
Evaluation the Electronic Properties of Glu-Ureido Template via Ab-Initio Study as Target Specific for PSMA †



Abstract
1. Introduction
2. Materials and Methods
Ab Initio Computational Calculation
3. Results and Discussion
3.1. Optimized Chemical Skeleton Parameters
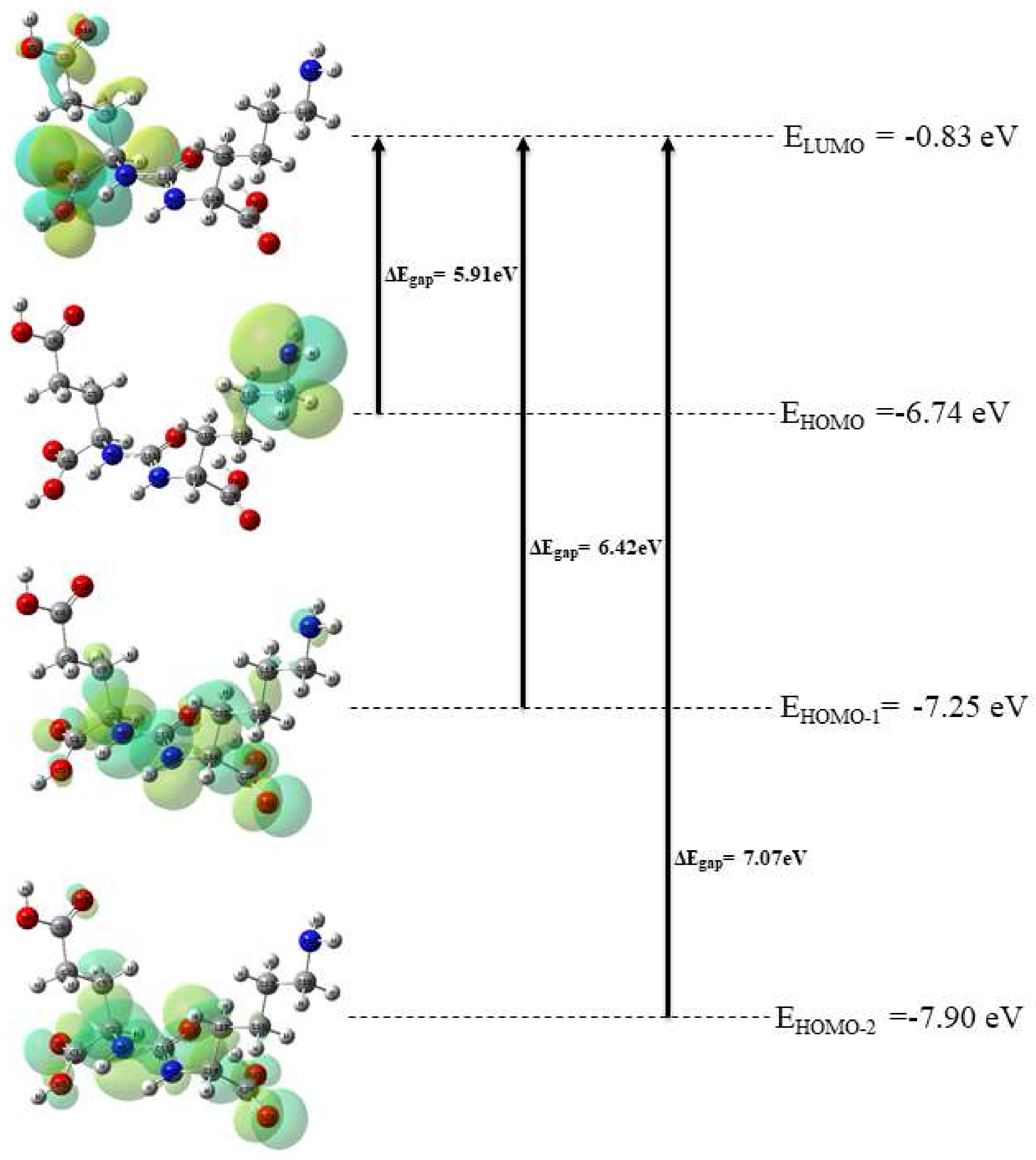
3.2. Electronic Properties
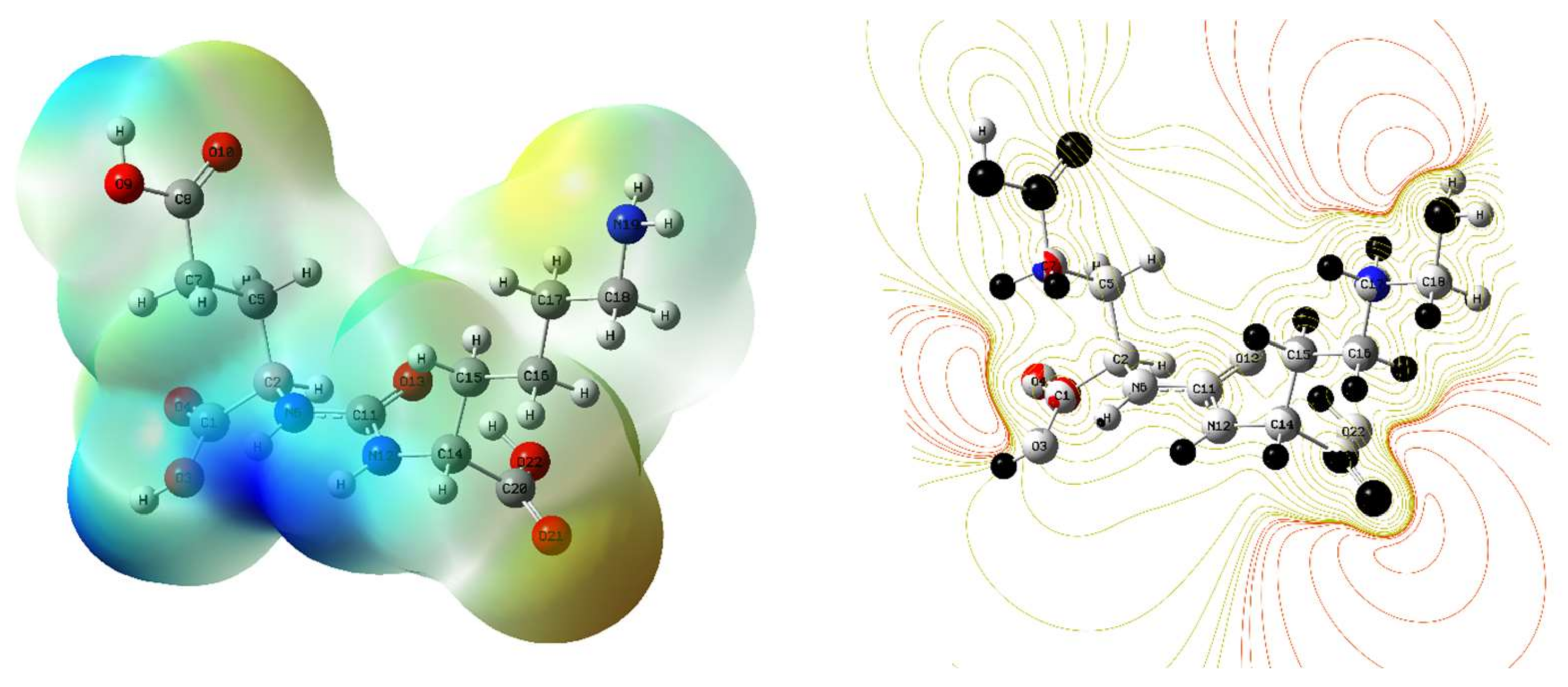
3.3. Molecular Electrostatic Potential Surface (MESP)
3.4. Global Reactivity Descriptors
3.5. Nonlinear Optical Properties
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Young, V.R.; Ajami, A.M. Glutamate: An Amino Acid of Particular Distinction. J. Nutr. 2000, 130, 892S–900S. [Google Scholar] [CrossRef] [PubMed]
- Wüstemann, T.; Bauder-Wüst, U.; Schäfer, M.; Eder, M.; Benesova, M.; Leotta, K.; Kratochwil, C.; Haberkorn, U.; Kopka, K.; Mier, W. Design of Internalizing PSMA-Specific Glu-Ureido-Based Radiotherapeuticals. Theranostics 2016, 6, 1085–1095. [Google Scholar] [CrossRef] [PubMed]
- Kopka, K.; Benešová, M.; Bařinka, C.; Haberkorn, U.; Babich, J. Glu-Ureido–Based Inhibitors of Prostate-Specific Membrane Antigen: Lessons Learned During the Development of a Novel Class of Low-Molecular-Weight Theranostic Radiotracers. J. Nucl. Med. 2017, 58 (Suppl. 2), 17S–26S. [Google Scholar] [CrossRef] [PubMed]
- Lowe, P.T.; Dall’Angelo, S.; Fleming, I.N.; Piras, M.; Zanda, M.; O’Hagan, D. Enzymatic Radiosynthesis of a 18 F-Glu-Ureido-Lys Ligand for the Prostate-Specific Membrane Antigen (PSMA). Org. Biomol. Chem. 2019, 17, 1480–1486. [Google Scholar] [CrossRef]
- Young, J.D.; Ma, M.T.; Eykyn, T.R.; Atkinson, R.A.; Abbate, V.; Cilibrizzi, A.; Hider, R.C.; Blower, P.J. Dipeptide Inhibitors of the Prostate Specific Membrane Antigen (PSMA): A Comparison of Urea and Thiourea Derivatives. Bioorg. Med. Chem. Lett. 2021, 42, 128044. [Google Scholar] [CrossRef]
- Hyväkkä, A.; Virtanen, V.; Kemppainen, J.; Grönroos, T.J.; Minn, H.; Sundvall, M. More Than Meets the Eye: Scientific Rationale behind Molecular Imaging and Therapeutic Targeting of Prostate-Specific Membrane Antigen (PSMA) in Metastatic Prostate Cancer and Beyond. Cancers 2021, 13, 2244. [Google Scholar] [CrossRef]
