
Encapsulation of Allergens into Core–Shell Chitosan Microparticles for Allergen-Specific Subcutaneous Immunotherapy

 , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Chitosan Succinylation
2.3. Physicochemical Characteristics of Chitosan Derivatives
2.4. Rhodamine Labeled Gal d1
2.5. Gel Electrophoresis
2.6. Preparation of Core Particles using Citric Acid and EDAC
2.7. Preparation of Core Particles Using CaCl2
2.8. Formation of Core–Shell Gal-MPs
2.9. Size and Charge of Gal-MPs
2.10. Cell Cultures
2.11. MTT-Assay
2.12. Bone Marrow Derived Macrophages and Dendritic Cells
2.13. Confocal Microscopy
2.14. Flow Cytometry
2.15. Mice
2.16. ASIT
2.17. Allergy Model
2.18. Enzyme Immunoassay (ELISA)
2.19. Histology
2.20. Statistical Analysis
3. Results
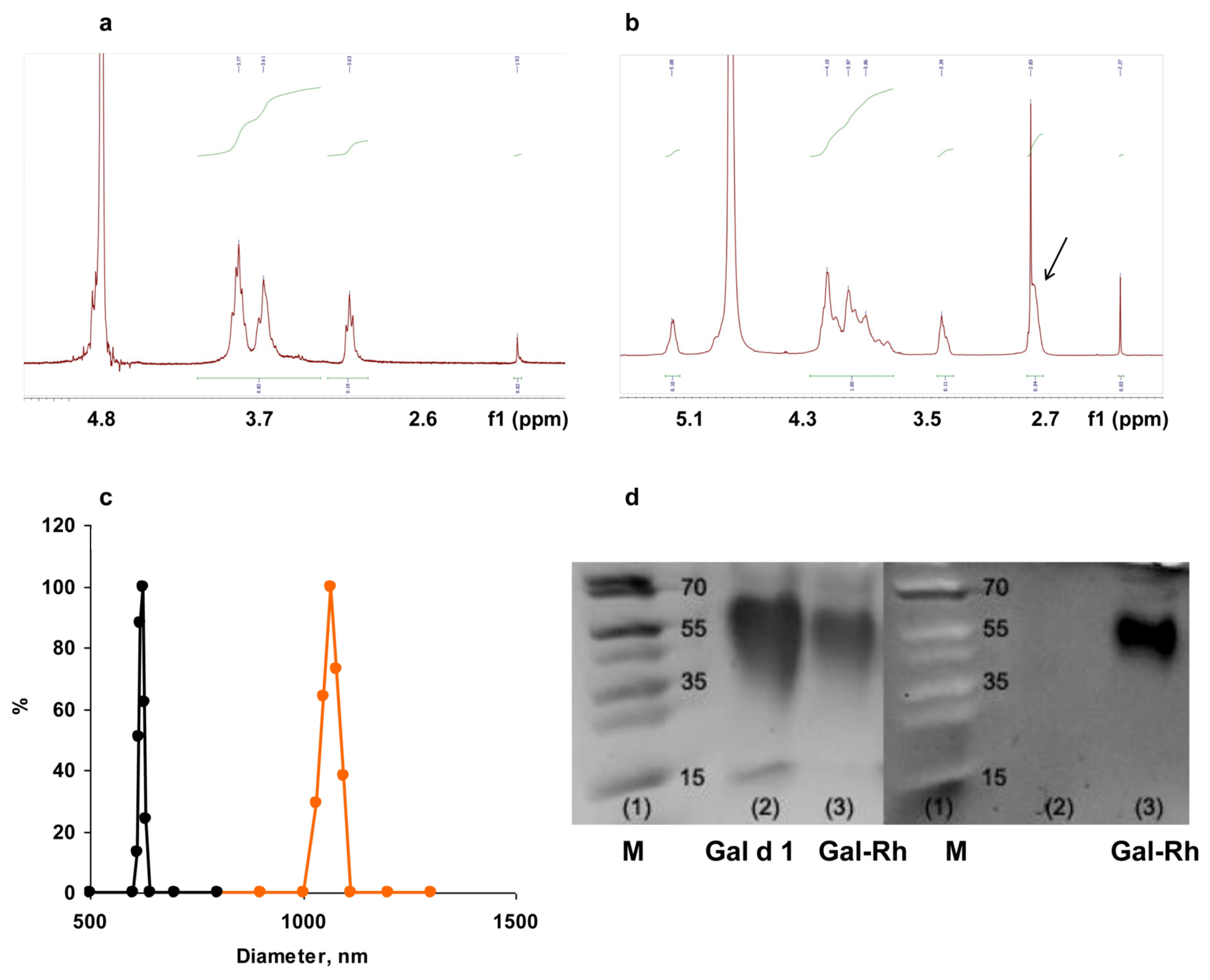
3.1. Characterization of Chitosan Derivatives and Core–Shell Particles
3.2. Cytotoxicity of Gal-MPs
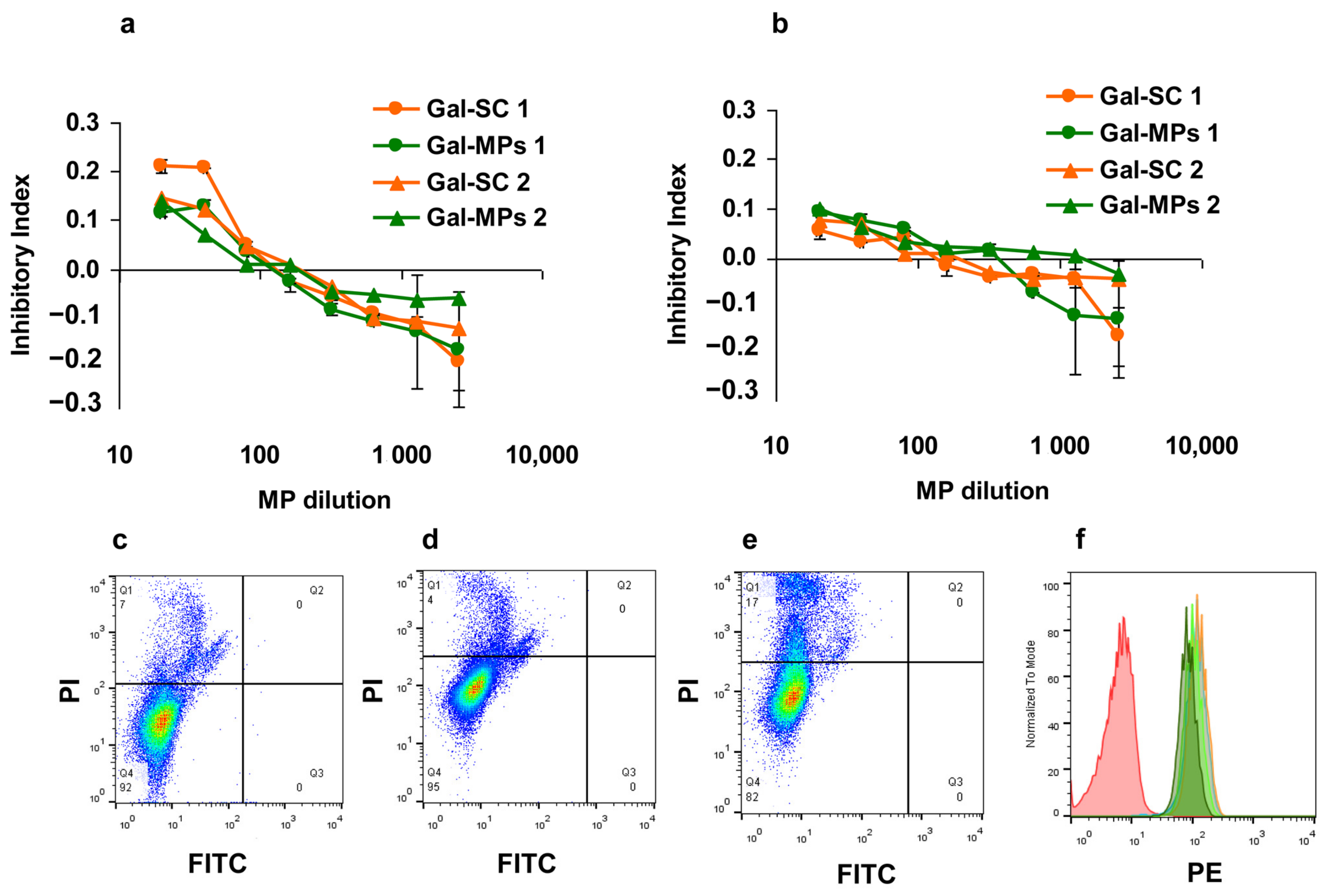
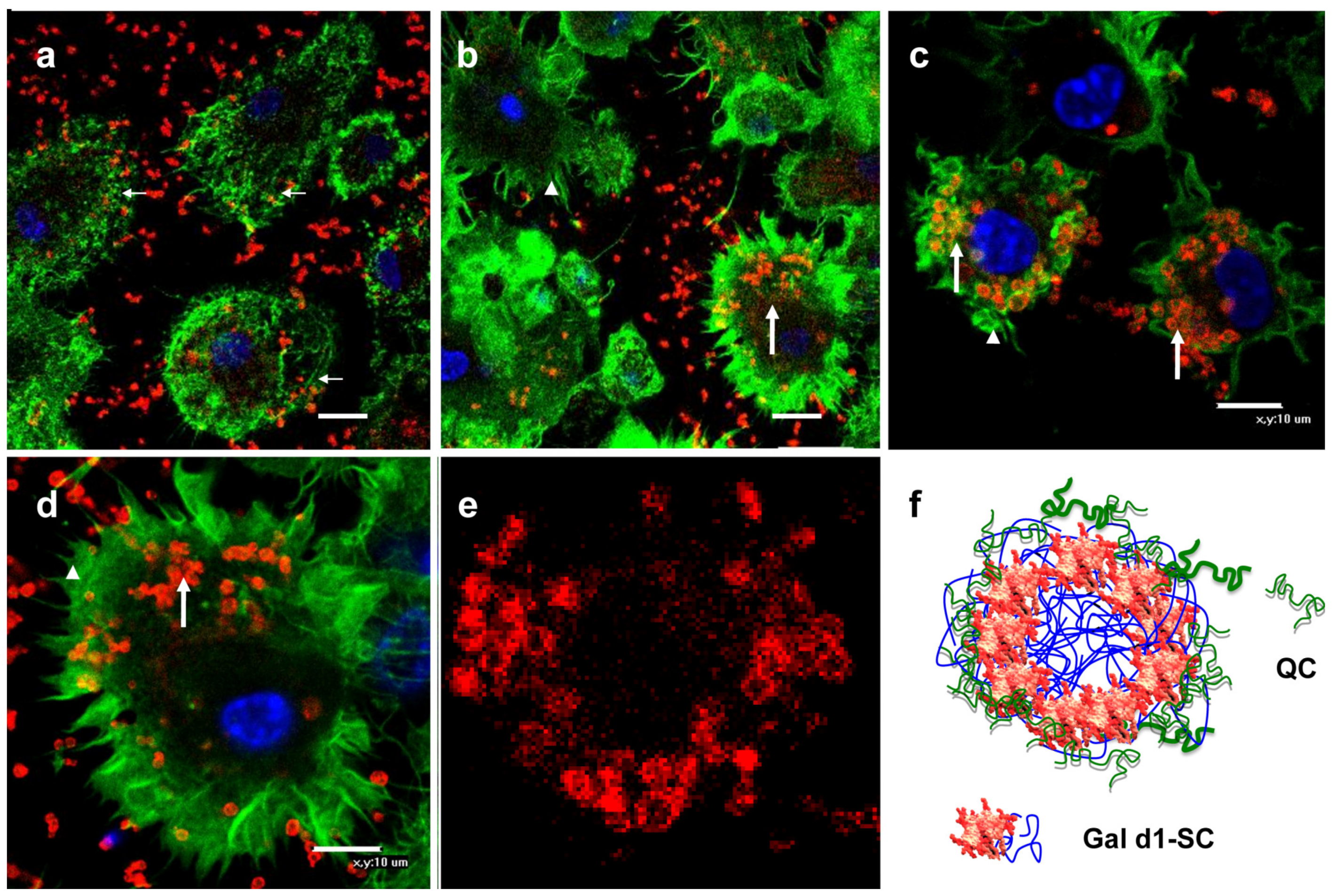
3.3. Binding of Gal-MPs to Bone Marrow Dendritic Cells
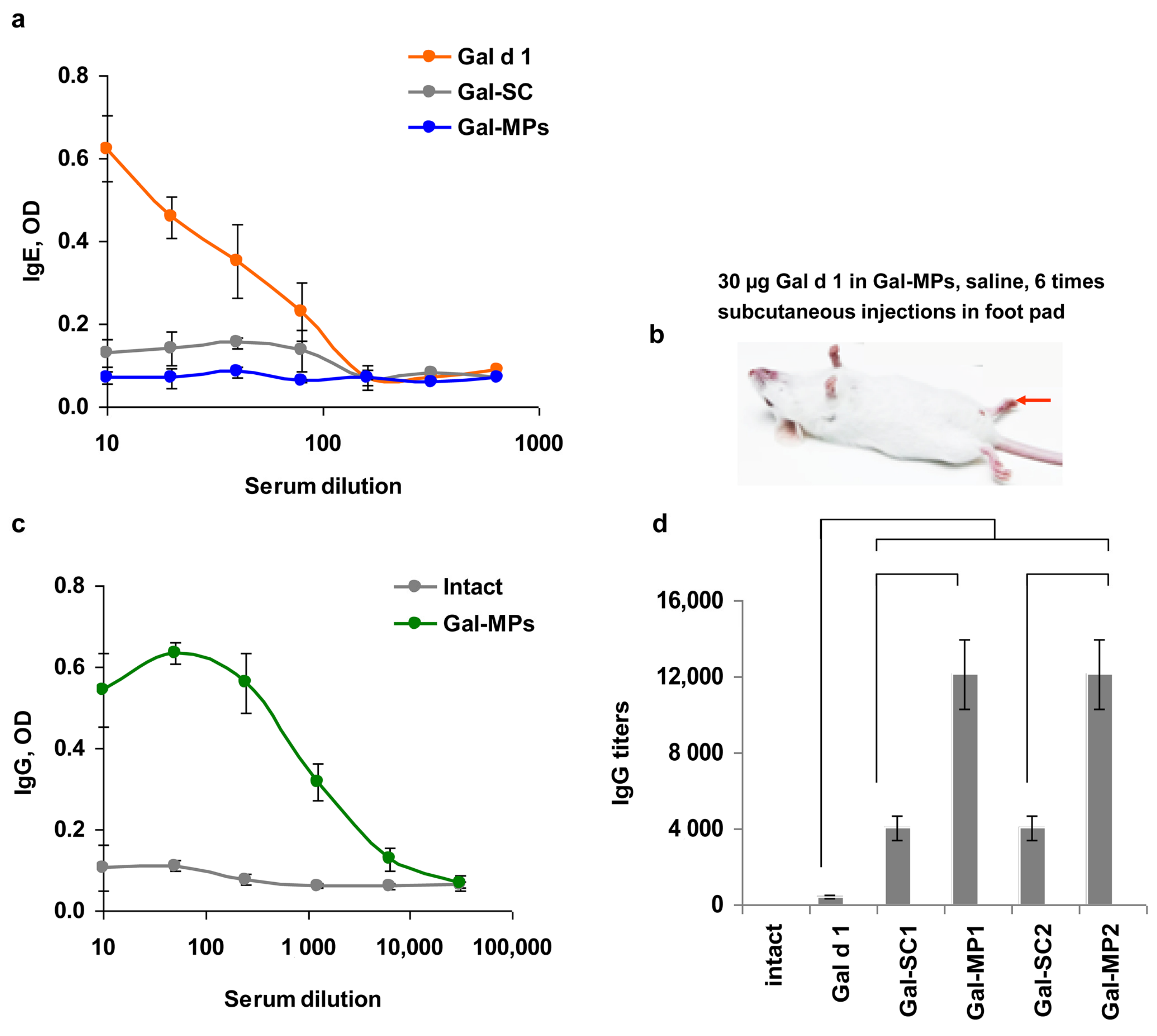
3.4. IgE Reactivity of Gal-MPs
3.5. Immunogenicity of the Encapsulated Gal d1
3.6. Can Subcutaneous ASIT with the Encapsulated Allergens Protect from Allergy?
