Abstract
Pancreatic neuroendocrine neoplasms (PNENs) are a diverse group of rare tumor subtypes, representing less than 2% of all pancreatic tumors. Often detected late in the clinical course, they are associated with high rates of morbidity and mortality. Hereditary syndromes such as multiple endocrine neoplasia type-1 and von Hippel–Lindau are associated with the development of PNENs, although only a small portion of total tumors have a genetic basis. This review aims to explore the recent advances in laboratory diagnostics, imaging modalities, medical management, and surgical approaches to hormone-producing PNENs (including some common, less common, and some rare subtypes), with the goal of assisting physicians in the integration of evidence-based information into their practice.
1. Introduction
Pancreatic neuroendocrine neoplasms (PNENs) are a fascinating group of diverse pancreatic tumor subtypes with both local and systemic effects [1,2,3]. PNENs are rare, representing <2% of all pancreatic tumors, and tend to have a longer estimated median life expectancy than the most commonly occurring adenocarcinomas (4.1 years vs. 6 months, respectively) [3]. PNENs can be classified as functional (F-PNENs) or non-functional (NF-PNENs), depending on their ability to produce biologically active hormones [4,5]. NF-PNENs characteristically either do not produce hormones, produce hormones at a low enough level to not cause symptoms, or are associated with hormones that are asymptomatic, such as pancreatic polypeptide [6]. This review will focus exclusively on F-PNENs. F-PNENs are further divided into subtypes based on the specific hormone they hypersecrete, leading to a distinct hormonal syndrome. Common F-PNENs include insulinoma and gastrinoma; less common subtypes include VIPoma, glucagonoma, and the extremely rare ACTHoma [4]. The diagnosis and treatment strategies for the very rare F-PNENs are akin to those of other more common F-PNENs [7]. NF-PNENs represent the majority of PNENs, and many of the data for PNENs are skewed towards this subcategory of neoplasms. Therefore, data that describe PNENs in an all-encompassing manner may be less applicable when the interest is focused solely on F-PNEN subtypes.
Genetics: While the following section will present a brief genetic overview, several excellent reviews have previously covered the genetic landscape associated with F-PNENs in detail [5,8]. Approximately 17% of PNENs occur in hereditary syndromes such as multiple endocrine neoplasia type 1 (MEN-1), von Hippel–Lindau syndrome (VHL), and, very rarely, tuberous sclerosis complex (TSC) and neurofibromatosis (NF-1). These disorders consist of germline mutations in tumor suppressor genes and follow an autosomal dominant pattern of inheritance [8,9,10]. Those with a hereditary syndrome-associated F-PNEN tend to be diagnosed at a younger age and have multiple extra-pancreatic endocrine tumors, as these syndromes affect several different organs. This highlights the importance of whole-body imaging and discussion of family history during clinical evaluations. PNENs are detected in up to 80–100% of MEN-1 syndromes, but only a small portion of all PNENs are related to MEN-1 syndromes [11], suggesting that the majority of PNENs are sporadic. Common genetic drivers of the formation of sporadic F-PNENs are somatic mutations in the MEN1, DAXX, ATRX, and select mammalian target of rapamycin (mTOR) pathway genes [5]. The mutational status of these genes has been described to have prognostic impact. Sporadic MEN1 mutations are the most frequent, having been reported in 37–44% of nonfamilial PNENs [10,12].
Histopathology: A definitive diagnosis of an F-PNEN requires cytologic and histologic analysis. F-PNEN tissue can be collected for histopathological analysis perioperatively. Endoscopic ultrasound (EUS) with fine needle aspiration (FNA) or biopsy (FNB) is the preferred method of pancreatic tissue collection, allowing for a small sampling of an area of the tumor without invasive surgery. This may be preferred in cases where tumor grade could alter treatment plans [13]. The drawback of this method is that tumors may have heterogeneity with varying levels of tissue involvement [14]. For this reason, histopathologic examination of whole tumor samples may be preferred, as it allows for a more inclusive representation of the total tumor tissue. Studies have reported approximately 80% concordance between EUS-FNA grading and surgical grading, with EUS-FNA more likely to undergrade in instances of discordance [15,16].
