
Integrated Computational Approach to Rational Drug Design Targeting SIK2/3: From Theory to Practice †


Abstract
1. Introduction
2. Methodology
2.1. Phase 1: De Novo Molecular Generation
- Single-Step Fine-Tuning: The model was fine-tuned using four SMILES;
- Two-Step Fine-Tuning: adjusted the model with 250 SMILES closely related to the reference;
- Random Samples: Variations of fine-tuning processes using randomly selected SMILES.
2.2. Phase 2: Fragment-Based Drug Design
- Fragment-Based Fine-Tuning: The model was further refined with fragment files;
- Lead Optimization: The lead was used as a basis for exploring optimization potential.
3. Experimental Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SMILE | Mode | Affinity (kcal/mol) | Dist from (rmsd l.b.) |
|---|---|---|---|
| Reference | 1 | −9.2 | 0 |
| 2 | −9.2 | 1.264 | |
| Lead | 1 | −8.2 | 0 |
| 2 | −7.9 | 2.367 |
| SMILE Number | Affinity (kcal/mol) |
|---|---|
| SMILE 10 | −8.788 |
| SMILE 29 | −8.786 |
| SMILE 30 | −8.763 |
| SMILE 8 | −8.719 |
| SMILE | Warhead Variant | Variant | RMSD (kcal/mol) | Log (kcal/mol) |
|---|---|---|---|---|
| SMILE 1 | Benzene sulfonyl Fluoride | 1.4 | −9.63 | −15.15 |
| SMILE 10 | 1.3 | −9.27 | −16.83 | |