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jacobsen, L.; Wahn, U.; Bilo, M.B. Allergen-Specific Immunotherapy Provides Immediate, Long-Term and Preventive Clinical Effects in Children and Adults: The Effects of Immunotherapy Can Be Categorised by Level of Benefit—The Centenary of Allergen Specific Subcutaneous Immunotherapy. Clin. Transl. Allergy 2012, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Ravetch, J.V. Fc Receptors. Curr. Opin. Immunol. 1997, 9, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Cooper, P.J. Interactions between Helminth Parasites and Allergy. Curr. Opin. Allergy Clin. Immunol. 2009, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Akdis, C.A.; Akdis, M. Mechanisms of Allergen-Specific Immunotherapy and Immune Tolerance to Allergens. World Allergy Organ. J. 2015, 8, 17. [Google Scholar] [CrossRef] [PubMed]
- Jutel, M.; Akdis, M.; Budak, F.; Aebischer-Casaulta, C.; Wrzyszcz, M.; Blaser, K.; Akdis, C.A. IL-10 and TGF-β Cooperate in the Regulatory T Cell Response to Mucosal Allergens in Normal Immunity and Specific Immunotherapy. Eur. J. Immunol. 2003, 33, 1205–1214. [Google Scholar] [CrossRef]
- Verhoef, A.; Alexander, C.; Kay, A.B.; Larché, M. T Cell Epitope Immunotherapy Induces a CD4+ T Cell Population with Regulatory Activity. PLoS Med. 2005, 2, 0253–0261. [Google Scholar] [CrossRef]
- Wen, Z.-S.; Xu, Y.-L.; Zou, X.-T.; Xu, Z.-R. Chitosan Nanoparticles Act as an Adjuvant to Promote Both Th1 and Th2 Immune Responses Induced by Ovalbumin in Mice. Mar. Drugs 2011, 9, 1038–1055. [Google Scholar] [CrossRef]