F-PNENs arise from islets of the pancreas, which embryologically derive from neuroendocrine lineage, and therefore, by definition, are well differentiated. Cells are epithelial and typically appear monomorphic with abundant eosinophilic cytoplasm and round nuclei with dense, salt-and-pepper chromatin. They are arranged similarly to islets of Langerhans in nests or trabeculae of cells surrounded by stroma rich in vasculature [14,17]. The 2022 WHO Classification endorses a three-tiered grading system for most NETs, particularly those in the gastrointestinal (GI) and pancreatobiliary tract, as well as in the upper aerodigestive tract and salivary glands. The grading system based on proliferation assessed by mitotic rate and Ki-67 labeling index stratifies NETs into grade 1 (G1), grade 2 (G2), or grade 3 (G3), corresponding to low-grade, intermediate-grade, and high-grade categories. Tumor necrosis, though recognized as a potential adverse prognostic factor, is not included in the grading parameters of tumors in the GI and pancreaticobiliary tract [18]. Low-grade (G1) PNENs are defined by <2 mitosis/2 mm2 and a Ki-67 index < 3%; intermediate-grade (G2) have 2–20 mitosis/2 mm2 and a Ki-67 index range of 3–20%; while the high-grade (G3) PNENs appear with >20 mitosis/2 mm2 and a Ki-67 > 20%.
At least two neuroendocrine biomarkers should appear with immunohistochemical (IHC) staining to diagnose F-PNEN. Commonly used neuroendocrine IHC markers are chromogranin A, synaptophysin, and INSM [19,20]. Islet 1 and PAX6 are highly expressed in primary and metastatic PNENs and can be used to differentiate between NEN of pancreatic origin from other NENs [20]. This is particularly beneficial when a liver metastasis of unknown origin is found [20,21]. Laboratory work-up and evaluation of F-PNENs vary with clinical presentation and suspected type of F-PNEN and will be further discussed below.
Imaging characteristics and location within the pancreas vary per type of F-PNEN, and these are discussed in more detail in the following sections. Generally, first-line imaging is performed with CT or MRI (Figure 1). F-PNENs generally exhibit homogenous hyperenhancement on CT and hyperintensity on T2-weighted MRI [22]. Functional imaging modalities that are useful for the evaluation and staging of F-PNENs consist of somatostatin receptor scintigraphy (SRS) using radiolabeled octreotide and PET with gallium-68-dotatate-somatostatin analog (68GA-DOTA-SSA) [23]. Additionally, 18F-NOTA-Octreotide is an emerging alternative to 68GA-DOTA-SSA imaging, which may be more efficacious in detecting small, or low somatostatin receptor expressing, F-PNENs [24]. These modalities take advantage of the fact that the majority of PNENs express high levels of somatostatin receptors. Their utility is in detecting tumors that could not be localized with cross-sectional imaging and providing insight regarding the utility of somatostatin-based therapies.
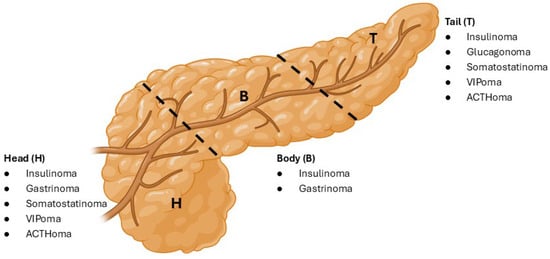
Figure 1.
Pancreatic localization of some F-PNEN subtypes. Consolidated from the current literature, this figure highlights the imaging-guided common localizations of F-PNET subtypes categorized into the pancreatic head, body and tail regions.
