
Reactive Oxygen Species: Angels and Demons in the Life of a Neuron



Abstract
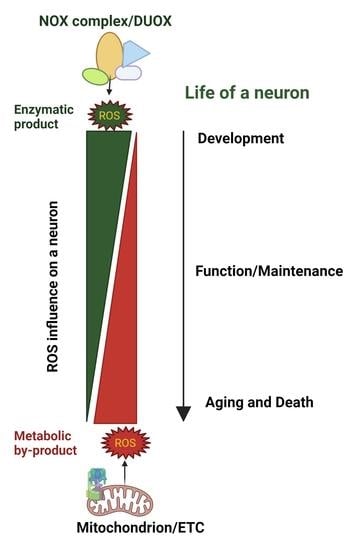
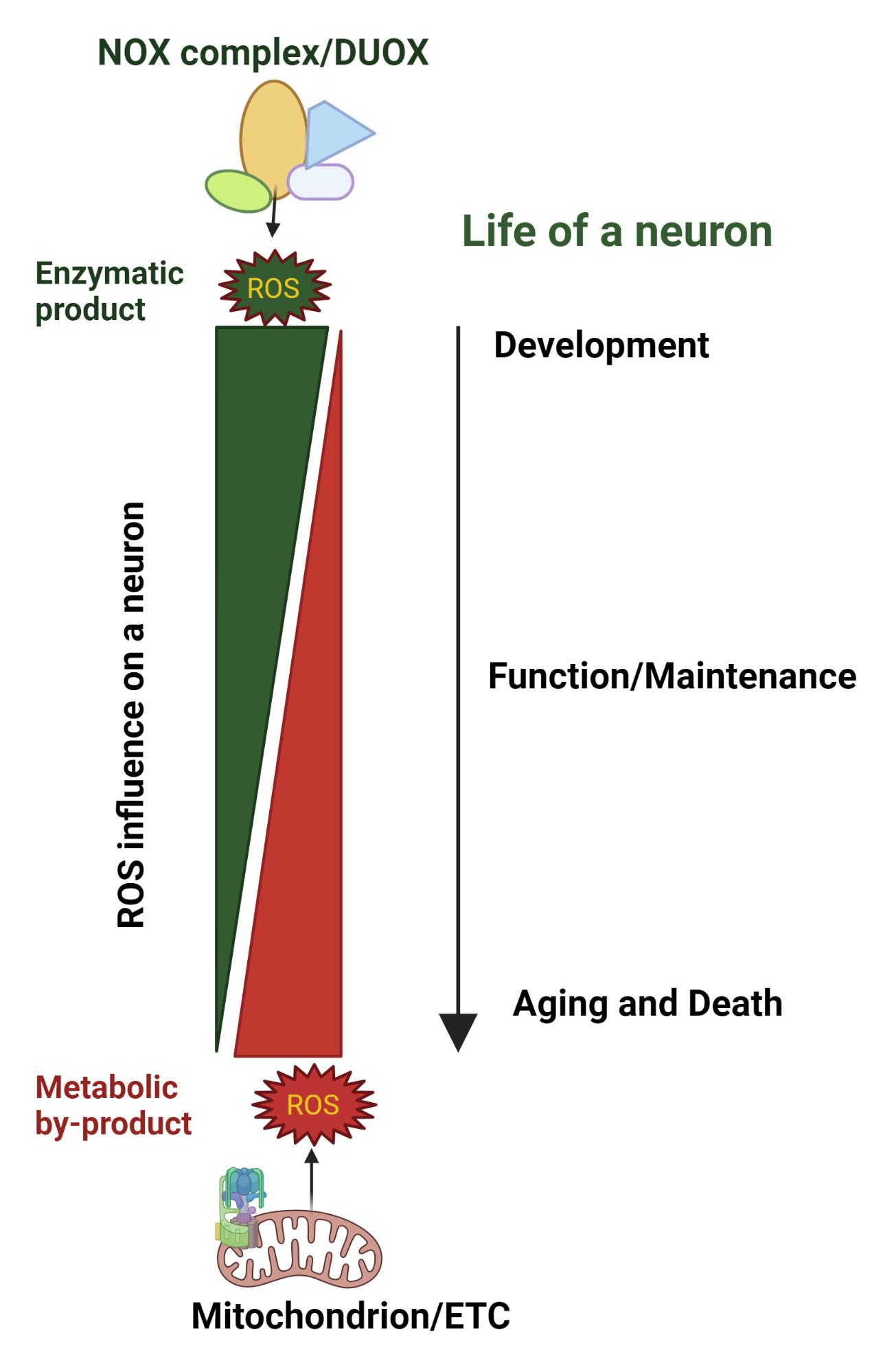{kind=link}
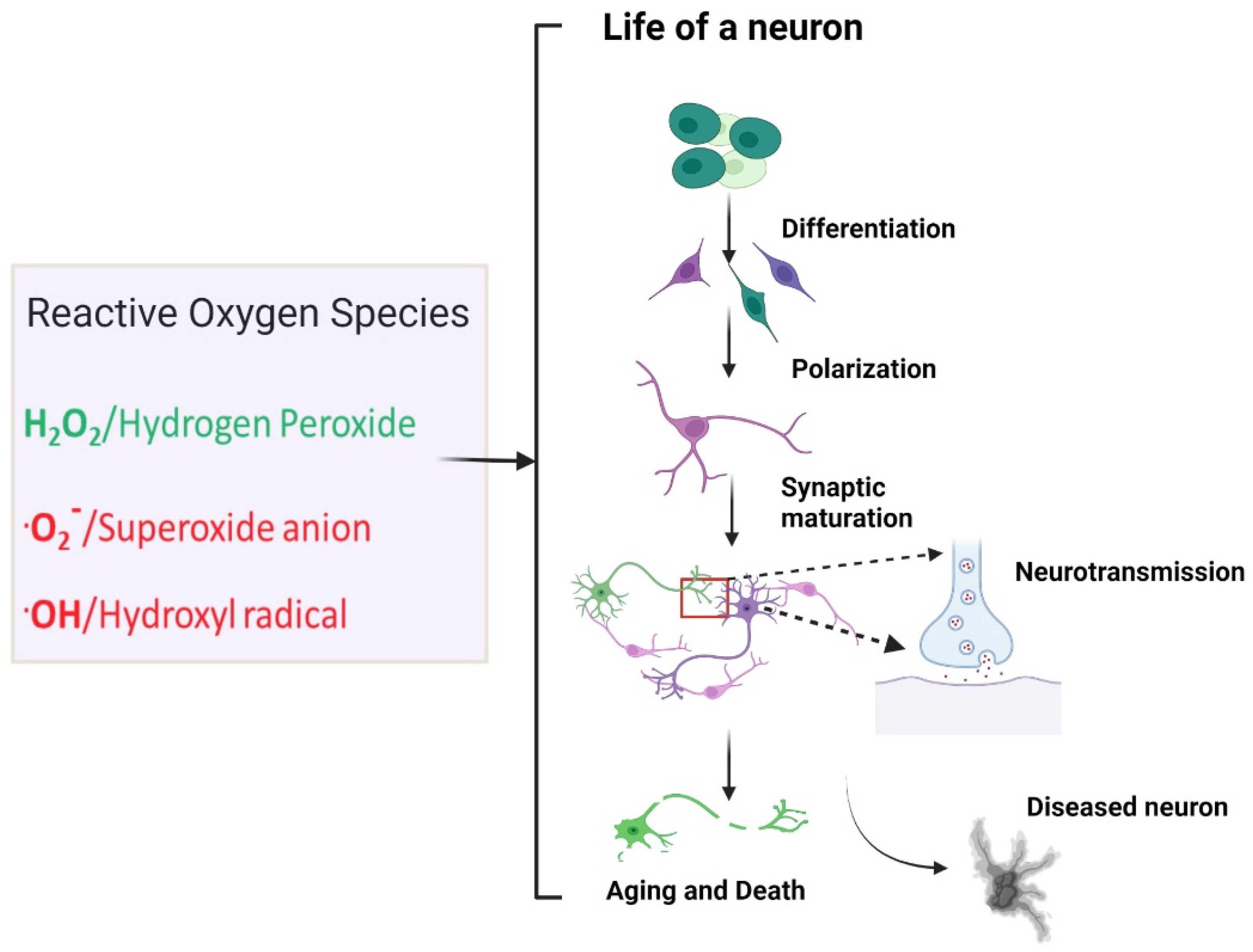{kind=link}
1. Introduction
Controlled Synthesis of ROS and Its Physiological Importance
2. ROS in Neurodevelopment
2.1. ROS in Neurogenesis and Differentiation
2.2. ROS in Neurite Outgrowth and Polarization
2.3. ROS Influence Growth Cone Guidance and Synaptic Maturation
2.4. Mitochondrial ROS Facilitate Synaptic Pruning by Intrinsic Apoptosis
2.5. Oxidative Damage in Neurodevelopmental Diseases
3. Roles for ROS in Mature Neurons
3.1. ROS in Synaptic Plasticity
3.2. ROS Influence Neurotransmission
4. The Dark Side of ROS in the Aging Brain
4.1. Oxidative Stress in Aging
4.2. Oxidative Damage in Neurodegenerative Disorders
4.3. ROS in Secondary Brain Injury
5. Emerging Concepts Linking Redox Biology and Neuroscience
6. Significance
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive Oxygen Species, Oxidative Damage, and Antioxidative Defense Mechanism in Plants under Stressful Conditions. J. Bot. 2012, 2012, 217037. [Google Scholar] [CrossRef]
- Riley, P.A. Free Radicals in Biology: Oxidative Stress and the Effects of Ionizing Radiation. Int. J. Radiat. Biol. 1994, 65, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Oswald, M.C.; Garnham, N.; Sweeney, S.T.; Landgraf, M. Regulation of Neuronal Development and Function by ROS. FEBS Lett. 2018, 592, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Fang, H.; Groom, L.; Cheng, A.; Zhang, W.; Liu, J.; Wang, X.; Li, K.; Han, P.; Zheng, M.; et al. Superoxide Flashes in Single Mitochondria. Cell 2008, 134, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Gross, E.; Sevier, C.S.; Heldman, N.; Vitu, E.; Bentzur, M.; Kaiser, C.A.; Thorpe, C.; Fass, D. Generating Disulfides Enzymatically: Reaction Products and Electron Acceptors of the Endoplasmic Reticulum Thiol Oxidase Ero1p. Proc. Natl. Acad. Sci. USA 2006, 103, 299–304. [Google Scholar] [CrossRef]
- del Río, L.A.; Sandalio, L.M.; Corpas, F.J.; Palma, J.M.; Barroso, J.B. Reactive Oxygen Species and Reactive Nitrogen Species in Peroxisomes. Production, Scavenging, and Role in Cell Signaling. Plant Physiol. 2006, 141, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Crane, F.L.; Low, H. Reactive Oxygen Species Generation at the Plasma Membrane for Antibody Control. Autoimmun. Rev. 2008, 7, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Kukreja, R.C.; Kontos, H.A.; Hess, M.L.; Ellis, E.F. PGH Synthase and Lipoxygenase Generate Superoxide in the Presence of NADH or NADPH. Circ. Res. 1986, 59, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, A.; Chattopadhyay, R.; Mitra, S.; Crowe, S.E. Oxidative Stress: An Essential Factor in the Pathogenesis of Gastrointestinal Mucosal Diseases. Physiol. Rev. 2014, 94, 329. [Google Scholar] [CrossRef]
- Georgiadou, E.C.; Kowalska, E.; Patla, K.; Kulbat, K.; Smolińska, B.; Leszczyńska, J.; Fotopoulos, V. Influence of Heavy Metals (Ni, Cu, and Zn) on Nitro-Oxidative Stress Responses, Proteome Regulation and Allergen Production in Basil (Ocimum basilicum L.) Plants. Front. Plant Sci. 2018, 9, 862. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Lopez-Delgado, H.; Dat, J.F.; Scott, I.M. Hydrogen Peroxide- and Glutathione-Associated Mechanisms of Acclimatory Stress Tolerance and Signalling. Physiol. Plant. 1997, 100, 241–254. [Google Scholar] [CrossRef]
- Pinto, E.; Sigaud-kutner, T.C.S.; Leitão, M.A.S.; Okamoto, O.K.; Morse, D.; Colepicolo, P. HEAVY METAL–INDUCED OXIDATIVE STRESS IN ALGAE1. J. Phycol. 2003, 39, 1008–1018. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [PubMed]
- Terrill, J.R.; Radley-Crabb, H.G.; Iwasaki, T.; Lemckert, F.A.; Arthur, P.G.; Grounds, M.D. Oxidative Stress and Pathology in Muscular Dystrophies: Focus on Protein Thiol Oxidation and Dysferlinopathies. FEBS J. 2013, 280, 4149–4164. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive Oxygen Species in Inflammation and Tissue Injury. Antioxid. Redox Signal. 2014, 20, 1126. [Google Scholar] [CrossRef] [PubMed]
- Green, K.; Brand, M.D.; Murphy, M.P. Prevention of Mitochondrial Oxidative Damage as a Therapeutic Strategy in Diabetes. Diabetes 2004, 53, S110–S118. [Google Scholar] [CrossRef]
- Aggarwal, V.; Tuli, H.S.; Varol, A.; Thakral, F.; Yerer, M.B.; Sak, K.; Varol, M.; Jain, A.; Khan, M.A.; Sethi, G. Role of Reactive Oxygen Species in Cancer Progression: Molecular Mechanisms and Recent Advancements. Biomolecules 2019, 9, 735. [Google Scholar] [CrossRef] [PubMed]