- Yang, W.; Dong, Z.; Li, Y.; Zhang, Y.; Fu, H.; Xie, Y. Therapeutic Efficacy of Chitosan Nanoparticles Loaded with BCG-Polysaccharide Nucleic Acid and Ovalbumin on Airway Inflammation in Asthmatic Mice. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 1. [Google Scholar] [CrossRef]
- Roy, K.; Mao, H.Q.; Huang, S.K.; Leong, K.W. Oral Gene Delivery with chitosan–DNA Nanoparticles Generates Immunologic Protection in a Murine Model of Peanut Allergy. Nat. Med. 1999, 5, 387–391. [Google Scholar] [CrossRef]
- Kashirina, E.; Reshetov, P.; Alekseeva, L.; Berzhets, V.; Ryazantsev, D.; Zubov, V.; Chudakov, D.; Svirshchevskaya, E. Encapsulation of Allergens into Chitosan-Alginate Nanoparticles Prevents IgE Binding. Jacobs J. Vaccines Vaccin. 2015, 1, 12. [Google Scholar]
- Konovalova, M.V.; Kurek, D.V.; Litvinets, S.G.; Martinson, E.A.; Varlamov, V.P. Preparation and Characterization of Cryogels Based on Pectin and Chitosan. Prog. Chem. Appl. Chitin Its Deriv. 2016, 21, 114–121. [Google Scholar] [CrossRef]
- Konovalova, M.; Shagdarova, B.T.; Zubov, V.; Svirshchevskaya, E. Express Analysis of Chitosan and Its Derivatives by Gel Electrophoresis. Prog. Chem. Appl. Chitin Its Deriv. 2019, XXIV, 84–95. [Google Scholar] [CrossRef]