- Sheehan, B.; Guo, C.; Neeb, A.; Paschalis, A.; Sandhu, S.; de Bono, J.S. Prostate-Specific Membrane Antigen Biology in Lethal Prostate Cancer and Its Therapeutic Implications. Eur. Urol. Focus. 2022, 8, 1157–1168. [Google Scholar] [CrossRef]
- Nikfarjam, Z.; Bavi, O.; Amini, S.K. Potential Effective Inhibitory Compounds against Prostate Specific Membrane Antigen (PSMA): A Molecular Docking and Molecular Dynamics Study. Arch. Biochem. Biophys. 2021, 699, 108747. [Google Scholar] [CrossRef]
- Pastorino, S.; Riondato, M.; Uccelli, L.; Giovacchini, G.; Giovannini, E.; Duce, V.; Ciarmiello, A. Toward the Discovery and Development of PSMA Targeted Inhibitors for Nuclear Medicine Applications. Curr. Radiopharm. 2020, 13, 63–79. [Google Scholar] [CrossRef]
- Liolios, C.; Patsis, C.; Lambrinidis, G.; Tzortzini, E.; Roscher, M.; Bauder-Wüst, U.; Kolocouris, A.; Kopka, K. Investigation of Tumor Cells and Receptor-Ligand Simulation Models for the Development of PET Imaging Probes Targeting PSMA and GRPR and a Possible Crosstalk between the Two Receptors. Mol. Pharm. 2022, 19, 2231–2247. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An Advanced Semantic Chemical Editor, Visualization, and Analysis Platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. I. The Effect of the Exchange-Only Gradient Correction. J. Chem. Phys. 1992, 96, 2155–2160. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian: Wallingford, CT, USA, 2009; pp. 150–166. [Google Scholar]
- Becke, A.D. Density functional thermochemistry. III The role of exact exchange. J. Chem. Phys. 1993, 93, 5648. [Google Scholar] [CrossRef]
- Kleinman, D.A. Nonlinear Dielectric Polarization in Optical Media. Phys. Rev. 1962, 126, 1977–1979. [Google Scholar] [CrossRef]
- Pipek, J.; Mezey, P.G. A Fast Intrinsic Localization Procedure Applicable for ab initio and Semiempirical Linear Combination of Atomic Orbital Wave Functions. J. Chem. Phys. 1989, 90, 4916–4926. [Google Scholar] [CrossRef]