- Poh Loh, K.; Huang, S.H.; De Silva, R.; Tan, B.K.H.; Zhu, Y.Z. Oxidative Stress: Apoptosis in Neuronal Injury. Curr. Alzheimer Res. 2006, 3, 327–337. [Google Scholar] [CrossRef]
- Balasaheb Nimse, S.; Pal, D. Free Radicals, Natural Antioxidants, and Their Reaction Mechanisms. RSC Adv. 2015, 5, 27986–28006. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef]
- Sumimoto, H. Structure, Regulation and Evolution of Nox-Family NADPH Oxidases That Produce Reactive Oxygen Species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef] [PubMed]
- El, J.; My, P.; Dang, C.; Gougerot, A.; Elbim, C. Phagocyte NADPH Oxidase: A Multicomponent Enzyme Essential for Host Defenses. Arch. Immunol. Ther. Exp. 2005, 53, 199–206. [Google Scholar]
- Wu, J.X.; Liu, R.; Song, K.; Chen, L. Structures of Human Dual Oxidase 1 Complex in Low-Calcium and High-Calcium States. Nat. Commun. 2021, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Muñoz-Palma, E.; González-Billault, C. From Birth to Death: A Role for Reactive Oxygen Species in Neuronal Development. Semin. Cell Dev. Biol. 2018, 80, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Denu, J.M.; Tanner, K.G. Specific and Reversible Inactivation of Protein Tyrosine Phosphatases by Hydrogen Peroxide: Evidence for a Sulfenic Acid Intermediate and Implications for Redox Regulation†. Biochemistry 1998, 37, 5633–5642. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Qu, C.K.; Maeng, J.S.; Falahati, R.; Lee, C.; Williams, M.S. Receptor-Stimulated Oxidation of SHP-2 Promotes T-Cell Adhesion through SLP-76–ADAP. EMBO J. 2005, 24, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, T.; Pogrebniak, A.; BelAiba, R.S.; Bonello, S.; Wotzlaw, C.; Acker, H.; Hess, J.; Görlach, A. The Expression of the NADPH Oxidase Subunit P22phox Is Regulated by a Redox-Sensitive Pathway in Endothelial Cells. Free Radic. Biol. Med. 2005, 38, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, M.C.; Sharma, R.V.; Davisson, R.L. Superoxide Mediates Angiotensin II-Induced Influx of Extracellular Calcium in Neural Cells. Hypertension 2005, 45, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Nayernia, Z.; Jaquet, V.; Krause, K.-H. New Insights on NOX Enzymes in the Central Nervous System. Antioxid. Redox Signal. 2014, 20, 2815. [Google Scholar] [CrossRef] [PubMed]
- Mizuki, K.; Kadomatsu, K.; Hata, K.; Ito, T.; Fan, Q.W.; Kage, Y.; Fukumaki, Y.; Sakaki, Y.; Takeshige, K.; Sumimoto, H. Functional Modules and Expression of Mouse P40(Phox) and P67(Phox), SH3-Domain-Containing Proteins Involved in the Phagocyte NADPH Oxidase Complex. Eur. J. Biochem. 1998, 251, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Tsatmali, M.; Walcott, E.C.; Makarenkova, H.; Crossin, K.L. Reactive Oxygen Species Modulate the Differentiation of Neurons in Clonal Cortical Cultures. Mol. Cell. Neurosci. 2006, 33, 345. [Google Scholar] [CrossRef] [PubMed]
- Forsberg, K.; Wuttke, A.; Quadrato, G.; Chumakov, P.M.; Wizenmann, A.; Di Giovanni, S. The Tumor Suppressor P53 Fine-Tunes Reactive Oxygen Species Levels and Neurogenesis via PI3 Kinase Signaling. J. Neurosci. 2013, 33, 14318. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, E.; McKay, R. Proliferation and Differentiation of Neuronal Stem Cells Regulated by Nerve Growth Factor. Nature 1990, 347, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Suzukawa, K.; Miura, K.; Mitsushita, J.; Resau, J.; Hirose, K.; Crystal, R.; Kamata, T. Nerve Growth Factor-Induced Neuronal Differentiation Requires Generation of Rac1-Regulated Reactive Oxygen Species. J. Biol. Chem. 2000, 275, 13175–13178. [Google Scholar] [CrossRef]
- Vaskovsky, A.; Lupowitz, Z.; Erlich, S.; Pinkas-Kramarski, R. ErbB-4 Activation Promotes Neurite Outgrowth in PC12 Cells. J. Neurochem. 2000, 74, 979–987. [Google Scholar] [CrossRef]
- Goldsmit, Y.; Erlich, S.; Pinkas-Kramarski, R. Neuregulin Induces Sustained Reactive Oxygen Species Generation to Mediate Neuronal Differentiation. Cell. Mol. Neurobiol. 2001, 21, 753–769. [Google Scholar] [CrossRef] [PubMed]
- Claude, J.; Linnartz-Gerlach, B.; Kudin, A.P.; Kunz, W.S.; Neumann, H. Microglial CD33-Related Siglec-E Inhibits Neurotoxicity by Preventing the Phagocytosis-Associated Oxidative Burst. J. Neurosci. 2013, 33, 18270–18276. [Google Scholar] [CrossRef] [PubMed]
