
Molecular Mobility of Different Forms of Ketoprofen Based on DFT Calculation Data †

 , , and
, , and
Abstract
1. Introduction
2. Methods
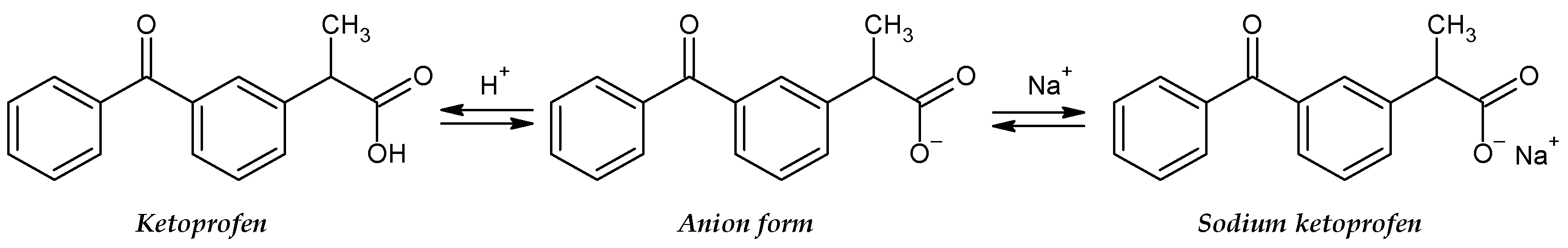
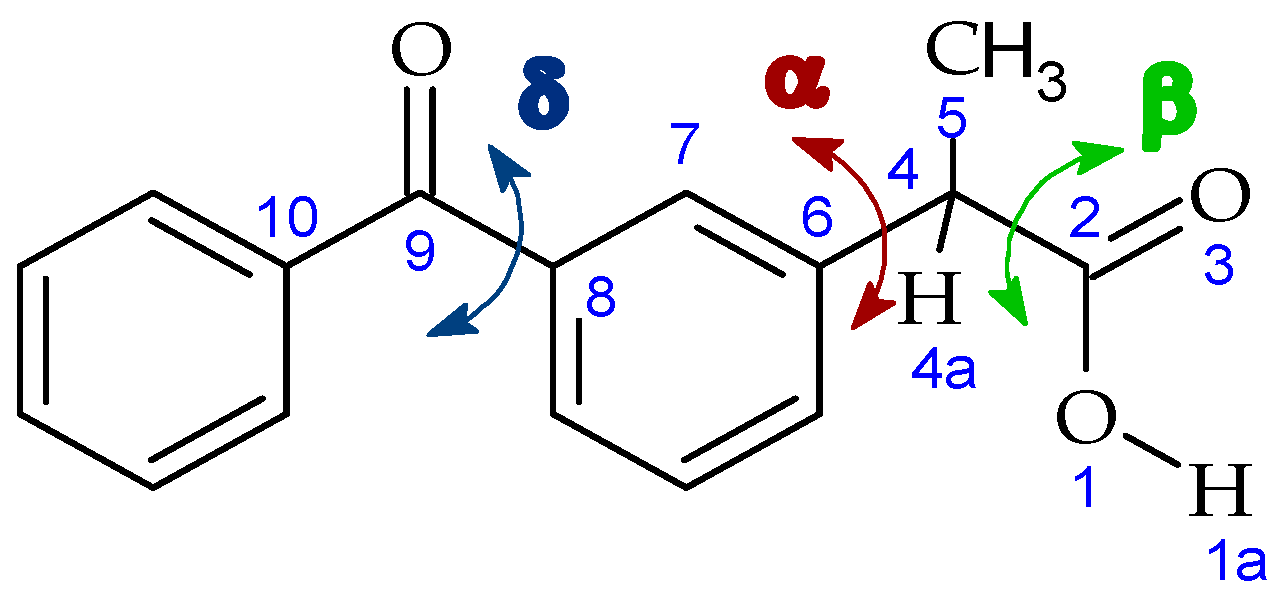
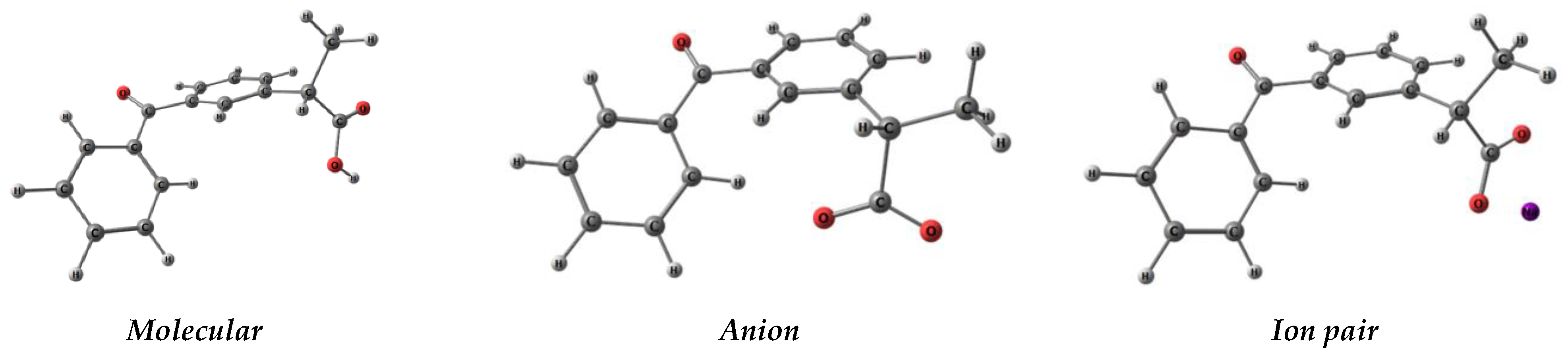
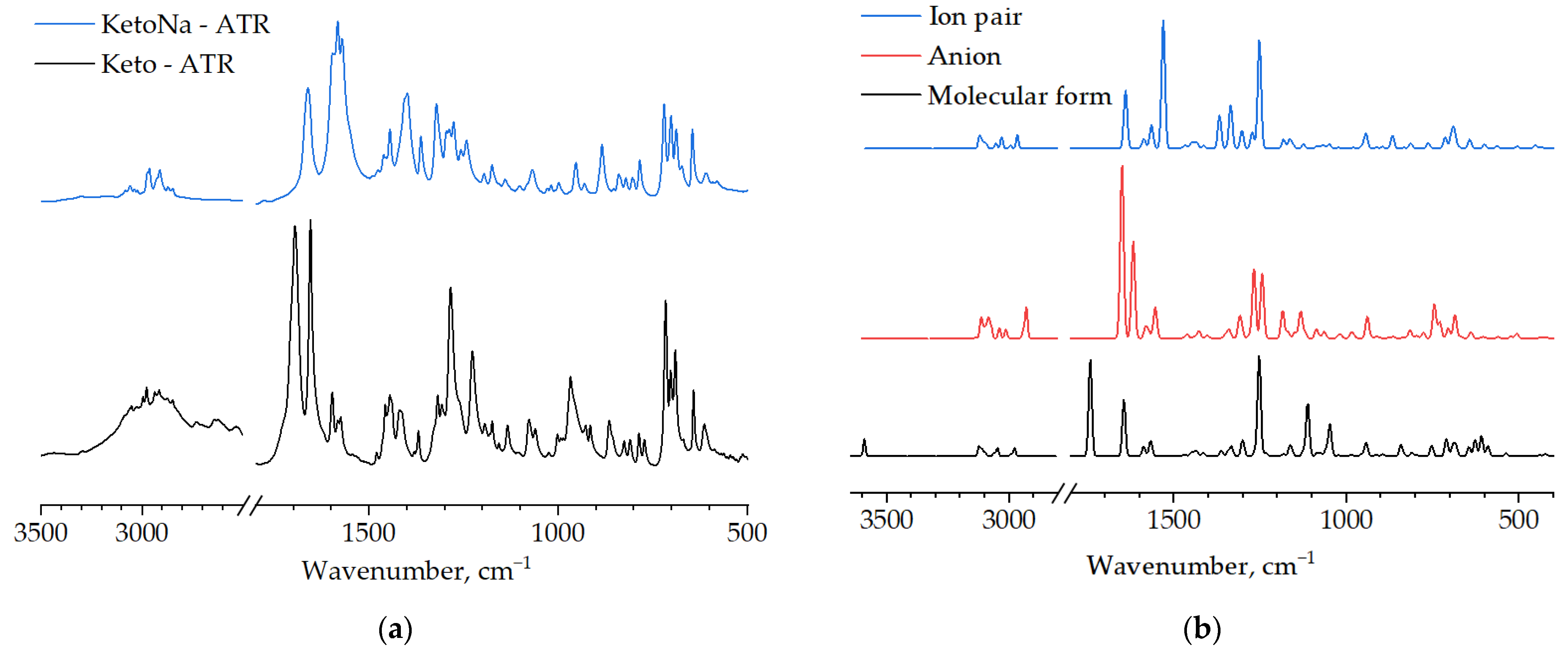
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tsoupras, A.; Gkika, D.A.; Siadimas, I.; Christodoulopoulos, I.; Efthymiopoulos, P.; Kyzas, G.Z. The Multifaceted Effects of Non-Steroidal and Non-Opioid Anti-Inflammatory and Analgesic Drugs on Platelets: Current Knowledge, Limitations, and Future Perspectives. Pharmaceuticals 2024, 17, 627. [Google Scholar] [CrossRef] [PubMed]
- Cunha, R.S.; Cruz, P.F.; Costa, T.; Almeida, Z.L.; Lima, M.E.F.d.; Serpa, C.; Chaves, O.A. Revisiting and Updating the Interaction between Human Serum Albumin and the Non-Steroidal Anti-Inflammatory Drugs Ketoprofen and Ketorolac. Molecules 2024, 29, 3001. