
Dysregulation of a Heme Oxygenase–Synuclein Axis in Parkinson Disease


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. α-Synuclein: A Key Player in Parkinson Disease
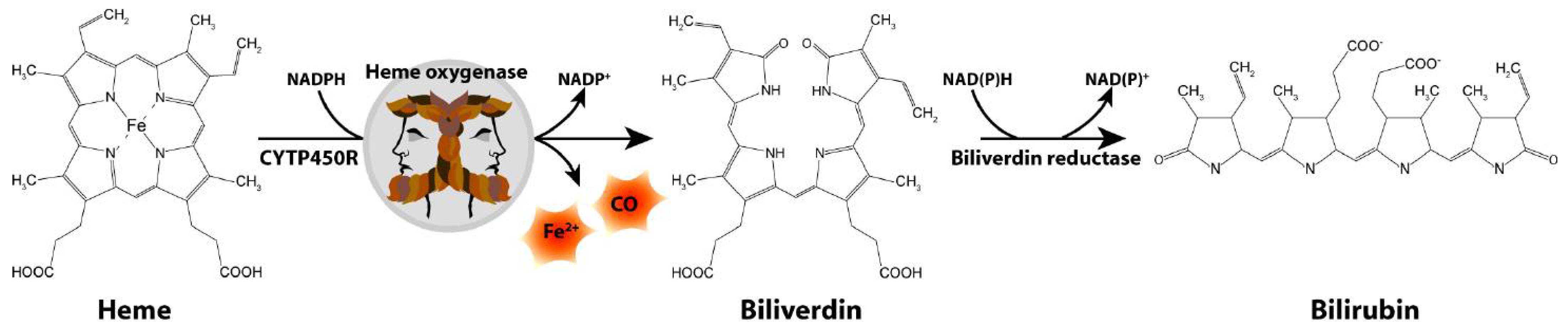
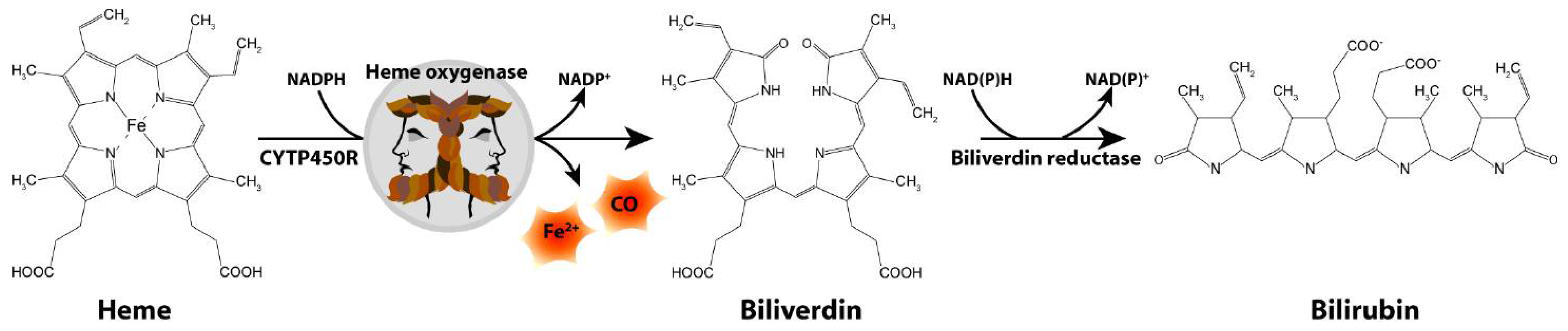
3. Involvement of Heme Oxygenase-1 in Parkinson Disease Pathology
4. GFAP.HMOX18.5−19 m Transgenic Mice
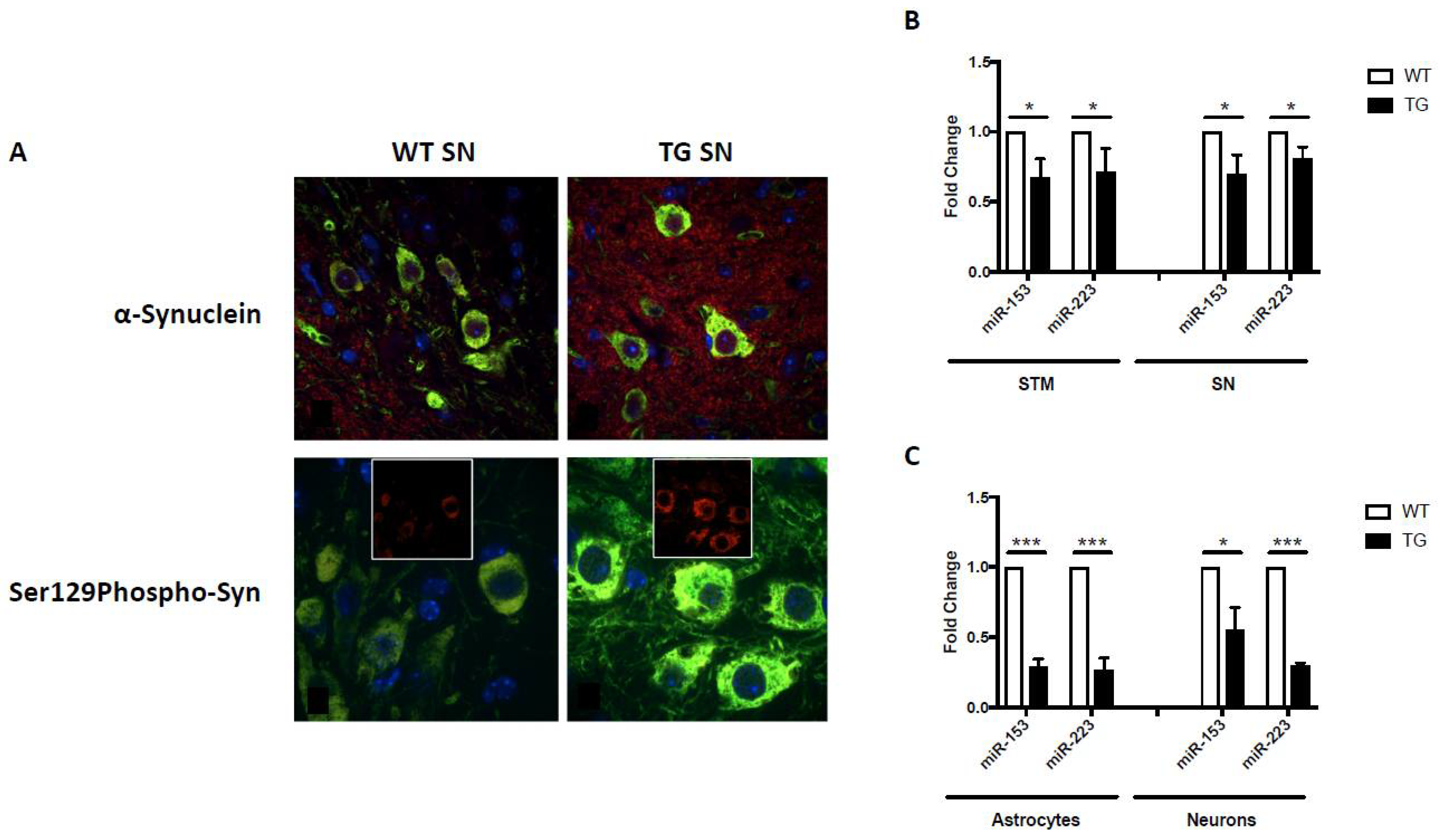
4.1. GFAP.HMOX18.5–19 m Mice and α-Synuclein
4.2. GFAP.HMOX1 Primary Cultures and α-Synuclein
4.3. MicroRNAs In Vitro and In Vivo
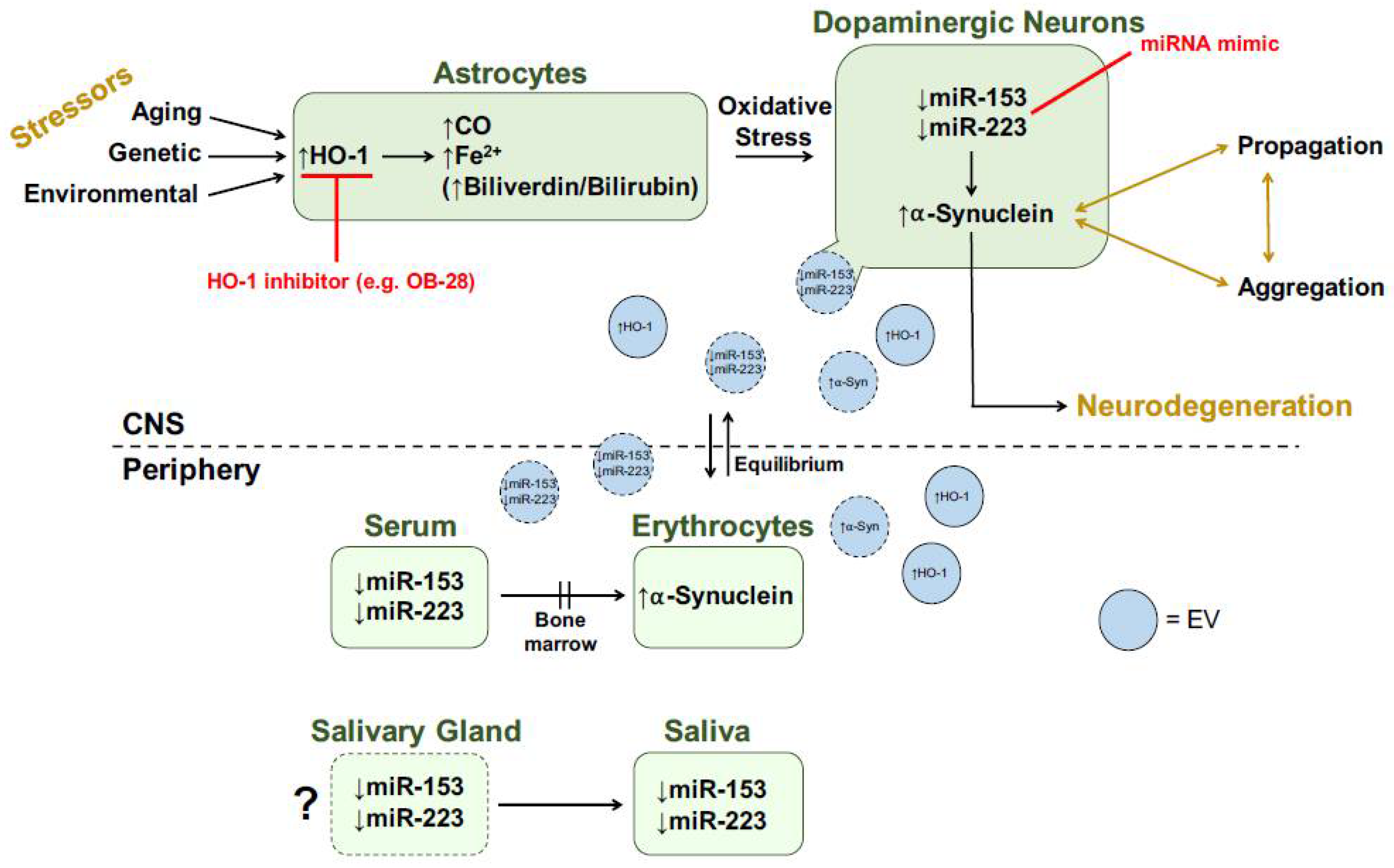
5. miR-153, miR-223 and Heme Oxygenase-1 as Biomarkers of Parkinson Disease
6. Concluding Remarks: HO-1, miR-153, miR-223 and α-Synuclein
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef] [PubMed]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.R.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. Alpha-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Takahashi, T.; Yamashita, H.; Nakamura, T.; Nagano, Y.; Nakamura, S. Tyrosine 125 of alpha-synuclein plays a critical role for dimerization following nitrative stress. Brain Res. 2002, 938, 73–80. [Google Scholar] [CrossRef]
- Kim, W.S.; Kagedal, K.; Halliday, G.M. Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Res. Ther. 2014, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Marques, O.; Outeiro, T.F. Alpha-synuclein: From secretion to dysfunction and death. Cell Death Dis. 2012, 3, e350. [Google Scholar] [CrossRef]
- Miranda, H.V.; Cassio, R.; Correia-Guedes, L.; Gomes, M.A.; Chegao, A.; Miranda, E.; Soares, T.; Coelho, M.; Rosa, M.M.; Ferreira, J.J.; et al. Posttranslational modifications of blood-derived alpha-synuclein as biochemical markers for Parkinson’s disease. Sci. Rep. 2017, 7, 13713. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [Green Version]
- Al-Nimer, M.S.; Mshatat, S.F.; Abdulla, H.I. Saliva alpha-Synuclein and A High Extinction Coefficient Protein: A Novel Approach in Assessment Biomarkers of Parkinson’s Disease. N. Am. J. Med. Sci. 2014, 6, 633–637. [Google Scholar] [CrossRef] [Green Version]
- Bougea, A.; Stefanis, L.; Paraskevas, G.P.; Emmanouilidou, E.; Vekrelis, K.; Kapaki, E. Plasma alpha-synuclein levels in patients with Parkinson’s disease: A systematic review and meta-analysis. Neurol. Sci. 2019, 40, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.; Costa, B.; Pietrobono, D.; Giacomelli, C.; Iofrida, C.; Trincavelli, M.L.; Fusi, J.; Franzoni, F.; Martini, C. Epigenetic Modifications of the alpha-Synuclein Gene and Relative Protein Content Are Affected by Ageing and Physical Exercise in Blood from Healthy Subjects. Oxidative Med. Cell. Longev. 2018, 2018, 3740345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daniele, S.; Frosini, D.; Pietrobono, D.; Petrozzi, L.; Lo Gerfo, A.; Baldacci, F.; Fusi, J.; Giacomelli, C.; Siciliano, G.; Trincavelli, M.L.; et al. alpha-Synuclein Heterocomplexes with beta-Amyloid Are Increased in Red Blood Cells of Parkinson’s Disease Patients and Correlate with Disease Severity. Front. Mol. Neurosci. 2018, 11, 53. [Google Scholar] [CrossRef] [PubMed]
- Devic, I.; Hwang, H.; Edgar, J.S.; Izutsu, K.; Presland, R.; Pan, C.; Goodlett, D.R.; Wang, Y.; Armaly, J.; Tumas, V.; et al. Salivary alpha-synuclein and DJ-1, potential biomarkers for Parkinson’s disease. Brain 2011, 134, e178. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Tang, H.; Nie, K.; Wang, L.; Zhao, J.; Gan, R.; Huang, J.; Zhu, R.; Feng, S.; Duan, Z.; et al. Cerebrospinal fluid alpha-synuclein as a biomarker for Parkinson’s disease diagnosis: A systematic review and meta-analysis. Int. J. Neurosci. 2015, 125, 645–654. [Google Scholar] [CrossRef]