- Stemple, D.L.; Anderson, D.J. Isolation of a Stem Cell for Neurons and Glia from the Mammalian Neural Crest. Cell 1992, 71, 973–985. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Peltier, J.; Stone, D.; Schaffer, D.V.; Chang, C.J. Nox2 Redox Signaling Maintains Essential Cell Populations in the Brain. Nat. Chem. Biol. 2010, 7, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Hameed, L.S.; Berg, D.A.; Belnoue, L.; Jensen, L.D.; Cao, Y.; Simon, A. Environmental Changes in Oxygen Tension Reveal ROS-Dependent Neurogenesis and Regeneration in the Adult Newt Brain. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Núñez, M.T.; González-Billault, C. Contribution of NADPH Oxidase to the Establishment of Hippocampal Neuronal Polarity in Culture. J. Cell Sci. 2015, 128, 2989–2995. [Google Scholar] [CrossRef]
- Munnamalai, V.; Suter, D.M. Reactive Oxygen Species Regulate F-Actin Dynamics in Neuronal Growth Cones and Neurite Outgrowth. J. Neurochem. 2009, 108, 644–661. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.H.; Jan, L.Y.; Jan, Y.N. Hippocampal Neuronal Polarity Specified by Spatially Localized MPar3/MPar6 and PI 3-Kinase Activity. Cell 2003, 112, 63–75. [Google Scholar] [CrossRef]
- Leslie, N.R.; Bennett, D.; Lindsay, Y.E.; Stewart, H.; Gray, A.; Downes, C.P. Redox Regulation of PI 3-Kinase Signalling via Inactivation of PTEN. EMBO J. 2003, 22, 5501. [Google Scholar] [CrossRef]
- Wilson, C.; Muñoz-Palma, E.; Henríquez, D.R.; Palmisano, I.; Núñez, M.T.; Di Giovanni, S.; González-Billault, C. A Feed-Forward Mechanism Involving the NOX Complex and RyR-Mediated Ca2+ Release During Axonal Specification. J. Neurosci. 2016, 36, 11107–11119. [Google Scholar] [CrossRef]
- Yazdani, U.; Terman, J.R. The Semaphorins. Genome Biol. 2006, 7, 211. [Google Scholar] [CrossRef] [PubMed]
- Goshima, Y.; Nakamura, F.; Strittmatter, P.; Strittmatter, S.M. Collapsin-Induced Growth Cone Collapse Mediated by an Intracellular Protein Related to UNC-33. Nature 1995, 376, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Morinaka, A.; Yamada, M.; Itofusa, R.; Funato, Y.; Yoshimura, Y.; Nakamura, F.; Yoshimura, T.; Kaibuchi, K.; Goshima, Y.; Hoshino, M.; et al. Thioredoxin Mediates Oxidation-Dependent Phosphorylation of CRMP2 and Growth Cone Collapse. Sci. Signal. 2011, 4, ra26. [Google Scholar] [CrossRef] [PubMed]
- Terzi, A.; Roeder, H.; Weaver, C.J.; Suter, D.M. Neuronal NADPH Oxidase 2 Regulates Growth Cone Guidance Downstream of Slit2/Robo2. Dev. Neurobiol. 2021, 81, 3. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.T.; Davis, G.W. Unrestricted Synaptic Growth in Spinster—A Late Endosomal Protein Implicated in TGF-β-Mediated Synaptic Growth Regulation. Neuron 2002, 36, 403–416. [Google Scholar] [CrossRef]
- Milton, V.J.; Jarrett, H.E.; Gowers, K.; Chalak, S.; Briggs, L.; Robinson, I.M.; Sweeney, S.T. Oxidative Stress Induces Overgrowth of the Drosophila Neuromuscular Junction. Proc. Natl. Acad. Sci. USA 2011, 108, 17521–17526. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, I. Schizophrenia: Caused by a Fault in Programmed Synaptic Elimination during Adolescence? J. Psychiatr. Res. 1982, 17, 319–334. [Google Scholar] [CrossRef]
- Sakai, J. Core Concept: How Synaptic Pruning Shapes Neural Wiring during Development and, Possibly, in Disease. Proc. Natl. Acad. Sci. USA 2020, 117, 16096–16099. [Google Scholar] [CrossRef] [PubMed]
- Cobley, J.N. Synapse Pruning: Mitochondrial ROS with Their Hands on the Shears. BioEssays 2018, 40, 1800031. [Google Scholar] [CrossRef]
- Sidlauskaite, E.; Gibson, J.W.; Megson, I.L.; Whitfield, P.D.; Tovmasyan, A.; Batinic-Haberle, I.; Murphy, M.P.; Moult, P.R.; Cobley, J.N. Mitochondrial ROS Cause Motor Deficits Induced by Synaptic Inactivity: Implications for Synapse Pruning. Redox Biol. 2018, 16, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Hongpaisan, J.; Winters, C.A.; Andrews, S.B. Strong Calcium Entry Activates Mitochondrial Superoxide Generation, Upregulating Kinase Signaling in Hippocampal Neurons. J. Neurosci. 2004, 24, 10878–10887. [Google Scholar] [CrossRef] [PubMed]
- Oswald, M.C.W.; Brooks, P.S.; Zwart, M.F.; Mukherjee, A.; West, R.J.H.; Giachello, C.N.G.; Morarach, K.; Baines, R.A.; Sweeney, S.T.; Landgraf, M. Reactive Oxygen Species Regulate Activity- Dependent Neuronal Plasticity in Drosophila. eLife 2018, 7, e39393. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Perez, C.; Roy, S.S.; Naghdi, S.; Lin, X.; Davies, E.; Hajnóczky, G. Bid-Induced Mitochondrial Membrane Permeabilization Waves Propagated by Local Reactive Oxygen Species (ROS) Signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 4497–4502. [Google Scholar] [CrossRef]