- Krishnakumar, V.; Keresztury, G.; Sundius, T.; Ramasamy, R. Simulation of IR and Raman Spectra Based on Scaled DFT Force Fields: A Case Study of 2-(Methylthio)Benzonitrile, with Emphasis on Band Assignment. J. Mol. Struct. 2004, 702, 9–21. [Google Scholar] [CrossRef]
- Gritsenko, O.V. Koopmans’ Theorem and Its Density-Functional-Theory Analog Assessed in Evaluation of the Red Shift of Vertical Ionization Potential upon Complexation. Chem. Phys. Lett. 2018, 691, 178–180. [Google Scholar] [CrossRef]
- Pearson, R.G. Absolute Electronegativity and Hardness: Applications to Organic Chemistry. J. Org. Chem. 1989, 54, 1423–1430. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.V.; Liu, S. Electrophilicity Index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Giri, S. Stability, Reactivity, and Aromaticity of Compounds of a Multivalent Superatom. J. Phys. Chem. A 2007, 111, 11116–11121. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, J.; Parthasarathi, R.; Subramanian, V.; Chattaraj, P.K. Electrophilicity-Based Charge Transfer Descriptor. J. Phys. Chem. A 2007, 111, 1358–1361. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
B.P. (K) | M.P. (K) | Critical Temp. (K) | Critical Pres. (bar) | Critical Vol (cm3/mol) | Gibbs Energy (KJ/mol) | LogP | tPSA | CLogP | LogS | pKa |
| 1157.4 | 978.4 | 1067.7 | 27.5 | 859.5 | −889.7 | −2.1 | 179.05 | −3.49 | −0.59 | 3.29, 4.30, 3.36 |
| Bond Length (A°) | Bond Angle (°) | Dihedral Angle (°) | |||
|---|---|---|---|---|---|
| Atoms Connectivity | B3LYP | Atoms Connectivity | B3LYP | Atoms Connectivity | B3LYP |
| C2C1 | 1.526 | O3C1C2 | 113.506 | O4C1C2O3 | 123.448 |
| O3C1 | 1.345 | O4C1C2 | 123.043 | C5C2C1O3 | −128.212 |
| O4C1 | 1.205 | C5C2C1 | 110.661 | N6C2C1C5 | 113.209 |
| C5C2 | 1.544 | N6C2C1 | 112.374 | C11N6C2C1 | 139.891 |
| N6C2 | 1.457 | C11N6C2 | 121.104 | C7C5C2C1 | 66.365 |
| C11N6 | 1.362 | C7C5C2 | 113.593 | C8C7C5C2 | 174.749 |
| C7C5 | 1.527 | C8C7C5 | 112.885 | O9C8C7C5 | 176.801 |
| C8C7 | 1.511 | O9C8C7 | 111.262 | O10C8C7O9 | 122.74 |
| O9C8 | 1.35 | O10C8C7 | 125.998 | N12C11N6C2 | −170.56 |