- Kang, W.; Chen, W.; Yang, Q.; Zhang, L.; Zhang, L.; Wang, X.; Dong, F.; Zhao, Y.; Chen, S.; Quinn, T.J.; et al. Salivary total alpha-synuclein, oligomeric alpha-synuclein and SNCA variants in Parkinson’s disease patients. Sci. Rep. 2016, 6, 28143. [Google Scholar] [CrossRef] [Green Version]
- Vivacqua, G.; Latorre, A.; Suppa, A.; Nardi, M.; Pietracupa, S.; Mancinelli, R.; Fabbrini, G.; Colosimo, C.; Gaudio, E.; Berardelli, A. Abnormal Salivary Total and Oligomeric Alpha-Synuclein in Parkinson’s Disease. PLoS ONE 2016, 11, e0151156. [Google Scholar] [CrossRef] [Green Version]
- Vivacqua, G.; Suppa, A.; Mancinelli, R.; Belvisi, D.; Fabbrini, A.; Costanzo, M.; Formica, A.; Onori, P.; Fabbrini, G.; Berardelli, A. Salivary alpha-synuclein in the diagnosis of Parkinson’s disease and Progressive Supranuclear Palsy. Parkinsonism Relat. Disord. 2019, 63, 143–148. [Google Scholar] [CrossRef]
- Cressatti, M.; Juwara, L.; Galindez, J.M.; Velly, A.M.; Nkurunziza, E.S.; Marier, S.; Canie, O.; Gornistky, M.; Schipper, H.M. Salivary microR-153 and microR-223 Levels as Potential Diagnostic Biomarkers of Idiopathic Parkinson’s Disease. Mov. Disord. 2019, 35, 468–477. [Google Scholar] [CrossRef]
- Galindez, J.M.; Juwara, L.; Cressatti, M.; Gornitsky, M.; Velly, A.M.; Schipper, H.M. Salivary heme oxygenase-1, a potential biomarker for central neurodegeneration. J. Cent. Nerv. Syst. Dis. 2021, 13, 11795735211029114. [Google Scholar] [CrossRef]
- Song, W.; Kothari, V.; Velly, A.M.; Cressatti, M.; Liberman, A.; Gornitsky, M.; Schipper, H.M. Evaluation of salivary heme oxygenase-1 as a potential biomarker of early Parkinson’s disease. Mov. Disord. 2018, 33, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W.; Tavitian, A.; Cressatti, M. The sinister face of heme oxygenase-1 in brain aging and disease. Prog. Neurobiol. 2019, 172, 40–70. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Song, W. A heme oxygenase-1 transducer model of degenerative and developmental brain disorders. Int. J. Mol. Sci. 2015, 16, 5400–5419. [Google Scholar] [CrossRef] [Green Version]
- Dore, S.; Takahashi, M.; Ferris, C.D.; Zakhary, R.; Hester, L.D.; Guastella, D.; Snyder, S.H. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc. Natl. Acad. Sci. USA 1999, 96, 2445–2450. [Google Scholar] [CrossRef] [Green Version]
- Llesuy, S.F.; Tomaro, M.L. Heme oxygenase and oxidative stress. Evidence of involvement of bilirubin as physiological protector against oxidative damage. Biochim. Biophys. Acta 1994, 1223, 9–14. [Google Scholar] [CrossRef]
- Nakagami, T.; Toyomura, K.; Kinoshita, T.; Morisawa, S. A beneficial role of bile pigments as an endogenous tissue protector: Anti-complement effects of biliverdin and conjugated bilirubin. Biochim. Biophys. Acta 1993, 1158, 189–193. [Google Scholar] [CrossRef]
- Stocker, R.; Yamamoto, Y.; McDonagh, A.F.; Glazer, A.N.; Ames, B.N. Bilirubin is an antioxidant of possible physiological importance. Science 1987, 235, 1043–1046. [Google Scholar] [CrossRef]
- Frankel, D.; Mehindate, K.; Schipper, H.M. Role of heme oxygenase-1 in the regulation of manganese superoxide dismutase gene expression in oxidatively-challenged astroglia. J. Cell. Physiol. 2000, 185, 80–86. [Google Scholar] [CrossRef]
- Piantadosi, C.A.; Carraway, M.S.; Suliman, H.B. Carbon monoxide, oxidative stress, and mitochondrial permeability pore transition. Free Radic. Biol. Med. 2006, 40, 1332–1339. [Google Scholar] [CrossRef]
- Ryter, S.W.; Tyrrell, R.M. The heme synthesis and degradation pathways: Role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic. Biol. Med. 2000, 28, 289–309. [Google Scholar] [CrossRef]