- Yamaguchi, R.; Arai, Y.; Itoh, T.; Hirano, S. Preparation of Partially N-Succinylated Chitosans and Their Cross-Linked Gels. Carbohydr. Res. 1981, 88, 172–175. [Google Scholar] [CrossRef]
- Lopatin, S.A.; Derbeneva, M.S.; Kulikov, S.N.; Varlamov, V.P.; Shpigun, O.A. Fractionation of Chitosan by Ultrafiltration. J. Anal. Chem. 2009, 64, 648–651. [Google Scholar] [CrossRef]
- Zubareva, A.; Shagdarova, B.; Varlamov, V.; Kashirina, E.; Svirshchevskaya, E. Penetration and Toxicity of Chitosan and Its Derivatives. Eur. Polym. J. 2017, 93, 743–749. [Google Scholar] [CrossRef]
- Chudakov, D.B.; Ryasantsev, D.Y.; Tsaregorotseva, D.S.; Kotsareva, O.D.; Fattakhova, G.V.; Svirshchevskaya, E.V. Tertiary Lymphoid Structure Related B-Cell IgE Isotype Switching and Secondary Lymphoid Organ Linked IgE Production in Mouse Allergy Model. BMC Immunol. 2020, 21, 45. [Google Scholar] [CrossRef]
- Durham, S.R.; Shamji, M.H. Allergen Immunotherapy: Past, Present and Future. Nat. Rev. Immunol. 2023, 23, 317–328. [Google Scholar] [CrossRef]
- Carreño-Gómez, B.; Duncan, R. Evaluation Of the Biological Properties of Soluble Chitosan and Chitosan Microspheres. Int. J. Pharm. 1997, 148, 231–240. [Google Scholar] [CrossRef]
- Mukhopadhyay, P.; Sarkar, K.; Bhattacharya, S.; Bhattacharyya, A.; Mishra, R.; Kundu, P.P. pH Sensitive N-Succinyl Chitosan Grafted Polyacrylamide Hydrogel for Oral Insulin Delivery. Carbohydr. Polym. 2014, 112, 627–637. [Google Scholar] [CrossRef]