| SMILE 1 | 1.1 | −8.99 | −15.78 |
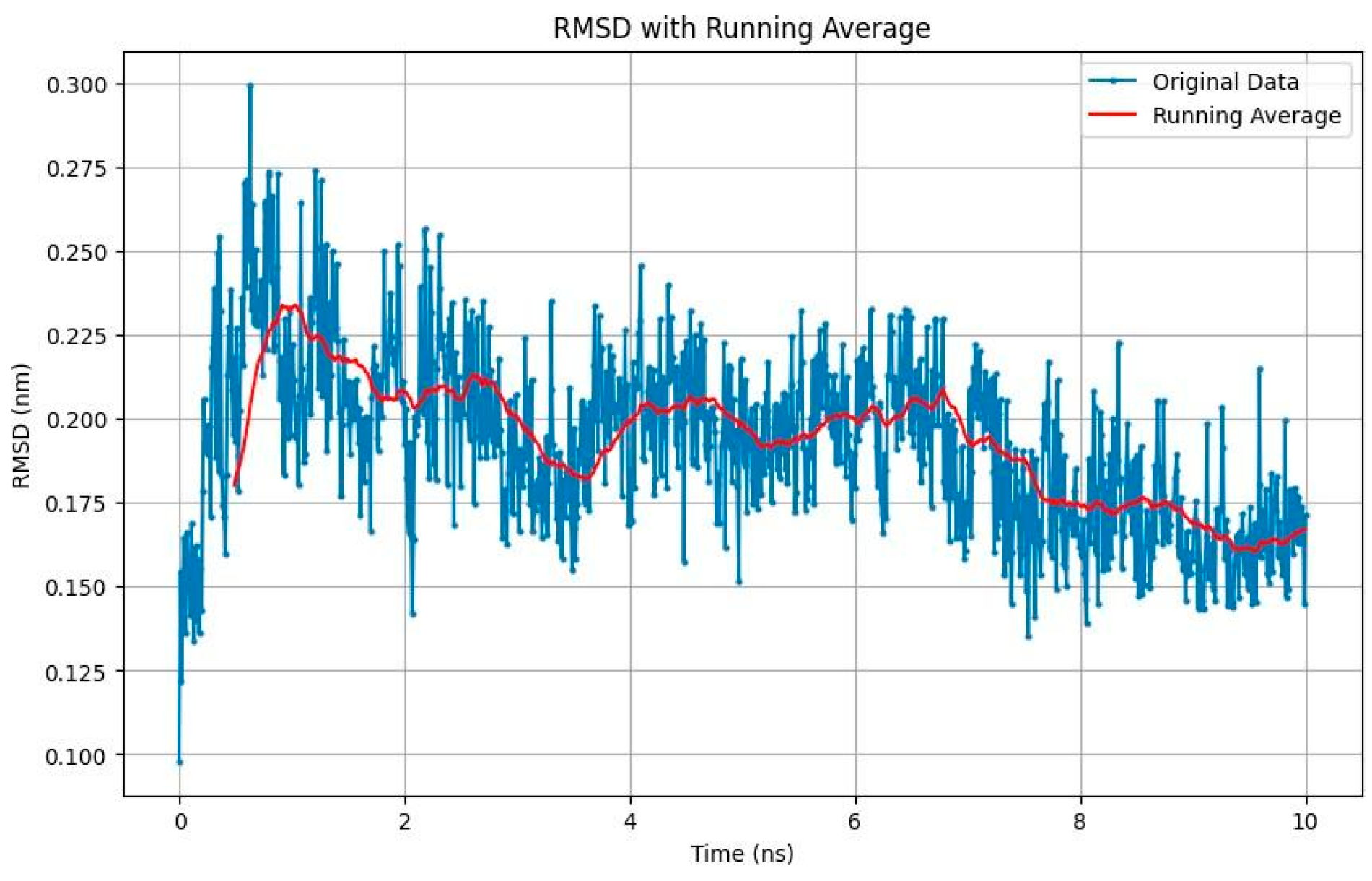
- SMILE 10 BSF Variant 1.3: Displayed relatively stable binding, with RMSD values stabilizing around 0.225 nm (Figure 3);
- Fragmenstein Ligand 2 Step 1: Exhibited larger conformational changes with RMSD peaks around 0.45 nm, suggesting a more flexible binding mode.
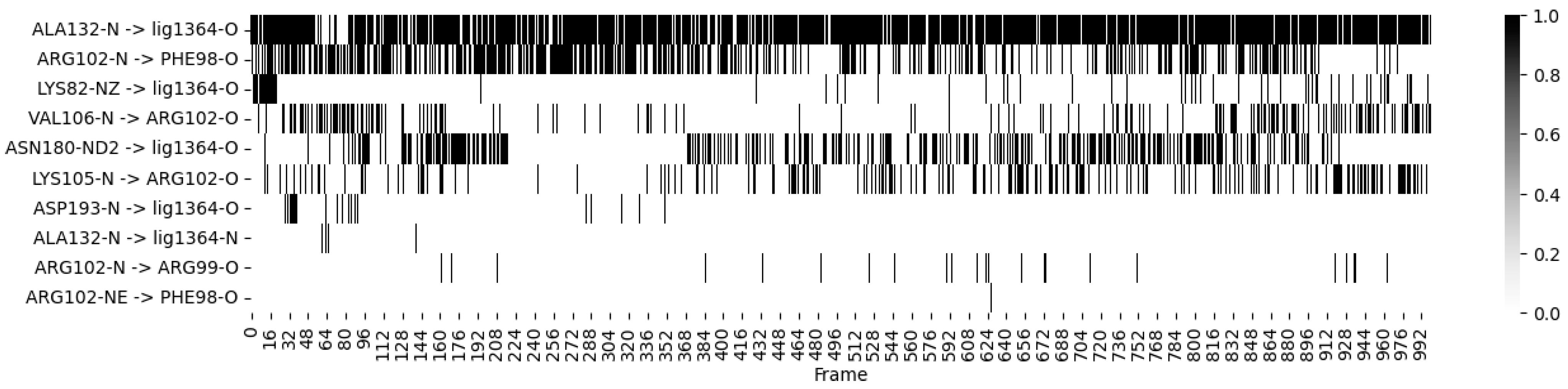
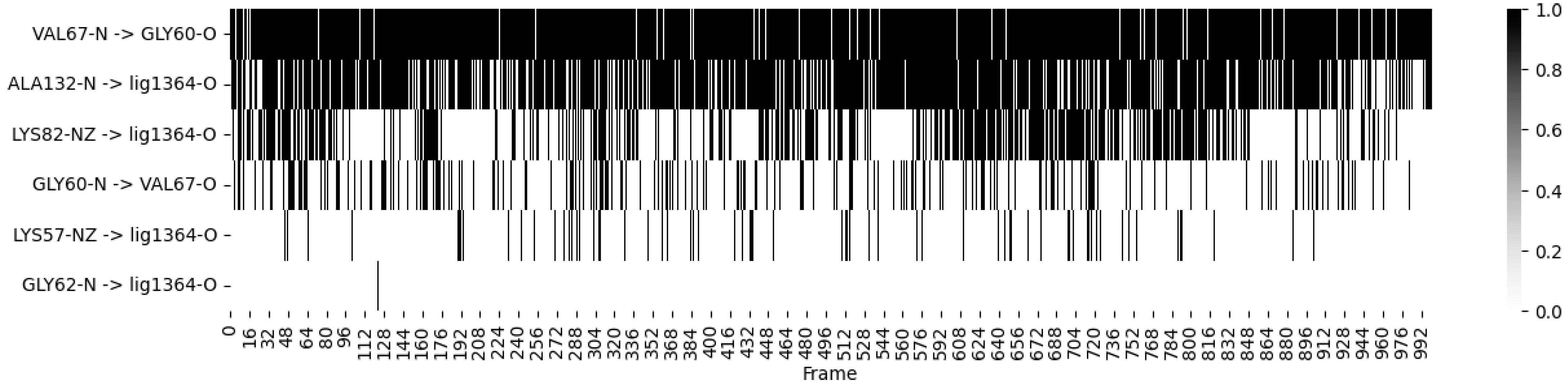
- Ligand 10 BSF Variant 1.3: This covalent inhibitor demonstrated consistent hydrogen bonding with critical residues, particularly MET 104 and GLU 120, throughout the simulation. The heatmap (Figure 4) visualizes these dynamics, showing stable interactions that reinforce the ligand’s strong binding affinity;
- Fragmenstein Ligand 2 Step 1: This ligand exhibited dynamic hydrogen bonding patterns, particularly with residues ARG 102 and ILE 119. Despite some fluctuation, the heatmap indicates overall stable interactions, suggesting the ligand maintains effective binding within the active site over the course of the simulation.