- Ricci, J.-E.; Gottlieb, R.A.; Green, D.R. Caspase-Mediated Loss of Mitochondrial Function and Generation of Reactive Oxygen Species during Apoptosis. J. Cell Biol. 2003, 160, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of Apoptosis Signalling Pathways by Reactive Oxygen Species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Meng, L.; Mulcahy, B.; Cook, S.J.; Neubauer, M.; Wan, A.; Jin, Y.; Yan, D. The Cell Death Pathway Regulates Synapse Elimination through Cleavage of Gelsolin in Caenorhabditis Elegans Neurons. Cell Rep. 2015, 11, 1737–1748. [Google Scholar] [CrossRef]
- Rose, S.; Melnyk, S.; Pavliv, O.; Bai, S.; Nick, T.G.; Frye, R.E.; James, S.J. Evidence of Oxidative Damage and Inflammation Associated with Low Glutathione Redox Status in the Autism Brain. Transl. Psychiatry 2012, 2, e134. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Jernigan, S.; Pavliv, O.; Trusty, T.; Lehman, S.; Seidel, L.; Gaylor, D.W.; Cleves, M.A. A Functional Polymorphism in the Reduced Folate Carrier Gene and DNA Hypomethylation in Mothers of Children with Autism. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153B, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Al-Gadani, Y.; El-Ansary, A.; Attas, O.; Al-Ayadhi, L. Metabolic Biomarkers Related to Oxidative Stress and Antioxidant Status in Saudi Autistic Children. Clin. Biochem. 2009, 42, 1032–1040. [Google Scholar] [CrossRef]
- Pangrazzi, L.; Balasco, L.; Bozzi, Y. Oxidative Stress and Immune System Dysfunction in Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3293. [Google Scholar] [CrossRef]
- Bitanihirwe, B.K.Y.; Woo, T.-U.W. Oxidative Stress in Schizophrenia: An Integrated Approach. Neurosci. Biobehav. Rev. 2011, 35, 878. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.D.; Yao, J.K. Free Radical Pathology in Schizophrenia: A Review. Prostaglandins Leukot. Essent. Fat. Acids 1996, 55, 33–43. [Google Scholar] [CrossRef]
- Martins-de-Souza, D.; Gattaz, W.F.; Schmitt, A.; Novello, J.C.; Marangoni, S.; Turck, C.W.; Dias-Neto, E. Proteome Analysis of Schizophrenia Patients Wernicke’s Area Reveals an Energy Metabolism Dysregulation. BMC Psychiatry 2009, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, S.; Swatton, J.E.; Ryan, M.M.; Huffaker, S.J.; Huang, J.T.J.; Griffin, J.L.; Wayland, M.; Freeman, T.; Dudbridge, F.; Lilley, K.S.; et al. Mitochondrial Dysfunction in Schizophrenia: Evidence for Compromised Brain Metabolism and Oxidative Stress. Mol. Psychiatry 2004, 9, 684–697. [Google Scholar] [CrossRef]
- Dadheech, G.; Mishra, S.; Gautam, S.; Sharma, P. Evaluation of Antioxidant Deficit in Schizophrenia. Indian J. Psychiatry 2008, 50, 16. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.K.; Leonard, S.; Reddy, R. Altered Glutathione Redox State in Schizophrenia. Dis. Markers 2006, 22, 83. [Google Scholar] [CrossRef] [PubMed]
- Kuloglu, M.; Ustundag, B.; Atmaca, M.; Canatan, H.; Ertan Tezcan, A.; Cinkilinc, N. Lipid Peroxidation and Antioxidant Enzyme Levels in Patients with Schizophrenia and Bipolar Disorder. Cell Biochem. Funct. 2002, 20, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and Biochemistry of 4-Hydroxynonenal, Malonaldehyde and Related Aldehydes. Free Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Bulut, M.; Selek, S.; Gergerlioglu, H.S.; Savas, H.A.; Yilmaz, H.R.; Yuce, M.; Ekici, G. Malondialdehyde Levels in Adult Attention-Deficit Hyperactivity Disorder. J. Psychiatry Neurosci. 2007, 32, 435. [Google Scholar]
- Popa-Wagner, A.; Mitran, S.; Sivanesan, S.; Chang, E.; Buga, A.M. ROS and Brain Diseases: The Good, the Bad, and the Ugly. Oxid. Med. Cell. Longev. 2013, 2013, 963520. [Google Scholar] [CrossRef]
- Bulut, M.; Selek, S.; Bez, Y.; Cemal Kaya, M.; Gunes, M.; Karababa, F.; Celik, H.; Asuman Savas, H. Lipid Peroxidation Markers in Adult Attention Deficit Hyperactivity Disorder: New Findings for Oxidative Stress. Psychiatry Res. 2013, 209, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Malenka, R.C. Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacology 2007, 33, 18–41. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Arias-Cavieres, A. Calcium, Reactive Oxygen Species, and Synaptic Plasticity. Physiology 2016, 31, 201–215. [Google Scholar] [CrossRef] [PubMed]
- Massaad, C.A.; Klann, E. Reactive Oxygen Species in the Regulation of Synaptic Plasticity and Memory. Antioxid. Redox Signal. 2011, 14, 2013. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.T.; Klann, D.E. Sources and Targets of Reactive Oxygen Species in Synaptic Plasticity and Memory. Antioxid. Redox Signal. 2006, 9, 233–244. [Google Scholar] [CrossRef]