- Schipper, H.M.; Song, W.; Zukor, H.; Hascalovici, J.R.; Zeligman, D. Heme oxygenase-1 and neurodegeneration: Expanding frontiers of engagement. J. Neurochem. 2009, 110, 469–485. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Liberman, A.; Stopa, E.G. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol. 1998, 150, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Béraud, D.; Hathaway, H.A.; Trecki, J.; Chasovskikh, S.; Johnson, D.A.; Johnson, J.A.; Federoff, H.J.; Shimoji, M.; Mhyre, T.R.; Maguire-Zeiss, K.A. Microglial activation and antioxidant responses induced by the Parkinson’s disease protein α-synuclein. J. Neuroimmune Pharmacol. 2013, 8, 94–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokes, A.H.; Hastings, T.G.; Vrana, K.E. Cytotoxic and genotoxic potential of dopamine. J. Neurosci. Res. 1999, 55, 659–665. [Google Scholar] [CrossRef]
- Castellani, R.; Smith, M.A.; Richey, P.L.; Perry, G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 1996, 737, 195–200. [Google Scholar] [CrossRef]
- Ayuso, P.; Martinez, C.; Pastor, P.; Lorenzo-Betancor, O.; Luengo, A.; Jimenez-Jimenez, F.J.; Alonso-Navarro, H.; Agundez, J.A.; Garcia-Martin, E. An association study between Heme oxygenase-1 genetic variants and Parkinson’s disease. Front. Cell. Neurosci. 2014, 8, 298. [Google Scholar] [CrossRef] [Green Version]
- Infante, J.; Sierra, M.; Sanchez-Juan, P.; Garcia-Gorostiaga, I.; Gonzalez-Aramburu, I.; Fernandez-Viadero, C.; Berciano, J.; Combarros, O. Interaction between heme oxygenase-1 genotypes and exposure to pesticides in Parkinson’s disease. Mov. Disord. 2011, 26, 916–917. [Google Scholar] [CrossRef]
- Ran, C.; Wirdefeldt, K.; Brodin, L.; Ramezani, M.; Westerlund, M.; Xiang, F.; Anvret, A.; Willows, T.; Sydow, O.; Johansson, A.; et al. Genetic Variations and mRNA Expression of NRF2 in Parkinson’s Disease. Parkinsons Dis. 2017, 2017, 4020198. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Campbell, M.R.; Lacher, S.E.; Cho, H.Y.; Wan, M.; Crowl, C.L.; Chorley, B.N.; Bond, G.L.; Kleeberger, S.R.; Slattery, M.; et al. A Polymorphic Antioxidant Response Element Links NRF2/sMAF Binding to Enhanced MAPT Expression and Reduced Risk of Parkinsonian Disorders. Cell Rep. 2016, 15, 830–842. [Google Scholar] [CrossRef] [Green Version]
- Kuter, K.; Olech, Ł.; Głowacka, U. Prolonged dysfunction of astrocytes and activation of microglia accelerate degeneration of dopaminergic neurons in the rat substantia nigra and block compensation of early motor dysfunction induced by 6-OHDA. Mol. Neurobiol. 2018, 55, 3049–3066. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Su, X.; Piao, L.; Jin, Z.; Jin, R. Involvement of astrocytes and microRNA dysregulation in neurodegenerative diseases: From pathogenesis to therapeutic potential. Front. Mol. Neurosci. 2021, 14, 36. [Google Scholar] [CrossRef] [PubMed]
- Mehindate, K.; Sahlas, D.J.; Frankel, D.; Mawal, Y.; Liberman, A.; Corcos, J.; Dion, S.; Schipper, H.M. Proinflammatory cytokines promote glial heme oxygenase-1 expression and mitochondrial iron deposition: Implications for multiple sclerosis. J. Neurochem. 2001, 77, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Glial HO-1 expression, iron deposition and oxidative stress in neurodegenerative diseases. Neurotox. Res. 1999, 1, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M. Mitochondrial Iron Deposition in Aging Astroglia: Mechanisms and Disease Implications. In Mitochondrial Ubiquinone (Coenzyme Q): Biochemical, Functional, Medical, and Therapeutic Aspects in Human Health and Disease; Ebadi, M.C.R.K., Ed.; Prominent Press: Shepparton, Australia, 2001; pp. 267–280. [Google Scholar]
- Di Monte, D.A.; Schipper, H.M.; Hetts, S.; Langston, J.W. Iron-mediated bioactivation of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in glial cultures. Glia 1995, 15, 203–206. [Google Scholar] [CrossRef]