| O10C8 | 1.207 | N12C11N6 | 116.468 | O13C11N6N12 | 122.231 |
| N12C11 | 1.362 | O13C11N6 | 121.299 | C14N12C11N6 | −165.519 |
| O13C11 | 1.247 | C14N12C11 | 124.945 | C20C14N12C11 | −70.661 |
| C14N12 | 1.472 | C20C14N12 | 113.469 | C15C14N12C20 | 111.687 |
| C20C14 | 1.543 | C15C14N12 | 112.893 | C16C15C14N12 | 171.736 |
| C15C14 | 1.545 | C16C15C14 | 113.794 | C17C16C15C14 | −175.901 |
| C16C15 | 1.533 | C17C16C15 | 112.325 | C18C17C16C15 | −179.36 |
| C17C16 | 1.532 | C18C17C16 | 113.117 | N19C18C17C16 | −177.917 |
| C18C17 | 1.529 | N19C18C17 | 110.733 | O22C20C14N12 | 53.82 |
| N19C18 | 1.472 | O22C20C14 | 117.038 | O21C20C14O22 | 121.537 |
| O22C20 | 1.332 | O21C20C14 | 121.364 | H23C2C1C5 | 107.029 |
| O21C20 | 1.21 | H23C2C1 | 105.817 | H25C5C2C7 | 109.987 |
| H23C2 | 1.093 | H25C5C2 | 107.477 | H26C5C2C7 | 110.19 |
| H25C5 | 1.091 | H26C5C2 | 108.129 | H27N6C2C11 | 119.479 |
| H26C5 | 1.091 | H27N6C2 | 117.913 | H28C7C5C8 | 107.378 |
| H27N6 | 1.008 | H28C7C5 | 111.811 | H29C7C5C8 | 107.167 |
| H28C7 | 1.095 | H29C7C5 | 111.113 | H31N12C11C14 | 115.574 |
| H29C7 | 1.095 | H31N12C11 | 118.302 | H32C14N12C15 | 108.495 |
| H31N12 | 1.008 | H32C14N12 | 105.154 | H33C15C14C16 | 110.047 |
| H32C14 | 1.089 | H33C15C14 | 109.476 | H34C15C14C16 | 109.514 |
| H33C15 | 1.093 | H34C15C14 | 106.803 | H35C16C15C17 | 109.366 |
| H34C15 | 1.095 | H35C16C15 | 109.414 | H36C16C15C17 | 109.284 |
| H35C16 | 1.097 | H36C16C15 | 110.013 | H37C17C16C18 | 109.011 |
| H36C16 | 1.095 | H37C17C16 | 109.306 | H38C17C16C18 | 108.609 |
| H37C17 | 1.098 | H38C17C16 | 110.075 | H39C18C17N19 | 107.759 |
| H38C17 | 1.096 | H39C18C17 | 109.197 | H40C18C17N19 | 113.323 |
| H39C18 | 1.096 | H40C18C17 | 109.033 | H24O3C1C2 | 179.702 |
| H40C18 | 1.102 | H24O3C1 | 108.026 | H30O9C8C7 | 179.545 |
| H24O3 | 0.971 | H30O9C8 | 107.559 | H41N19C18C17 | 177.261 |
| H30O9 | 0.97 | H41N19C18 | 109.427 | H42N19C18H41 | 105.495 |
| H41N19 | 1.016 | H42N19C18 | 109.132 | H43O22C20C14 | −5.235 |
| H42N19 | 1.017 | H43O22C20 | 110.328 | --- | --- |
| SN | Electronic Transitions (Molecular Orbitals Involved) | Energy (eV) | Oscillatory Strength (f) | Calculated λmax in nm (B3LYP) |