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.O.; Al-Hetlani, E.; Lednev, I.K. Detection and identification of drug traces in latent fingermarks using Raman spectroscopy. Sci. Rep. 2022, 12, 3136. [Google Scholar] [CrossRef] [PubMed]
- Isik, I.B.; Tekin, N.; Sagdinc, S.G. The analyses of solvent effects on infrared spectra and thermodynamic parameters, Hirshfeld surface, reduced density gradient and molecular docking of ketoprofen as a member of nonsteroidal anti-inflammatory drugs. J. Molec. Struct. 2022, 1250, 131861. [Google Scholar] [CrossRef]
- Rodríguez-Ortega, P.G.; Sánchez-Valera, M.; López-González, J.J.; Montejo, M. Fourier Transform Infrared Spectroscopy and Vibrational Circular Dichroism Assisted Elucidation of the Solution-State Supramolecular Speciation in Racemic and Enantiopure Ketoprofen. Appl. Spectr. 2022, 76, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Mastova, A.V.; Selyutina, O.Y.; Polyakov, N.E. Stereoselectivity of Interaction of Nonsteroidal Anti-Inflammatory Drug S-Ketoprofen with L/D-Tryptophan in Phospholipid Membranes. Membranes 2022, 12, 460. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Wennmohs, F.; Becker, U.; Riplinger, C. The ORCA quantum chemistry program package. J. Chem. Phys. 2020, 152, 224108. [Google Scholar] [CrossRef] [PubMed]
- Aberg, G.; Ciofalo, V.B.; Pendleton, R.G.; Ray, G.; Weddle, D. Inversion of (R)- to (S)-ketoprofen in eight animal species. Chirality 1995, 7, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Zhurko, G.A. Chemcraft—Graphical Program for Visualization of Quantum Chemistry Computations. Ivanovo, Russia. 2005. Available online: https://chemcraftprog.com (accessed on 1 September 2024).