- Knapp, L.T.; Klann, E. Role of Reactive Oxygen Species in Hippocampal Long-Term Potentiation: Contributory or Inhibitory? J. Neurosci. Res. 2002, 70, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, S.; Myers, P.; Bailey, D.M.D.; Ostrovsky, A.D.; Evers, J.F.; Landgraf, M. Reactive Oxygen Species Mediate Activity-Regulated Dendritic Plasticity Through NADPH Oxidase and Aquaporin Regulation. Front. Cell. Neurosci. 2021, 15, 237. [Google Scholar] [CrossRef] [PubMed]
- Doser, R.L.; Amberg, G.C.; Hoerndli, F.J. Reactive Oxygen Species Modulate Activity-Dependent AMPA Receptor Transport in C. Elegans. J. Neurosci. 2020, 40, 7405–7420. [Google Scholar] [CrossRef] [PubMed]
- Hongpaisan, J.; Winters, C.A.; Andrews, S.B. Calcium-Dependent Mitochondrial Superoxide Modulates Nuclear CREB Phosphorylation in Hippocampal Neurons. Mol. Cell. Neurosci. 2003, 24, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A.; Colton, J.S.; Gilbert, D.L. Changes in Synaptic Transmission Produced by Hydrogen Peroxide. J. Free Radic. Biol. Med. 1986, 2, 141–148. [Google Scholar] [CrossRef]
- Beltrán González, A.N.; López Pazos, M.I.; Calvo, D.J. Reactive Oxygen Species in the Regulation of the GABA Mediated Inhibitory Neurotransmission. Neuroscience 2020, 439, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Penna, A.; Wang, D.S.; Yu, J.; Lecker, I.; Brown, P.M.G.E.; Bowie, D.; Orser, B.A. Hydrogen Peroxide Increases GABAA Receptor-Mediated Tonic Current in Hippocampal Neurons. J. Neurosci. 2014, 34, 10624. [Google Scholar] [CrossRef] [PubMed]
- Tsentsevitsky, A.N.; Zakyrjanova, G.F.; Petrov, A.M. Cadmium Desynchronizes Neurotransmitter Release in the Neuromuscular Junction: Key Role of ROS. Free Radic. Biol. Med. 2020, 155, 19–28. [Google Scholar] [CrossRef]
- Jia, Q.; Sieburth, D. Mitochondrial Hydrogen Peroxide Positively Regulates Neuropeptide Secretion during Diet-Induced Activation of the Oxidative Stress Response. Nat. Commun. 2021, 12, 1–22. [Google Scholar] [CrossRef]
- Zhao, T.; Hao, Y.; Kaplan, J.M. Axonal Mitochondria Modulate Neuropeptide Secretion Through the Hypoxic Stress Response in Caenorhabditis Elegans. Genetics 2018, 210, 275–285. [Google Scholar] [CrossRef] [PubMed]
- Sparaco, M.; Gaeta, L.M.; Santorelli, F.M.; Passarelli, C.; Tozzi, G.; Bertini, E.; Simonati, A.; Scaravilli, F.; Taroni, F.; Duyckaerts, C.; et al. Friedreich’s Ataxia: Oxidative Stress and Cytoskeletal Abnormalities. J. Neurol. Sci. 2009, 287, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Rolfe, D.F.S.; Brown, G.C. Cellular Energy Utilization and Molecular Origin of Standard Metabolic Rate in Mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef]
- Weil, A. Metabolism of the Nervous System. J. Neuropathol. Exp. Neurol. 1958, 17, 661. [Google Scholar] [CrossRef][Green Version]
- Attwell, D.; Laughlin, S.B. An Energy Budget for Signaling in the Grey Matter of the Brain. J. Cereb. Blood Flow Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial Proton and Electron Leaks. Essays Biochem. 2010, 47, 53. [Google Scholar] [CrossRef] [PubMed]
- Addabbo, F.; Montagnani, M.; Goligorsky, M.S. Mitochondria and Reactive Oxygen Species. Hypertension 2009, 53, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, M.; Ben-Shachar, D.; Riederer, P.; Youdim, M.B.H. Altered Brain Metabolism of Iron as a Cause of Neurodegenerative Diseases? J. Neurochem. 1994, 63, 793–807. [Google Scholar] [CrossRef] [PubMed]
- Case, A.J.; Li, S.; Basu, U.; Tian, J.; Zimmerman, M.C. Mitochondria in Cardiovascular Physiology and Disease: Mitochondrial-Localized NADPH Oxidase 4 Is a Source of Superoxide in Angiotensin II-Stimulated Neurons. Am. J. Physiol. Hear. Circ. Physiol. 2013, 305, H19. [Google Scholar] [CrossRef]
- Kozieł, R.; Pircher, H.; Kratochwil, M.; Lener, B.; Hermann, M.; Dencher, N.A.; Jansen-Dürr, P. Mitochondrial Respiratory Chain Complex I Is Inactivated by NADPH Oxidase Nox4. Biochem. J. 2013, 452, 231–239. [Google Scholar] [CrossRef] [PubMed]
- HARMAN, D. The Free Radical Theory of Aging: Effect of Age on Serum Copper Levels. J. Gerontol. 1965, 20, 151–153. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Gralla, E.B.; Valentine, J.S. Superoxide Dismutase Activity Is Essential for Stationary Phase Survival in Saccharomyces Cerevisiae. Mitochondrial Production of Toxic Oxygen Species in Vivo. J. Biol. Chem. 1996, 271, 12275–12280. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, P.; Liou, L.L.; Moy, V.N.; Diaspro, A.; Valentine, J.S.; Gralla, E.B.; Longo, V.D. SOD2 Functions Downstream of Sch9 to Extend Longevity in Yeast. Genetics 2003, 163, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Wawryn, J.; Krzepiłko, A.; Myszka, A.; Biliński, T. Deficiency in Superoxide Dismutases Shortens Life Span of Yeast Cells. Acta Biochim. Pol. 1999, 46, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Kirby, K.; Hu, J.; Hilliker, A.J.; Phillips, J.P. RNA Interference-Mediated Silencing of Sod2 in Drosophila Leads to Early Adult-Onset Mortality and Elevated Endogenous Oxidative Stress. Proc. Natl. Acad. Sci. USA 2002, 99, 16162–16167. [Google Scholar] [CrossRef] [PubMed]