- These results highlight the stability of both ligands in their respective binding pockets, suggesting strong potential as inhibitors targeting SIK2/ SIK3. The Root Mean Square Deviation (RMSD) analysis offers insight into the stability of ligands within the binding pocket:
- SMILE 10 BSF Variant 1.3: This exhibited fluctuations in interaction energy between 4650 kJ/mol and 5000 kJ/mol, with a running average around 4850 kJ/mol, indicating stable binding;
- Fragmenstein Ligand 2 Step 1: This showed a wider range of energy fluctuations (4700–5050 kJ/mol) but maintained an overall stable interaction, with the running average around 4850 kJ/mol. The interaction energy profiles of both ligands indicate generally stable interactions with the target protein despite some transient destabilization.
- Variant 48: Intermittent hydrogen bonding, with RMSD values stabilizing around 0.2 nm;
- Variant 5: Displayed better hydrogen bond stability. RMSD fluctuated more, reaching 0.6 nm (Figure 5).
- Ligand 1 Variant 1.1 New: Displayed intermittent hydrogen bonding with residues, with RMSD values fluctuating between 0.22 nm and 0.3 nm;
- Ligand 1 Variant 1.4: Demonstrated significant H-bond stability. RMSD stabilizing 0.22 nm;
- Ligand 1 Variant 1.4 New 2: Demonstrated stable hydrogen bonding, with RMSD stabilizing around 0.25 nm.