- Yan, C.; Chen, D.; Gu, J.; Qin, J. Nanoparticles of 5-Fluorouracil (5-FU) Loaded N-Succinyl-Chitosan (Suc-Chi) for Cancer Chemotherapy: Preparation, Characterization—In-Vitro Drug Release and Anti-Tumour Activity. J. Pharm. Pharmacol. 2010, 58, 1177–1181. [Google Scholar] [CrossRef]
- Toh, E.K.W.; Chen, H.Y.; Lo, Y.L.; Huang, S.J.; Wang, L.F. Succinated Chitosan as a Gene Carrier for Improved Chitosan Solubility and Gene Transfection. Nanomed. Nanotechnol. Biol. Med. 2011, 7, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.A.; Syeda, J.T.M.; Wasan, K.M.; Wasan, E.K. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9, 53. [Google Scholar] [CrossRef] [PubMed]
- Iacob, A.T.; Lupascu, F.G.; Apotrosoaei, M.; Vasincu, I.M.; Tauser, R.G.; Lupascu, D.; Giusca, S.E.; Caruntu, I.D.; Profire, L. Recent Biomedical Approaches for Chitosan Based Materials as Drug Delivery Nanocarriers. Pharmaceutics 2021, 13, 587. [Google Scholar] [CrossRef] [PubMed]
- Urisu, A.; Kondo, Y.; Tsuge, I. Hen’s Egg Allergy. Chem. Immunol. Allergy 2015, 101, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Svirshchevskaya, E.V.; Zubareva, A.A.; Boyko, A.A.; Shustova, O.A.; Grechikhina, M.V.; Shagdarova, B.T.; Varlamov, V.P. Analysis of Toxicity and Biocompatibility of Chitosan Derivatives with Different Physico-Chemical Properties. Appl. Biochem. Microbiol. 2016, 52, 483–490. [Google Scholar] [CrossRef]
