeyó, M.; Martinez-Vicente, M. The emerging role of the lysosome in Parkinson’s disease. Cells 2020, 9, 2399. [Google Scholar] [CrossRef]
- Duda, J.; Pötschke, C.; Liss, B. Converging roles of ion channels, calcium, metabolic stress, and activity pattern of Substantia nigra dopaminergic neurons in health and Parkinson’s disease. J. Neurochem. 2016, 139, 156–178. [Google Scholar] [CrossRef]
- Yagami, T.; Kohma, H.; Yamamoto, Y. L-type voltage-dependent calcium channels as therapeutic targets for neurodegenerative diseases. Curr. Med. Chem. 2012, 19, 4816–4827. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.L.; Miller, R.J. Excitatory amino acid receptors, second messengers and regulation of intracellular Ca2+ in mammalian neurons. Trends Pharmacol. Sci. 1990, 11, 254–260. [Google Scholar] [CrossRef]
- Ritz, B.; Rhodes, S.L.; Qian, L.; Schernhammer, E.; Olsen, J.H.; Friis, S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann. Neurol. 2010, 67, 600–606. [Google Scholar] [PubMed]
- Anitha, M.; Nandhu, M.S.; Anju, T.R.; Jes, P.; Paulose, C.S. Targeting glutamate mediated excitotoxicity in Huntington’s disease: Neural progenitors and partial glutamate antagonist-memantine. Med. Hypotheses. 2011, 76, 138–140. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Guzman, J.N.; Sanchez, J.; Schumacker, P.T. Physiological phenotype and vulnerability in Parkinson’s disease. Cold Spring Harbor perspectives in medicine 2012, 2, a009290. [Google Scholar] [CrossRef]
- Schuster, S.; Doudnikoff, E.; Rylander, D.; Berthet, A.; Aubert, I.; Ittrich, C.; Bloch, B.; Cenci, M.A.; Surmeier, D.J.; Hengerer, B.; et al. Antagonizing L-type Ca2+ channel reduces development of abnormal involuntary movement in the rat model of L-3,4-dihydroxyphenylalanine-induced dyskinesia. Biol. Psychiatry 2009, 65, 518–526. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Halliday, G.M.; Simuni, T. Calcium, mitochondrial dysfunction and slowing the progression of Parkinson’s disease. Exp. Neurol. 2017, 298, 202–209. [Google Scholar] [CrossRef]
- Kshatri, A.S.; Gonzalez-Hernandez, A.; Giraldez, T. Physiological roles and therapeutic potential of Ca2+ activated potassium channels in the nervous system. Front. Mol. Neurosci. 2018, 11, 258. [Google Scholar] [CrossRef]
- Berkefeld, H.; Sailer, C.A.; Bildl, W.; Rohde, V.; Thumfart, J.O.; Eble, S.; Klugbauer, N.; Reisinger, E.; Bischofberger, J.; Oliver, D.; et al. BKCa-Cav channel complexes mediate rapid and localized Ca2+-activated K+ signaling. Science 2006, 314, 615–620. [Google Scholar] [CrossRef]
- Higgins, J.J.; Hao, J.; Kosofsky, B.E.; Rajadhyaksha, A.M. Dysregulation of large-conductance Ca2+-activated K+ channel expression in nonsyndromal mental retardation due to a cereblon p. R419X mutation. Neurogenetics 2008, 9, 219–223. [Google Scholar] [CrossRef]
- Küng, C.F.; Lüscher, T.F. Different mechanisms of endothelial dysfunction with aging and hypertension in rat aorta. Hypertension 1995, 25, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Guzmán, J.N.; Sánchez-Padilla, J.; Goldberg, J.A. W s in Brain Research; Elsevier: Amsterdam, The Netherlands, 2010; Volume 183, pp. 59–77. [Google Scholar]
- Vijiaratnam, N.; Simuni, T.; Bandmann, O.; Morris, H.R.; Foltynie, T. Progress towards therapies for disease modification in Parkinson’s disease. Lancet Neurol. 2021, 20, 559–572. [Google Scholar] [CrossRef]
- Hurley, M.J.; Dexter, D.T. Voltage-gated calcium channels and Parkinson’s disease. Pharmacol. & Therapeutics 2012, 133, 324–333. [Google Scholar]
- Hunter, A.J. Calcium Antagonists: Their Role in Neuroprotection. In International Review of Neurobiology; Academic Press: Cambridge, MA, USA, 1996; Volume 40, pp. 95–108. [Google Scholar]
- Sanguinetti, M.C.; Kass, R.S. Voltage-dependent block of calcium channel current in the calf cardiac Purkinje fiber by dihydropyridine calcium channel antagonists. Circ. Res. 1984, 55, 336–348. [Google Scholar] [CrossRef]
- Stefani, A.; Spadoni, F.; Bernardi, G. Voltage-activated calcium channels: Targets of antiepileptic drug therapy? Epilepsia 1997, 38, 959–965. [Google Scholar] [CrossRef]
- Gudala, K.; Kanukula, R.; Bansal, D. Reduced risk of Parkinson’s disease in users of calcium channel blockers: A meta-analysis. Int. J. Chronic Dis. 2015, 2015, 697404. [Google Scholar] [CrossRef]
- Leandrou, E.; Emmanouilidou, E.; Vekrellis, K. Voltage-gated calcium channels and α-synuclein: Implications in Parkinson’s disease. Front. Mol. Neurosci. 2019, 12, 237. [Google Scholar] [CrossRef]
- Winklhofer, K.F.; Haass, C. Mitochondrial dysfunction in Parkinson’s disease. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Disease. 2010, 1802, 29–44. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef]
- Marjina Singh, A.; Sharma, A.; Narang, R.K.; Singh, G. Management of Hypertension with Conventional and Herbals Drugs. J. Drug Deliv. Ther. 2020, 10, 280–287. [Google Scholar] [CrossRef]
- Joshi, N.J.; Shelke, S.A. Medicinal Plants as Calcium-channel Blockers Against Hypertension. Vascular 2021, 1, 4–5. [Google Scholar]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).