- Duttaroy, A.; Paul, A.; Kundu, M.; Belton, A. A Sod2 Null Mutation Confers Severely Reduced Adult Life Span in Drosophila. Genetics 2003, 165, 2295–2299. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Huang, T.T.; Carlson, E.J.; Melov, S.; Ursell, P.C.; Olson, J.L.; Noble, L.J.; Yoshimura, M.P.; Berger, C.; Chan, P.H.; et al. Dilated Cardiomyopathy and Neonatal Lethality in Mutant Mice Lacking Manganese Superoxide Dismutase. Nat. Genet. 1995, 11, 376–381. [Google Scholar] [CrossRef]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, Myocardial Injury, and Perinatal Death in Mitochondrial Superoxide Dismutase-Deficient Mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Hekimi, S. Reactive Oxygen Species and Aging in Caenorhabditis Elegans: Causal or Casual Relationship? Antioxid. Redox Signal. 2010, 13, 1911–1953. [Google Scholar] [CrossRef] [PubMed]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a “Good” Look at Free Radicals in the Aging Process. Trends Cell Biol. 2011, 21, 569. [Google Scholar] [CrossRef]
- Van Raamsdonk, J.M.; Hekimi, S. Deletion of the Mitochondrial Superoxide Dismutase Sod-2 Extends Lifespan in Caenorhabditis Elegans. PLoS Genet. 2009, 5, e1000361. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Hekimi, S. Superoxide Dismutase Is Dispensable for Normal Animal Lifespan. Proc. Natl. Acad. Sci. USA 2012, 109, 5785–5790. [Google Scholar] [CrossRef] [PubMed]
- Van Remmen, H.; Ikeno, Y.; Hamilton, M.; Pahlavani, M.; Wolf, N.; Thorpe, S.R.; Alderson, N.L.; Baynes, J.W.; Epstein, C.J.; Huang, T.T.; et al. Life-Long Reduction in MnSOD Activity Results in Increased DNA Damage and Higher Incidence of Cancer but Does Not Accelerate Aging. Physiol. Genom. 2004, 16, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ikeno, Y.; Qi, W.; Chaudhuri, A.; Li, Y.; Bokov, A.; Thorpe, S.R.; Baynes, J.W.; Epstein, C.; Richardson, A.; et al. Mice Deficient in Both Mn Superoxide Dismutase and Glutathione Peroxidase-1 Have Increased Oxidative Damage and a Greater Incidence of Pathology but No Reduction in Longevity. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2009, 64, 1212. [Google Scholar] [CrossRef]
- Kujoth, C.C.; Hiona, A.; Pugh, T.D.; Someya, S.; Panzer, K.; Wohlgemuth, S.E.; Hofer, T.; Seo, A.Y.; Sullivan, R.; Jobling, W.A.; et al. Medicine: Mitochondrial DNA Mutations, Oxidative Stress, and Apoptosis in Mammalian Aging. Science 2005, 309, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Stefanatos, R.; Sanz, A. The Role of Mitochondrial ROS in the Aging Brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Finkel, T. Mitohormesis. Cell Metab. 2014, 19, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose-Response 2014, 12, 288–341. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Senchuk, M.M.; Dues, D.J.; Johnson, B.K.; Cooper, J.F.; Lew, L.; Machiela, E.; Schaar, C.E.; DeJonge, H.; Blackwell, T.K.; et al. Mitochondrial Unfolded Protein Response Transcription Factor ATFS-1 Promotes Longevity in a Long-Lived Mitochondrial Mutant through Activation of Stress Response Pathways. BMC Biol. 2018, 16, 147. [Google Scholar] [CrossRef] [PubMed]
- Lakowski, B.; Hekimi, S. Determination of Life-Span in Caenorhabditis Elegans by Four Clock Genes. Science 1996, 272, 1010–1013. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Bussière, F.; Hekimi, S. Mitochondrial Electron Transport Is a Key Determinant of Life Span in Caenorhabditis Elegans. Dev. Cell 2001, 1, 633–644. [Google Scholar] [CrossRef]
- Lee, S.J.; Hwang, A.B.; Kenyon, C. Inhibition of Respiration Extends C. Elegans Life Span via Reactive Oxygen Species That Increase HIF-1 Activity. Curr. Biol. 2010, 20, 2131–2136. [Google Scholar] [CrossRef]
- Aranda-Anzaldo, A.; Dent, M.A.R. Why Cortical Neurons Cannot Divide, and Why Do They Usually Die in the Attempt? J. Neurosci. Res. 2017, 95, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Castelli, V.; Benedetti, E.; Antonosante, A.; Catanesi, M.; Pitari, G.; Ippoliti, R.; Cimini, A.; d’Angelo, M. Neuronal Cells Rearrangement During Aging and Neurodegenerative Disease: Metabolism, Oxidative Stress and Organelles Dynamic. Front. Mol. Neurosci. 2019, 12, 132. [Google Scholar] [CrossRef] [PubMed]