Treatment: Surgical resection remains the gold standard treatment, performed in 52–67% of cases [2,3]. Of PNENs removed surgically, approximately 42% are located in the tail, 33% in the head, and 23% in the body of the pancreas [1]. The most common surgical procedures are distal pancreatectomy or pancreaticoduodenectomy. According to recent studies, PNEN recurrence rates after surgical resection are 20–24%. This highlights the importance of continued surveillance [1,2]. For advanced unresectable or metastatic F-PNENs, cytoreductive debulking surgery may be considered in cases where 70–90% of visible metastasis can be resected [22]. Radiofrequency ablation (RFA), trans-arterial embolization (TAE), trans-arterial chemoembolization (TACE), or trans-arterial radio-embolization (TARE) offer options for reducing liver metastasis [22,25]. RFA may be considered for patients with less extensive, yet unresectable, liver metastasis due to relatively low morbidity and mortality risk or in combination with surgical resection [26,27]. In cases of high tumor burden or disease refractory to RFA, TAE/TACE/TARE is effective for reducing symptoms and increasing overall survival [26,28]. It should be noted that TAE/TACE/TARE come with more significant morbidity risk, with liver failure being an absolute contraindication to TACE [22,26]. Furthermore, TARE should be avoided in cases where future peptide receptor radionuclide therapy (PRRT) is planned, as it may preclude treatment [28]. Systemic therapies, including somatostatin analogues (SSA), chemotherapies, targeted therapies (everolimus and sunitinib), and PRRT, are a few of the treatments available for recurrent or unresectable F-PNEN [29]. These medications have varying efficacies based on the PNEN subtype. PRRT is the newest modality with continually emerging data exploring its efficacy [30]. It should be acknowledged that no dedicated subgroup analysis for F-NENs was present in the NETTER-1 trial, which led to FDA approval of PRRT treatment in advanced NENs [31]. However, there is growing evidence that PRRT is effective in managing the hormone-excess state, independent of its anti-growth effect [32,33]. Furthermore, PRRT is generally safe, although 10% of patients may suffer a hormonal crisis [32,34,35]. SSAs have shown evidence of synergistic effects in the treatment of NENs when combined with PRRT in the NETTER-2 trial and everolimus in the RADIANT-2 trial [24,36,37]. Some F-PNEN subtypes have specific therapies targeting the symptom-causing hormones, which are further discussed in the sections below.
PNENs have a variety of clinical presentations, prognoses, treatment options, and surgical approaches. This review will focus on F-PNENs, including the common, less common, and some extremely rare subtypes, with an aim to emphasize the most-up-to-date distinct characteristics for each, discussed with the intention of helping physicians integrate evidence-based research into their practice (Table 1).

Table 1.
A brief summarization of valuable rates, statistics and clinical symptoms for each F-PNEN subtype.
2. Insulinoma
Insulinoma is the most common F-PNEN [38]. It can occur at any age, although it tends to be most prevalent in the fifth decade of life [38]. Female sex appears more prevalent for low-grade cases, although malignant insulinoma shows no sex preference [39]. Insulinoma may have genetic associations with MEN-1 syndrome, TSC, and NF-1; thus, it is important to assess if there are family members diagnosed with hereditary endocrine disorders [22,39].
2.1. Morbidity and Mortality
Non-metastatic insulinoma is significantly more common, with 10–15% of insulinomas being classified as metastatic [39]. The 5-year survival rate for patients with low-grade insulinoma is reported as high as 94–100%, whereas malignant insulinoma survival rates range between 24 and 67% [40]. For surgically resected insulinoma, morbidity rates of 35.4% and 32.8% have been reported for open and laparoscopic resection, respectively, with the most common postoperative complications being fistula formation and diabetes mellitus [38].
2.2. Clinical Presentation
Whipple’s Triad, a combination of low blood glucose levels (<45 mg/dL), symptoms of hypoglycemia, and resolution of symptoms with carbohydrate ingestion, establishes a hypoglycemic disorder [22,41]. Patients with hypoglycemia may present with autonomic symptoms of nervousness, sweating, palpitations, shaking, or neuroglycopenic symptoms such as confusion, blurred vision, seizure, or coma [40,42,43]. Many patients present with weight gain corresponding to the onset of symptoms [43]. For diagnosis, timing of symptoms is a key clinical indicator, with hypoglycemia occurring during times of fasting, such as at night [22].
2.3. Laboratory Diagnostic Factors
If the patient is asymptomatic at presentation and alternative diagnoses to insulinoma have been excluded, a 72 h fasting glucose test should be performed [22]. At the onset of hypoglycemic symptoms, fasting plasma glucose, pro-insulin, insulin, and C-peptide should be measured [22,44]. With insulinoma, the concentrations of plasma insulin and C-peptide are paradoxically elevated in the setting of hypoglycemia [22]. Hypoglycemia can be defined as a recorded spontaneous blood glucose level of less than 45 mg/dL, and elevated insulin and C-peptide are characterized by laboratory values greater than 20.8 pmol/L and 0.2 pmol/L, respectively [22]. Recent changes to the NCCN guidelines include the addition of a fasting blood glucose in the initial laboratory evaluation to confirm hypoglycemia [44].