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
Appendix B
Appendix C
Appendix D
References
- Chen, F.; Chen, L.; Qin, Q.; Sun, X. Salt-Inducible Kinase 2: An Oncogenic Signal Transmitter and Potential Target for Cancer Therapy. Front. Oncol. 2019, 9, 18. [Google Scholar] [CrossRef] [PubMed]
- Ballarotto, M.; Willems, S.; Stiller, T.; Nawa, F.; Marschner, J.A.; Grisoni, F.; Merk, D. De novo design of Nurr1 agonists via fragment-augmented generative deep learning in low-data regime. J. Med. Chem. 2023, 66, 8170–8177. [Google Scholar] [CrossRef] [PubMed]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Loeffler, H.H.; He, J.; Tibo, A.; Janet, J.P.; Voronov, A.; Mervin, L.H.; Engkvist, O. REINVENT4: Modern AI–Driven Generative Molecule Design. ChemRxiv 2023. [Google Scholar] [CrossRef]
- Forli, S. Meeko: Preparation of Small Molecules for AutoDock [Software]. Available online: https://github.com/forlilab/Meeko (accessed on 15 March 2024).
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations Through Multi-Level Parallelism from Laptops to Supercomputers. GROMACS. Available online: https://manual.gromacs.org/current/index.html (accessed on 1 April 2024).
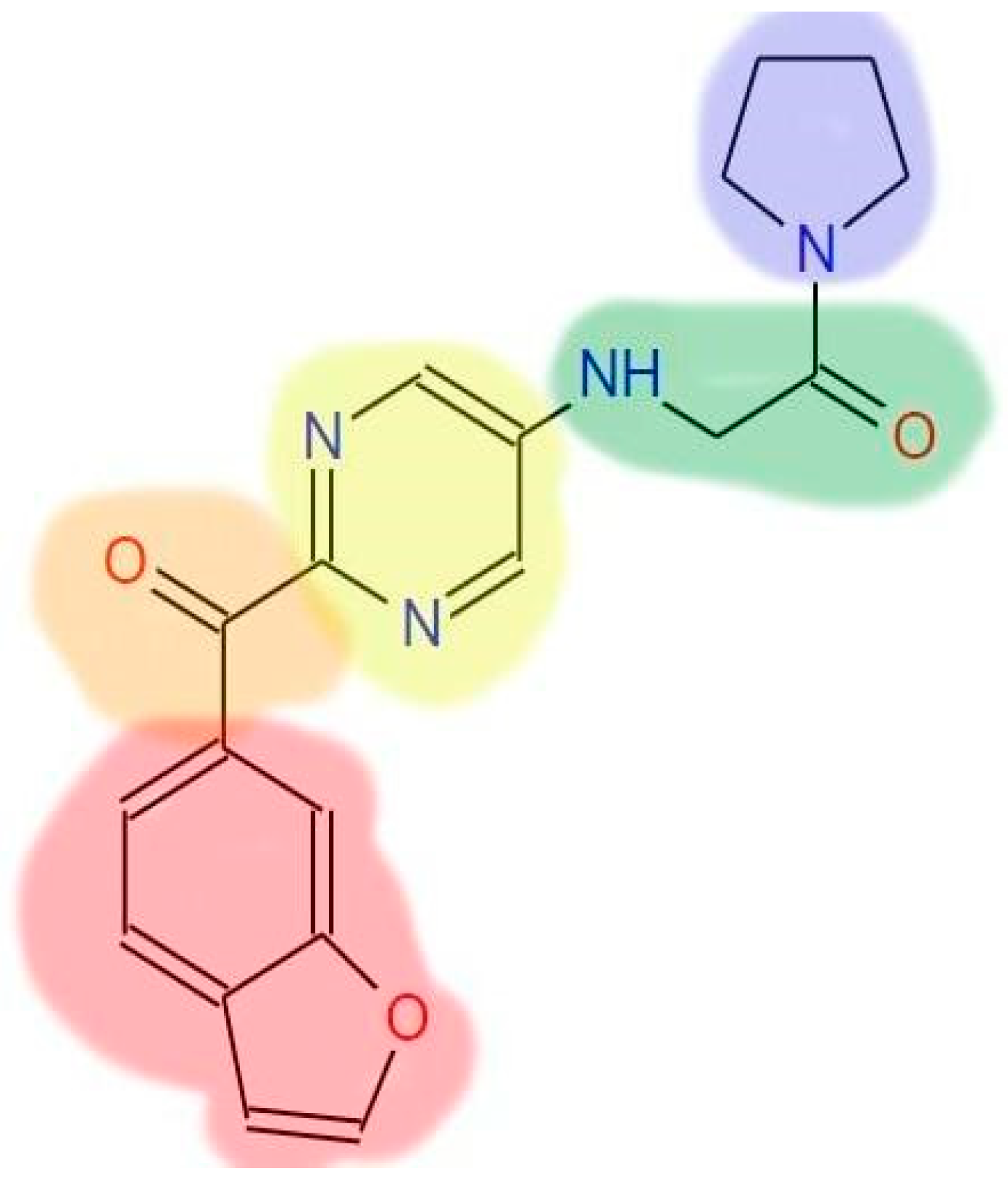
| STEP | SMILE | RMSD (kcal/mol) |
|---|---|---|
| Step 1 | SMILE13 | −10.7 |
| SMILE2 | −9.4 | |
| Step 2 | SMILE8 | −9.3 |
| SMILE15 | −9.2 |
| Trial | Variant | Binding Energy (kcal/mol) | SMILE |
|---|---|---|---|
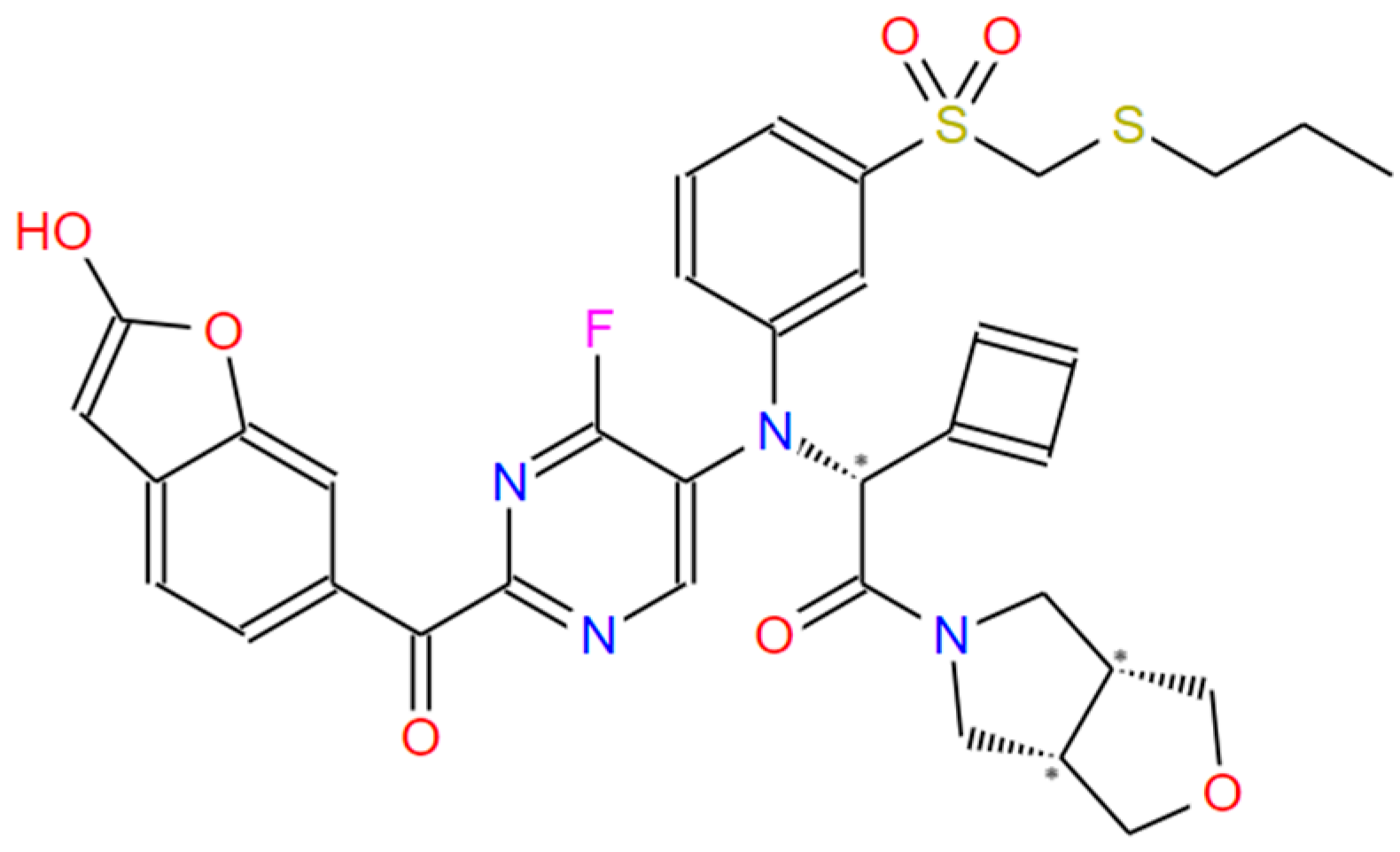
| 1 | 18 | −10.42 | CCSCS(=O)(=O)c1cccc(N(c2cnc(C(=O)c3ccc4cc(O)oc4c3)nc2F)C@HN2CCCC2)c1 |
| 1 | 5 | −9.95 | CCCSCS(=O)(=O)c1cccc(N(CC(=O)N2C[C@H]3COC[C@H]3C2)c2cnc(C(=O) c3ccc4cc(O)oc4c3)nc2F)c1 |
| 1 | 44 | −10.39 | CCSCS(=O)(=O)c1cccc(N(CC(=O)N2C[C@H]3COC[C@H]3C2)c2cnc(C(=O)c3ccc4cc(N5CCCC5)oc4c3)nc2F)c1 |
| 1 | 48 | −10.79 | CCSCS(=O)(=O)c1cccc(N(c2cnc(C(=O)c3ccc4cc(O)oc4c3)nc2F)C@@HC2=CC=C2)c1 |
| Ligand | RMSD (kcal/mol) | SMILE |
|---|---|---|
| Ligand 1 variant 1.4 New 2 | −10.17 | CCSC=S(=O)(ON(CC(=O)N1CCCC1)c4cnc(C(=O)c3ccc2cc[nH]c2c3)nc4)c5ccccc5 |
| Ligand 10 variant 1.3 new | −9.95 | CCSC=S(=O)(ON(CC(=O)N2C[C@@H]1COC[C@@H]1C2)c5cnc(C(=O)c4ccc3ccoc3c4)nc5)c6ccccc6 |