- Morale, M.C.; Serra, P.A.; L’Episcopo, F.; Tirolo, C.; Caniglia, S.; Testa, N.; Gennuso, F.; Giaquinta, G.; Rocchitta, G.; Desole, M.S.; et al. Estrogen, neuroinflammation and neuroprotection in Parkinson’s disease: Glia dictates resistance versus vulnerability to neurodegeneration. Neuroscience 2006, 138, 869–878. [Google Scholar] [CrossRef]
- Wang, J.L.; Xu, C.J. Astrocytes autophagy in aging and neurodegenerative disorders. Biomed. Pharmacother. 2020, 122, 109691. [Google Scholar] [CrossRef]
- Yang, C.M.; Lin, C.C.; Hsieh, H.L. High-glucose-derived oxidative stress-dependent heme oxygenase-1 expression from astrocytes contributes to the neuronal apoptosis. Mol. Neurobiol. 2017, 54, 470–483. [Google Scholar] [CrossRef]
- Song, W.; Patel, A.; Qureshi, H.Y.; Han, D.; Schipper, H.M.; Paudel, H.K. The Parkinson disease-associated A30P mutation stabilizes alpha-synuclein against proteasomal degradation triggered by heme oxygenase-1 over-expression in human neuroblastoma cells. J. Neurochem. 2009, 110, 719–733. [Google Scholar] [CrossRef]
- Volles, M.J.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41, 4595–4602. [Google Scholar] [CrossRef]
- He, Q.; Song, N.; Jia, F.; Xu, H.; Yu, X.; Xie, J.; Jiang, H. Role of α-synuclein aggregation and the nuclear factor E2-related factor 2/heme oxygenase-1 pathway in iron-induced neurotoxicity. Int. J. Biochem. Cell Biol. 2013, 45, 1019–1030. [Google Scholar] [CrossRef]
- Xu, J.; Xiao, C.; Song, W.; Cui, X.; Pan, M.; Wang, Q.; Feng, Y.; Xu, Y. Elevated heme oxygenase-1 correlates with increased brain iron deposition measured by quantitative susceptibility mapping and decreased hemoglobin in patients with Parkinson’s disease. Front. Aging Neurosci. 2021, 13, 122. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Song, N.; Guo, X.; Jiang, H.; Zhang, H.; Xie, J. Differences in vulnerability of neurons and astrocytes to heme oxygenase-1 modulation: Implications for mitochondrial ferritin. Sci. Rep. 2016, 6, 24200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zukor, H.; Song, W.; Liberman, A.; Mui, J.; Vali, H.; Fillebeen, C.; Pantopoulos, K.; Wu, T.D.; Guerquin-Kern, J.L.; Schipper, H.M. HO-1-mediated macroautophagy: A mechanism for unregulated iron deposition in aging and degenerating neural tissues. J. Neurochem. 2009, 109, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Cressatti, M.; Zukor, H.; Liberman, A.; Galindez, C.; Schipper, H.M. Parkinsonian features in aging GFAP.HMOX1 transgenic mice overexpressing human HO-1 in the astroglial compartment. Neurobiol. Aging 2017, 58, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, B.E.; Nishimura, R.N.; Lu, S.Y. Differential expression of heme oxygenase-1 in cultured cortical neurons and astrocytes determined by the aid of a new heme oxygenase antibody. Response to oxidative stress. Mol. Brain Res. 1995, 30, 37–47. [Google Scholar] [CrossRef]
- Manganaro, F.; Chopra, V.S.; Mydlarski, M.B.; Bernatchez, G.; Schipper, H.M. Redox perturbations in cysteamine-stressed astroglia: Implications for inclusion formation and gliosis in the aging brain. Free Radic. Biol. Med. 1995, 19, 823–835. [Google Scholar] [CrossRef]
- Snyder, S.H.; Jaffrey, S.R.; Zakhary, R. Nitric oxide and carbon monoxide: Parallel roles as neural messengers. Brain Res. Rev. 1998, 26, 167–175. [Google Scholar] [CrossRef]
- Schipper, H.M. Brain iron deposition and the free radical-mitochondrial theory of ageing. Ageing Res. Rev. 2004, 3, 265–301. [Google Scholar] [CrossRef]