- Chollakup, R.; Uttayarat, P.; Chworos, A.; Smitthipong, W. Noncovalent Sericin-Chitosan Scaffold: Physical Properties and Low Cytotoxicity Effect. Int. J. Mol. Sci. 2020, 21, 775. [Google Scholar] [CrossRef]
- Li, D.; Liu, P.; Hao, F.; Lv, Y.; Xiong, W.; Yan, C.; Wu, Y.; Luo, H. Preparation and Application of Silver/chitosan-Sepiolite Materials with Antimicrobial Activities and Low Cytotoxicity. Int. J. Biol. Macromol. 2022, 210, 337–349. [Google Scholar] [CrossRef]
- Walter, F.; Winter, E.; Rahn, S.; Heidland, J.; Meier, S.; Struzek, A.M.; Lettau, M.; Philipp, L.M.; Beckinger, S.; Otto, L.; et al. Chitosan Nanoparticles as Antigen Vehicles to Induce Effective Tumor Specific T Cell Responses. PLoS ONE 2020, 15, e0239369. [Google Scholar] [CrossRef]
- Ahmed, T.A.; Aljaeid, B.M. Preparation, Characterization, and Potential Application of Chitosan, Chitosan Derivatives, and Chitosan Metal Nanoparticles in Pharmaceutical Drug Delivery. Drug Des. Devel. Ther. 2016, 10, 483–507. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diameter, nm | ζ Potential, mV | Gal d1 Concentration, µg/mL | |
|---|---|---|---|
| EDAC/citric acid | |||
| Core, Gal-SC 1 | 710 ± 12 | −11 ± 2 | 350 ± 50 |
| Core–shell, Gal-MPs 1 | 1230 ± 44 | +9 ± 5 | 350 ± 65 |
| CaCl2 precipitation | |||