- Angelova, P.R.; Abramov, A.Y. Role of Mitochondrial ROS in the Brain: From Physiology to Neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Elesela, S.; Lukacs, N.W. Role of Mitochondria in Viral Infections. Life 2021, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- White, M.G.; Saleh, O.; Nonner, D.; Barrett, E.F.; Moraes, C.T.; Barrett, J.N. Mitochondrial Dysfunction Induced by Heat Stress in Cultured Rat CNS Neurons. J. Neurophysiol. 2012, 108, 2203. [Google Scholar] [CrossRef]
- Kam, W.W.Y.; Banati, R.B. Effects of Ionizing Radiation on Mitochondria. Free Radic. Biol. Med. 2013, 65, 607–619. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Berezhnov, A.V.; Fedotova, E.I.; Zinchenko, V.P.; Dolgacheva, L.P. Interaction of Misfolded Proteins and Mitochondria in Neurodegenerative Disorders. Biochem. Soc. Trans. 2017, 45, 1025–1033. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S. Du Mitochondrial Aβ: A Potential Focal Point for Neuronal Metabolic Dysfunction in Alzheimer’s Disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Hoeffer, C.A.; Wong, H.; Massaad, C.A.; Zhou, P.; Iadecola, C.; Murphy, M.P.; Pautler, R.G.; Klann, E. Amyloid β-Induced Impairments in Hippocampal Synaptic Plasticity Are Rescued by Decreasing Mitochondrial Superoxide. J. Neurosci. 2011, 31, 5589. [Google Scholar] [CrossRef]
- Dias, V.; Junn, E.; Mouradian, M.M. The Role of Oxidative Stress in Parkinson’s Disease. J. Parkinsons. Dis. 2013, 3, 461. [Google Scholar] [CrossRef]
- Sulzer, D.; Surmeier, D.J. Neuronal Vulnerability, Pathogenesis, and Parkinson’s Disease. Mov. Disord. 2013, 28, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.D.; Zhao, X.; Li, Y.; Li, G.R.; Liu, X.L. Damage to Dopaminergic Neurons by Oxidative Stress in Parkinson’s Disease (Review). Int. J. Mol. Med. 2018, 41, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F. Oxidatively Modified Proteins in Aging and Disease. Free Radic. Biol. Med. 2002, 32, 797–803. [Google Scholar] [CrossRef]
- Cristóvão, A.C.; Guhathakurta, S.; Bok, E.; Je, G.; Yoo, S.D.; Choi, D.H.; Kim, Y.S. NADPH Oxidase 1 Mediates α-Synucleinopathy in Parkinson’s Disease. J. Neurosci. 2012, 32, 14465. [Google Scholar] [CrossRef]
- Browne, S.E.; Ferrante, R.J.; Beal, M.F. Oxidative Stress in Huntington’s Disease. Brain Pathol. 1999, 9, 147–163. [Google Scholar] [CrossRef]
- Kumar, A.; Ratan, R.R. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J. Huntingtons. Dis. 2016, 5, 217. [Google Scholar] [CrossRef] [PubMed]
- Schulte, J.; Littleton, J.T. The Biological Function of the Huntingtin Protein and Its Relevance to Huntington’s Disease Pathology. Curr. Trends Neurol. 2011, 5, 65–78. [Google Scholar]
- Quinti, L.; Naidu, S.D.; Träger, U.; Chen, X.; Kegel-Gleason, K.; Llères, D.; Connolly, C.; Chopra, V.; Low, C.; Moniot, S.; et al. KEAP1-Modifying Small Molecule Reveals Muted NRF2 Signaling Responses in Neural Stem Cells from Huntington’s Disease Patients. Proc. Natl. Acad. Sci. USA 2017, 114, E4676–E4685. [Google Scholar] [CrossRef] [PubMed]
- Shaw, I.C.; Fitzmaurice, P.S.; Mitchell, J.D.; Lynch, P.G. Studies on Cellular Free Radical Protection Mechanisms in the Anterior Horn from Patients with Amyotrophic Lateral Sclerosis. Neurodegeneration 1995, 4, 391–396. [Google Scholar] [CrossRef]
- Julien, J.P.; Kriz, J. Transgenic Mouse Models of Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 1013–1024. [Google Scholar] [CrossRef] [PubMed]
- Shibata, N.; Nagai, R.; Uchida, K.; Horiuchi, S.; Yamada, S.; Hirano, A.; Kawaguchi, M.; Yamamoto, T.; Sasaki, S.; Kobayashi, M. Morphological Evidence for Lipid Peroxidation and Protein Glycoxidation in Spinal Cords from Sporadic Amyotrophic Lateral Sclerosis Patients. Brain Res. 2001, 917, 97–104. [Google Scholar] [CrossRef]
- Rodrigo, R.; Fernandez-Gajardo, R.; Gutierrez, R.; Matamala, J.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative Stress and Pathophysiology of Ischemic Stroke: Novel Therapeutic Opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714. [Google Scholar] [CrossRef]
- Peters, O.; Back, T.; Lindauer, U.; Busch, C.; Megow, D.; Dreier, J.; Dirnagl, U. Increased Formation of Reactive Oxygen Species after Permanent and Reversible Middle Cerebral Artery Occlusion in the Rat. J. Cereb. Blood Flow Metab. 1998, 18, 196–205. [Google Scholar] [CrossRef]
- Kim, M.; Byun, J.; Chung, Y.; Lee, S.U.; Park, J.E.; Park, W.; Park, J.C.; Ahn, J.S.; Lee, S. Reactive Oxygen Species Scavenger in Acute Intracerebral Hemorrhage Patients: A Multicenter, Randomized Controlled Trial. Stroke 2021, 52, 1172–1181. [Google Scholar] [CrossRef]