- Gupta, A.; Lacoste, B.; Pistell, P.J.; Ingram, D.K.; Hamel, E.; Alaoui-Jamali, M.A.; Szarek, W.A.; Vlahakis, J.Z.; Jie, S.; Song, W.; et al. Neurotherapeutic effects of novel HO-1 inhibitors in vitro and in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2014, 131, 778–790. [Google Scholar] [CrossRef]
- Emir, U.E.; Tuite, P.J.; Oz, G. Elevated pontine and putamenal GABA levels in mild-moderate Parkinson disease detected by 7 tesla proton MRS. PLoS ONE 2012, 7, e30918. [Google Scholar] [CrossRef] [Green Version]
- Kish, S.J.; Rajput, A.; Gilbert, J.; Rozdilsky, B.; Chang, L.J.; Shannak, K.; Hornykiewicz, O. Elevated gamma-aminobutyric acid level in striatal but not extrastriatal brain regions in Parkinson’s disease: Correlation with striatal dopamine loss. Ann. Neurol. 1986, 20, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Oz, G.; Terpstra, M.; Tkac, I.; Aia, P.; Lowary, J.; Tuite, P.J.; Gruetter, R. Proton MRS of the unilateral substantia nigra in the human brain at 4 tesla: Detection of high GABA concentrations. Magn. Reson. Med. 2006, 55, 296–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cressatti, M.; Song, W.; Turk, A.Z.; Garabed, L.R.; Benchaya, J.A.; Galindez, C.; Liberman, A.; Schipper, H.M. Glial HMOX1 expression promotes central and peripheral alpha-synuclein dysregulation and pathogenicity in parkinsonian mice. Glia 2019, 67, 1730–1744. [Google Scholar] [PubMed]
- Tavitian, A.; Cressatti, M.; Song, W.; Turk, A.Z.; Galindez, C.; Smart, A.; Liberman, A.; Schipper, H.M. Strategic timing of glial HMOX1 expression results in either schizophrenia-like or parkinsonian behavior in mice. Antioxid. Redox Signal. 2019, 32, 1259–1272. [Google Scholar] [CrossRef]
- Chen, P.C.; Vargas, M.R.; Pani, A.K.; Smeyne, R.J.; Johnson, D.A.; Kan, Y.W.; Johnson, J.A. Nrf2-mediated neuroprotection in the MPTP mouse model of Parkinson’s disease: Critical role for the astrocyte. Proc. Natl. Acad. Sci. USA 2009, 106, 2933–2938. [Google Scholar] [CrossRef] [Green Version]
- Innamorato, N.G.; Jazwa, A.; Rojo, A.I.; Garcia, C.; Fernandez-Ruiz, J.; Grochot-Przeczek, A.; Stachurska, A.; Jozkowicz, A.; Dulak, J.; Cuadrado, A. Different susceptibility to the Parkinson’s toxin MPTP in mice lacking the redox master regulator Nrf2 or its target gene heme oxygenase-1. PLoS ONE 2010, 5, e11838. [Google Scholar] [CrossRef] [Green Version]
- Pan, P.K.; Qiao, L.Y.; Wen, X.N. Safranal prevents rotenone-induced oxidative stress and apoptosis in an in vitro model of Parkinson’s disease through regulating Keap1/Nrf2 signaling pathway. Cell. Mol. Biol. 2016, 62, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Tsou, Y.H.; Shih, C.T.; Ching, C.H.; Huang, J.Y.; Jen, C.J.; Yu, L.; Kuo, Y.M.; Wu, F.S.; Chuang, J.I. Treadmill exercise activates Nrf2 antioxidant system to protect the nigrostriatal dopaminergic neurons from MPP+ toxicity. Exp. Neurol. 2015, 263, 50–62. [Google Scholar] [CrossRef]
- Kapturczak, M.H.; Wasserfall, C.; Brusko, T.; Campbell-Thompson, M.; Ellis, T.M.; Atkinson, M.A.; Agarwal, A. Heme oxygenase-1 modulates early inflammatory responses: Evidence from the heme oxygenase-1-deficient mouse. Am. J. Pathol. 2004, 165, 1045–1053. [Google Scholar] [CrossRef]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010, 80, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Gunter, K.; Maines, M.D. Neurons overexpressing heme oxygenase-1 resist oxidative stress-mediated cell death. J. Neurochem. 2000, 75, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Panahian, N.; Yoshiura, M.; Maines, M.D. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J. Neurochem. 1999, 72, 1187–1203. [Google Scholar] [CrossRef] [PubMed]