| Ligand 10 variant 1.3 New 2 | −10.14 | CCS(C)=S(=O)(ON(CC(=O)N2C[C@H]1COC[C@H]1C2)c5cnc(C(=O)c4ccc3ccoc3c4)nc5)c6ccccc6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashed, E.Y.E.; Gorislav, A. Integrated Computational Approach to Rational Drug Design Targeting SIK2/3: From Theory to Practice. Chem. Proc. 2024, 16, 3. https://doi.org/10.3390/ecsoc-28-20241
Rashed EYE, Gorislav A. Integrated Computational Approach to Rational Drug Design Targeting SIK2/3: From Theory to Practice. Chemistry Proceedings. 2024; 16(1):3. https://doi.org/10.3390/ecsoc-28-20241
Chicago/Turabian StyleRashed, Eid Youssef Eid, and Alisa Gorislav. 2024. "Integrated Computational Approach to Rational Drug Design Targeting SIK2/3: From Theory to Practice" Chemistry Proceedings 16, no. 1: 3. https://doi.org/10.3390/ecsoc-28-20241
APA StyleRashed, E. Y. E., & Gorislav, A. (2024). Integrated Computational Approach to Rational Drug Design Targeting SIK2/3: From Theory to Practice. Chemistry Proceedings, 16(1), 3. https://doi.org/10.3390/ecsoc-28-20241