| Core, Gal-SC 2 | 680 ± 15 | −10 ± 3 | 340 ± 35 |
| Core–shell, Gal-MPs 2 | 1180 ± 53 | +9 ± 5 | 340 ± 55 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konovalova, M.; Kashirina, E.; Beltsova, K.; Kotsareva, O.; Fattakhova, G.; Svirshchevskaya, E. Encapsulation of Allergens into Core–Shell Chitosan Microparticles for Allergen-Specific Subcutaneous Immunotherapy. Polysaccharides 2023, 4, 142-155. https://doi.org/10.3390/polysaccharides4020011
Konovalova M, Kashirina E, Beltsova K, Kotsareva O, Fattakhova G, Svirshchevskaya E. Encapsulation of Allergens into Core–Shell Chitosan Microparticles for Allergen-Specific Subcutaneous Immunotherapy. Polysaccharides. 2023; 4(2):142-155. https://doi.org/10.3390/polysaccharides4020011
Chicago/Turabian StyleKonovalova, Mariya, Elena Kashirina, Kseniya Beltsova, Olga Kotsareva, Gulnar Fattakhova, and Elena Svirshchevskaya. 2023. "Encapsulation of Allergens into Core–Shell Chitosan Microparticles for Allergen-Specific Subcutaneous Immunotherapy" Polysaccharides 4, no. 2: 142-155. https://doi.org/10.3390/polysaccharides4020011
APA StyleKonovalova, M., Kashirina, E., Beltsova, K., Kotsareva, O., Fattakhova, G., & Svirshchevskaya, E. (2023). Encapsulation of Allergens into Core–Shell Chitosan Microparticles for Allergen-Specific Subcutaneous Immunotherapy. Polysaccharides, 4(2), 142-155. https://doi.org/10.3390/polysaccharides4020011