- More, S.; Choi, D.K. Neuroprotective role of atractylenolide-I in an in vitro and in vivo model of Parkinson’s disease. Nutrients 2017, 9, 451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.A.; Kim, J.H.; Woo, S.Y.; Son, H.J.; Han, S.H.; Jang, B.K.; Choi, J.W.; Kim, D.J.; Park, K.D.; Hwang, O. A novel compound VSC 2 has anti-inflammatory and antioxidant properties in microglia and in Parkinson’s disease animal model. Br. J. Pharmacol. 2015, 172, 1087–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen-Roetling, J.; Song, W.; Schipper, H.M.; Regan, C.S.; Regan, R.F. Astrocyte overexpression of heme oxygenase-1 improves outcome after intracerebral hemorrhage. Stroke 2015, 46, 1093–1098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen-Roetling, J.; Kamalapathy, P.; Cao, Y.; Song, W.; Schipper, H.M.; Regan, R.F. Astrocyte heme oxygenase-1 reduces mortality and improves outcome after collagenase-induced intracerebral hemorrhage. Neurobiol. Dis. 2017, 102, 140–146. [Google Scholar] [CrossRef]
- Gai, W.P.; Yuan, H.X.; Li, X.Q.; Power, J.T.; Blumbergs, P.C.; Jensen, P.H. In situ and in vitro study of colocalization and segregation of alpha-synuclein, ubiquitin, and lipids in Lewy bodies. Exp. Neurol. 2000, 166, 324–333. [Google Scholar] [CrossRef]
- Mahul-Mellier, A.L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Zukor, H.; Lin, S.H.; Hascalovici, J.; Liberman, A.; Tavitian, A.; Mui, J.; Vali, H.; Tong, X.K.; Bhardwaj, S.K.; et al. Schizophrenia-like features in transgenic mice overexpressing human HO-1 in the astrocytic compartment. J. Neurosci. 2012, 32, 10841–10853. [Google Scholar] [CrossRef] [Green Version]
- Basak, I.; Patil, K.S.; Alves, G.; Larsen, J.P.; Moller, S.G. microRNAs as neuroregulators, biomarkers and therapeutic agents in neurodegenerative diseases. Cell. Mol. Life Sci. 2016, 73, 811–827. [Google Scholar] [CrossRef]
- Lukiw, W.J. Micro-RNA speciation in fetal, adult and Alzheimer’s disease hippocampus. Neuroreport 2007, 18, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.; Peplow, P.V. MicroRNAs in Parkinson’s disease and emerging therapeutic targets. Neural Regen. Res. 2017, 12, 1945. [Google Scholar] [PubMed]
- Lin, S.H.; Song, W.; Cressatti, M.; Zukor, H.; Wang, E.; Schipper, H.M. Heme oxygenase-1 modulates microRNA expression in cultured astroglia: Implications for chronic brain disorders. Glia 2015, 63, 1270–1284. [Google Scholar] [CrossRef] [PubMed]
- Doxakis, E. Post-transcriptional regulation of alpha-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fragkouli, A.; Doxakis, E. miR-7 and miR-153 protect neurons against MPP(+)-induced cell death via upregulation of mTOR pathway. Front. Cell. Neurosci. 2014, 8, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Je, G.; Kim, Y.S. Mitochondrial ROS-mediated post-transcriptional regulation of alpha-synuclein through miR-7 and miR-153. Neurosci. Lett. 2017, 661, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Ridzon, D.; Wong, L.; Chen, C. Characterization of microRNA expression profiles in normal human tissues. BMC Genom. 2007, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Long, J.M.; Ray, B.; Lahiri, D.K. MicroRNA-153 physiologically inhibits expression of amyloid-beta precursor protein in cultured human fetal brain cells and is dysregulated in a subset of Alzheimer disease patients. J. Biol. Chem. 2012, 287, 31298–31310. [Google Scholar] [CrossRef] [Green Version]
- Ji, Q.; Gao, J.; Zheng, Y.; Liu, X.; Zhou, Q.; Shi, C.; Yao, M.; Chen, X. Inhibition of microRNA-153 protects neurons against ischemia/reperfusion injury in an oxygen-glucose deprivation and reoxygenation cellular model by regulating Nrf2/HO-1 signaling. J. Biochem. Mol. Toxicol. 2017, 31, e21905. [Google Scholar] [CrossRef]