|---|---|---|---|---|
| 01 | HOMO → LUMO | 5.91 | 0.0036 | 222.68 |
| 02 | HOMO-1 → LUMO | 6.42 | 0.0036 | 222.68 |
| 03 | HOMO-2 → LUMO | 7.07 | 0.0036 | 222.68 |
| 04 | Urea | 7.36 | -- | -- |
| 05 | 2M4NA (2-methyl-4-nitroaniline) | 6.36 | -- | -- |
| Compound | εHOMO | εLUMO | εLUMO-εHOMO | IP | EA | χ | η | μ | ω | δNmax |
|---|---|---|---|---|---|---|---|---|---|---|
| Glu-Ureido | −6.74 | −0.83 | 5.91 | 6.74 | 0.83 | 3.78 | 2.95 | −3.78 | 2.42 | 1.28 |
| Dipole Moment | Polarizability | Hyperpolarizability | |||
|---|---|---|---|---|---|
| µx | 1.290 | αxx | 224.385 | βxxx | −3.811 |
| µy | 0.674 | αyy | 1.813 | βyyy | −17.940 |
| µz | −2.083 | αzz | 187.583 | βzzz | 104.721 |
| µtot(D) | 2.54 | αxy | 1.360 | βxyy | 33.355 |
| αxz | 5.798 | βxxy | 24.754 | ||
| αyz | 166.295 | βxxz | 43.385 | ||
| α0 (esu) | 137.927 | βxzz | −33.447 | ||
| βyzz | 20.029 | ||||
| βyyz | −6.349 | ||||
| βxyz | −71.173 | ||||
| β0 (esu) | 144.328 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faheem, M.; Pandey, V.; Shrivastav, A.; Prasad, M.; Dixit, M. Evaluation the Electronic Properties of Glu-Ureido Template via Ab-Initio Study as Target Specific for PSMA. Chem. Proc. 2024, 16, 9. https://doi.org/10.3390/ecsoc-28-20204
Faheem M, Pandey V, Shrivastav A, Prasad M, Dixit M. Evaluation the Electronic Properties of Glu-Ureido Template via Ab-Initio Study as Target Specific for PSMA. Chemistry Proceedings. 2024; 16(1):9. https://doi.org/10.3390/ecsoc-28-20204
Chicago/Turabian StyleFaheem, Mohd., Vaibhav Pandey, Anjli Shrivastav, Manisha Prasad, and Manish Dixit. 2024. "Evaluation the Electronic Properties of Glu-Ureido Template via Ab-Initio Study as Target Specific for PSMA" Chemistry Proceedings 16, no. 1: 9. https://doi.org/10.3390/ecsoc-28-20204
APA StyleFaheem, M., Pandey, V., Shrivastav, A., Prasad, M., & Dixit, M. (2024). Evaluation the Electronic Properties of Glu-Ureido Template via Ab-Initio Study as Target Specific for PSMA. Chemistry Proceedings, 16(1), 9. https://doi.org/10.3390/ecsoc-28-20204