- Urday, S.; Kimberly, W.T.; Beslow, L.A.; Vortmeyer, A.O.; Selim, M.H.; Rosand, J.; Simard, J.M.; Sheth, K.N. Targeting Secondary Injury in Intracerebral Haemorrhage—Perihaematomal Oedema. Nat. Rev. Neurol. 2015, 11, 111–122. [Google Scholar] [CrossRef]
- Wadsworth, T.L.; Bishop, J.A.; Pappu, A.S.; Woltjer, R.L.; Quinn, J.F. Evaluation of Coenzyme Q as an Antioxidant Strategy for Alzheimer’s Disease. J. Alzheimer’s Dis. 2008, 14, 225. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Wang, H.; Zhou, J.; Dai, W.; Zhu, Y.; Zhou, Y.; Wang, X.; Zhou, M. Baicalin Provides Neuroprotection in Traumatic Brain Injury Mice Model through Akt/Nrf2 Pathway. Drug Des. Devel. Ther. 2018, 12, 2497. [Google Scholar] [CrossRef]
- Fisher, D.W.; Bennett, D.A.; Dong, H. Sexual Dimorphism in Predisposition to Alzheimer’s Disease. Neurobiol. Aging 2018, 70, 308. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.; Laws, K.R.; Gale, T.M.; Kondel, T.K. Greater Cognitive Deterioration in Women than Men with Alzheimer’s Disease: A Meta Analysis. J. Clin. Exp. Neuropsychol. 2012, 34, 989–998. [Google Scholar] [CrossRef]
- Baldereschi, M.; Di Carlo, A.; Rocca, W.A.; Vanni, P.; Maggi, S.; Perissinotto, E.; Grigoletto, F.; Amaducci, L.; Inzitari, D. Parkinson’s Disease and Parkinsonism in a Longitudinal Study. Neurology 2000, 55, 1358–1363. [Google Scholar] [CrossRef]
- Ermakova, Y.G.; Bilan, D.S.; Matlashov, M.E.; Mishina, N.M.; Markvicheva, K.N.; Subach, O.M.; Subach, F.V.; Bogeski, I.; Hoth, M.; Enikolopov, G.; et al. Red Fluorescent Genetically Encoded Indicator for Intracellular Hydrogen Peroxide. Nat. Commun. 2014, 5, 5222. [Google Scholar] [CrossRef]
- Kirstein, J.; Morito, D.; Kakihana, T.; Sugihara, M.; Minnen, A.; Hipp, M.S.; Nussbaum-Krammer, C.; Kasturi, P.; Hartl, F.U.; Nagata, K.; et al. Proteotoxic Stress and Ageing Triggers the Loss of Redox Homeostasis across Cellular Compartments. EMBO J. 2015, 34, 2334–2349. [Google Scholar] [CrossRef]
- Gauron, C.; Meda, F.; Dupont, E.; Albadri, S.; Quenech’Du, N.; Ipendey, E.; Volovitch, M.; Del Bene, F.; Joliot, A.; Rampon, C.; et al. Hydrogen Peroxide (H2O2) Controls Axon Pathfinding during Zebrafish Development. Dev. Biol. 2016, 414, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Hanson, K.M.; Clegg, R.M. Two-Photon Fluorescence Imaging and Reactive Oxygen Species Detection Within the Epidermis. Methods Mol. Biol. 2005, 289, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Tang, G.; Zhu, J.; Wu, W.; Li, G.; Lin, X.; Liang, L.; Chai, C.; Zeng, Y.; Wang, F.; et al. Single-Cell Brain Atlas of Parkinson’s Disease Mouse Model. J. Genet. Genom. 2021, 48, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Mendiola, A.S.; Ryu, J.K.; Bardehle, S.; Meyer-Franke, A.; Ang, K.K.H.; Wilson, C.; Baeten, K.M.; Hanspers, K.; Merlini, M.; Thomas, S.; et al. Transcriptional Profiling and Therapeutic Targeting of Oxidative Stress in Neuroinflammation. Nat. Immunol. 2020, 21, 513. [Google Scholar] [CrossRef] [PubMed]
- Tain, R.-W.; Scotti, A.M.; Li, W.; Zhou, X.J.; Cai, K. Imaging Short-Lived Reactive Oxygen Species (ROS) with Endogenous Contrast MRI. J. Magn. Reson. Imaging 2018, 47, 222. [Google Scholar] [CrossRef]
- Hossain, I.; Tan, C.; Doughty, P.T.; Dutta, G.; Murray, T.A.; Siddiqui, S.; Iasemidis, L.; Arumugam, P.U. A Novel Microbiosensor Microarray for Continuous Ex Vivo Monitoring of Gamma-Aminobutyric Acid in Real-Time. Front. Neurosci. 2018, 12, 500. [Google Scholar] [CrossRef] [PubMed]
- Nash, K.M.; Ahmed, S. Nanomedicine in the ROS-Mediated Pathophysiology: Applications and Clinical Advances. Nanomedicine 2015, 11, 2033. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biswas, K.; Alexander, K.; Francis, M.M. Reactive Oxygen Species: Angels and Demons in the Life of a Neuron. NeuroSci 2022, 3, 130-145. https://doi.org/10.3390/neurosci3010011
Biswas K, Alexander K, Francis MM. Reactive Oxygen Species: Angels and Demons in the Life of a Neuron. NeuroSci. 2022; 3(1):130-145. https://doi.org/10.3390/neurosci3010011
Chicago/Turabian StyleBiswas, Kasturi, Kellianne Alexander, and Michael M. Francis. 2022. "Reactive Oxygen Species: Angels and Demons in the Life of a Neuron" NeuroSci 3, no. 1: 130-145. https://doi.org/10.3390/neurosci3010011
APA StyleBiswas, K., Alexander, K., & Francis, M. M. (2022). Reactive Oxygen Species: Angels and Demons in the Life of a Neuron. NeuroSci, 3(1), 130-145. https://doi.org/10.3390/neurosci3010011