- Liang, C.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Deng, W.; Liu, Y.; Qin, C. MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 2012, 1455, 103–113. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, S.; Qi, W.; Xu, X.; Liang, Y. Overexpression of miR-153 promotes oxidative stress in MPP(+)-induced PD model by negatively regulating the Nrf2/HO-1 signaling pathway. Int. J. Clin. Exp. Pathol. 2018, 11, 4179–4187. [Google Scholar] [PubMed]
- Matsumoto, J.; Stewart, T.; Sheng, L.; Li, N.; Bullock, K.; Song, N.; Shi, M.; Banks, W.A.; Zhang, J. Transmission of alpha-synuclein-containing erythrocyte-derived extracellular vesicles across the blood-brain barrier via adsorptive mediated transcytosis: Another mechanism for initiation and progression of Parkinson’s disease? Acta Neuropathol. Commun. 2017, 5, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mateo, I.; Infante, J.; Sanchez-Juan, P.; Garcia-Gorostiaga, I.; Rodriguez-Rodriguez, E.; Vazquez-Higuera, J.L.; Berciano, J.; Combarros, O. Serum heme oxygenase-1 levels are increased in Parkinson’s disease but not in Alzheimer’s disease. Acta Neurol. Scand. 2010, 121, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Zheng, J.; Ma, J.; Wang, Z.; Shi, X.; Li, M.; Huang, S.; Hu, S.; Zhao, Z.; Li, D. Increased Plasma Heme Oxygenase-1 Levels in Patients with Early-Stage Parkinson’s Disease. Front. Aging Neurosci. 2021, 39. [Google Scholar] [CrossRef]
- Roi, A.; Rusu, L.C.; Roi, C.I.; Luca, R.E.; Boia, S.; Munteanu, R.I. A new approach for the diagnosis of systemic and oral diseases based on salivary biomolecules. Dis. Markers 2019, 2019, 8761860. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Schipper, H.M.; Velly, A.M.; Mohit, S.; Gornitsky, M. Salivary biomarkers of oxidative stress: A critical review. Free Radic. Biol. Med. 2015, 85, 95–104. [Google Scholar] [CrossRef]
- Orive, G.; Lopera, F.; Carro, E. Saliva is a Good Candidate to be the New Gold-Standard Sample for Neurodegenerative Diseases. J. Alzheimer’s Dis. JAD 2022. [Google Scholar] [CrossRef]
- Schipper, H.M. Heme oxygenase-1, role in brain aging and neurodegeneration. Exp. Gerontol. 2000, 35, 821–830. [Google Scholar] [CrossRef]
- Schipper, H.M.; Cisse, S.; Stopa, E.G. Expression of heme oxygenase-1 in the senescent and Alzheimer-diseased brain. Ann. Neurol. 1995, 37, 758–768. [Google Scholar] [CrossRef]
- Smith, M.A.; Kutty, R.K.; Richey, P.L.; Yan, S.D.; Stern, D.; Chader, G.J.; Wiggert, B.; Petersen, R.B.; Perry, G. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am. J. Pathol. 1994, 145, 42–47. [Google Scholar]
- Junn, E.; Lee, K.W.; Jeong, B.S.; Chan, T.W.; Im, J.Y.; Mouradian, M.M. Repression of alpha-synuclein expression and toxicity by microRNA-7. Proc. Natl. Acad. Sci. USA 2009, 106, 13052–13057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farh, K.K.; Grimson, A.; Jan, C.; Lewis, B.P.; Johnston, W.K.; Lim, L.P.; Burge, C.B.; Bartel, D.P. The widespread impact of mammalian MicroRNAs on mRNA repression and evolution. Science 2005, 310, 1817–1821. [Google Scholar] [CrossRef] [Green Version]
- Tagliafierro, L.; Glenn, O.C.; Zamora, M.E.; Beach, T.G.; Woltjer, R.L.; Lutz, M.W.; Chiba-Falek, O. Genetic analysis of alpha-synuclein 3′ untranslated region and its corresponding microRNAs in relation to Parkinson’s disease compared to dementia with Lewy bodies. Alzheimer’s Dement. 2017, 13, 1237–1250. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043–37053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.S.; Jakymiw, A.; Yao, B.; Pauley, B.A.; Carcamo, W.C.; Katz, J.; Cheng, J.Q.; Chan, E.K. High resolution of microRNA signatures in human whole saliva. Arch. Oral Biol. 2011, 56, 1506–1513. [Google Scholar] [CrossRef] [Green Version]