- Liu, L.; Gao, H. First principles study on the molecular structure and vibrational spectra of ketoprofen. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2012, 97, 329–339. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Junior, H.; Borges, B.A.; Barbosa, T.W.L.; Batista, A.; Braga, M.T.L.; de Araújo, M.B.; Bonfilio, R. A New Crystalline Ketoprofen Sodium Salt: Solid-State Characterization, Solubility, and Stability. J. Pharm. Sci. 2022, 111, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Briard, P.; Rossi, J.C. Ketoprofene. Acta Cryst. C 1990, 46, 1036. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | Molecular | Anion | Ion Pair |
|---|---|---|---|
| C2-O3, Å | 1.215 | 1.260 | 1.274 |
| C2-O1, Å | 1.365 | 1.250 | 1.276 |
| C2-C4, Å | 1.526 | 1.632 | 1.547 |
| C4-H4a, Å | 1.100 | 1.104 | 1.099 |
| C4-C6, Å | 1.536 | 1.526 | 1.537 |
| C4-C5, Å | 1.528 | 1.498 | 1.522 |
| O3-C2-O1, ° | 122.59 | 130.34 | 124.31 |
| O3-C2-C4, ° | 125.88 | 114.28 | 117.52 |
| α (O3-C2-C4-C6), ° | 92.2 | −54.3 | −96.3 |
| β (C2-C4-C6-C7), ° | 122.8 | 70.2 | 116.8 |
| γ (C7-C8-C9-C10), ° | −32.3 | −27.3 | −29.8 |
| μ, D | 3.07 | 7.22 | 9.19 |
| EHOMO, eV | −5.84 | −1.02 | −5.43 |
| ELUMO, eV | −2.93 | 0.06 | −2.53 |
| ΔE (LUMO-HOMO), eV | 2.91 | 1.08 | 2.90 |
| Q (O1), e | −0.343 | −0.421 | −0.543 |
| Q (O3), e | −0.286 | −0.454 | −0.524 |
| Δq (O1-O3), e | 0.057 | 0.033 | 0.019 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Logacheva, K.A.; Belushkin, A.V.; Gergelezhiu, P.A.; Eresko, A.B.; Malakhov, S.N.; Raksha, E.V.; Savostina, L.I.; Chudoba, D.M. Molecular Mobility of Different Forms of Ketoprofen Based on DFT Calculation Data. Chem. Proc. 2024, 16, 60. https://doi.org/10.3390/ecsoc-28-20162
Logacheva KA, Belushkin AV, Gergelezhiu PA, Eresko AB, Malakhov SN, Raksha EV, Savostina LI, Chudoba DM. Molecular Mobility of Different Forms of Ketoprofen Based on DFT Calculation Data. Chemistry Proceedings. 2024; 16(1):60. https://doi.org/10.3390/ecsoc-28-20162
Chicago/Turabian StyleLogacheva, Kapitolina A., Alexander V. Belushkin, Polina A. Gergelezhiu, Alexander B. Eresko, Sergey N. Malakhov, Elena V. Raksha, Ludmila I. Savostina, and Dorota M. Chudoba. 2024. "Molecular Mobility of Different Forms of Ketoprofen Based on DFT Calculation Data" Chemistry Proceedings 16, no. 1: 60. https://doi.org/10.3390/ecsoc-28-20162
APA StyleLogacheva, K. A., Belushkin, A. V., Gergelezhiu, P. A., Eresko, A. B., Malakhov, S. N., Raksha, E. V., Savostina, L. I., & Chudoba, D. M. (2024). Molecular Mobility of Different Forms of Ketoprofen Based on DFT Calculation Data. Chemistry Proceedings, 16(1), 60. https://doi.org/10.3390/ecsoc-28-20162