- Vallelunga, A.; Ragusa, M.; Di Mauro, S.; Iannitti, T.; Pilleri, M.; Biundo, R.; Weis, L.; Di Pietro, C.; De Iuliis, A.; Nicoletti, A.; et al. Identification of circulating microRNAs for the differential diagnosis of Parkinson’s disease and Multiple System Atrophy. Front. Cell. Neurosci. 2014, 8, 156. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yang, R.; Hu, B.L.; Lu, P.; Zhou, L.L.; He, Z.Y.; Wu, H.M.; Zhu, J.H. Reduced Circulating Levels of miR-433 and miR-133b Are Potential Biomarkers for Parkinson’s Disease. Front. Cell. Neurosci. 2017, 11, 170. [Google Scholar] [CrossRef] [Green Version]
- Fan, W.; Huang, F.; Zhu, X.; Li, D.; Fu, S.; He, H. The heme oxygenase system and oral diseases. Oral Dis. 2011, 17, 252–257. [Google Scholar] [CrossRef]
- Del Tredici, K.; Hawkes, C.H.; Ghebremedhin, E.; Braak, H. Lewy pathology in the submandibular gland of individuals with incidental Lewy body disease and sporadic Parkinson’s disease. Acta Neuropathol. 2010, 119, 703–713. [Google Scholar] [CrossRef]
- Roberts, H.L.; Brown, D.R. Seeking a mechanism for the toxicity of oligomeric alpha-synuclein. Biomolecules 2015, 5, 282–305. [Google Scholar] [CrossRef] [Green Version]
- Cressatti, M.; Galindez, J.M.; Juwara, L.; Orlovetskie, N.; Velly, A.M.; Eintracht, S.; Liberman, A.; Gornitsky, M.; Schipper, H.M. Characterization and heme oxygenase-1 content of extracellular vesicles in human biofluids. J. Neurochem. 2021, 157, 2195–2209. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.M.; Wiklander, P.B.; Nordin, J.Z.; Al-Shawi, R.; Wood, M.J.; Vithlani, M.; Schapira, A.H.; Simons, J.P.; El-Andaloussi, S.; Alvarez-Erviti, L. Systemic exosomal siRNA delivery reduced alpha-synuclein aggregates in brains of transgenic mice. Mov. Disord. 2014, 29, 1476–1485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galieva, L.R.; James, V.; Mukhamedshina, Y.O.; Rizvanov, A.A. Therapeutic Potential of Extracellular Vesicles for the Treatment of Nerve Disorders. Front. Neurosci. 2019, 13, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef] [PubMed]
- Haney, M.J.; Klyachko, N.L.; Zhao, Y.; Gupta, R.; Plotnikova, E.G.; He, Z.; Patel, T.; Piroyan, A.; Sokolsky, M.; Kabanov, A.V.; et al. Exosomes as drug delivery vehicles for Parkinson’s disease therapy. J. Control. Release 2015, 207, 18–30. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, X.; Xiang, X.; Grizzle, W.; Sun, D.; Zhang, S.; Axtell, R.C.; Ju, S.; Mu, J.; Zhang, L.; Steinman, L.; et al. Treatment of brain inflammatory diseases by delivering exosome encapsulated anti-inflammatory drugs from the nasal region to the brain. Mol. Ther. 2011, 19, 1769–1779. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Chu, Y.; Kordower, J.H. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol. Dis. 2007, 25, 134–149. [Google Scholar] [CrossRef]
- Harada, C.N.; Natelson Love, M.C.; Triebel, K.L. Normal cognitive aging. Clin. Geriatr. Med. 2013, 29, 737–752. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cressatti, M.; Schipper, H.M. Dysregulation of a Heme Oxygenase–Synuclein Axis in Parkinson Disease. NeuroSci 2022, 3, 284-299. https://doi.org/10.3390/neurosci3020020
Cressatti M, Schipper HM. Dysregulation of a Heme Oxygenase–Synuclein Axis in Parkinson Disease. NeuroSci. 2022; 3(2):284-299. https://doi.org/10.3390/neurosci3020020
Chicago/Turabian StyleCressatti, Marisa, and Hyman M. Schipper. 2022. "Dysregulation of a Heme Oxygenase–Synuclein Axis in Parkinson Disease" NeuroSci 3, no. 2: 284-299. https://doi.org/10.3390/neurosci3020020
APA StyleCressatti, M., & Schipper, H. M. (2022). Dysregulation of a Heme Oxygenase–Synuclein Axis in Parkinson Disease. NeuroSci, 3(2), 284-299. https://doi.org/10.3390/neurosci3020020