2.4. Imaging Features
A multiphasic CT or MRI is indicated in the initial evaluation of all insulinomas [44]. CT scans are between 83% and 88% sensitive for the detection of small F-PNENs during the arterial phase, and biphasic thin-section CT has been reported as 94% sensitive for insulinomas, which appear as circular, hyperenhancing lesions [45]. On MRI, PNENs are characterized by hypointensity in T1-weighted and hyperintensity in T2-weighted images [22]. CT in combination with EUS is the most sensitive imaging method for insulinoma, at 100% [45]. Targeted PET/CT or PET/MRI may also be useful in the evaluation of insulinoma. GLP-1R-based PET/CT and PET/MRI are more effective at both detection and localization of indolent insulinoma compared to somatostatin receptor-targeted imaging [46]. Yet, it is important to consider that only 36% of malignant insulinomas overexpress GLP-1R; thus, 68GA-DOTA-SSA PET is superior in these cases [47]. Arterial calcium stimulation with hepatic venous sampling may be used to help with localization if other imaging tests are inconclusive, although this method provides poor tumor localization [48].
2.5. Medical Management
In localized disease, initial management of hypoglycemia with diet, diazoxide, and/or everolimus is indicated before resection [44]. In unresectable or metastatic insulinoma, medical management is indicated. Therapy includes dietary management with sufficient complex carbohydrates in combination with diazoxide, and continuous glucose monitoring may be helpful for patients managing hypoglycemia through diet [22]. Long-acting octreotide or lanreotide SSA may also be used, although short-acting analogues should be first used to demonstrate somatostatin responsiveness [22,44,49]. Pasireotide has also demonstrated efficacy in the treatment of insulinoma [22,50]. In treating progressive insulinoma, everolimus and sunitinib are the first choice of therapies, particularly if refractory to SSA treatment [44,51,52]. Advanced insulinoma refractory to treatment with SSA may also be treated with 177Lu-DOTATATE PRRT [44,53]. Temozolomide in combination with capecitabine has recently shown efficacy in advanced NEN management, particularly for progression-free survival (PFS), and may additionally be utilized for progressive insulinoma [44,54]. Finally, cytotoxic chemotherapy may also be considered for symptomatic or progressive, unresectable insulinomas [44].
2.6. Surgical Management
Surgical management is the gold standard for localized insulinoma, with sparing of pancreatic parenchyma and relationship to the main pancreatic duct being important considerations [22,55]. Peripheral or exophytic tumors should undergo enucleation, whereas tumors near the main pancreatic duct or with invasion may be resected with pancreatoduodenectomy or distal pancreatectomy, depending on their location [44]. A minimally invasive approach should be considered for preoperatively localized insulinoma, whereas an open procedure in combination with intraoperative US is appropriate for tumors that cannot be preoperatively identified [22,44]. EUS-RFA has shown promising success rates of 95.5% for tumors less than 2 cm in diameter, with a reduced incidence of adverse events compared to surgical resection, although this technique is currently confined to centers with specific expertise [56].
2.7. Recent Changes and Emerging Therapies
EUS-RFA is a promising alternative to surgical resection in the management of small localized insulinomas, as it may significantly reduce the incidence of postoperative morbidity [56]. Additionally, pasireotide is emerging as an efficacious SSA in the medical treatment of refractory insulinoma, although its use in the disease remains off-label [40,50]. Another treatment option for metastatic insulinoma is liver-directed radioembolization or chemoembolization, which has shown high effectiveness in the clinical management of hypoglycemia in advanced metastatic insulinoma with a success rate of 100% [57]. Additionally, the option of cytoreductive debulking surgery in cases of metastatic and advanced regional disease is now recommended when complete resection is not possible [44].
3. Gastrinoma
Gastrinomas are the second most common F-PNEN following insulinoma and the most common duodenal NET [58,59,60]. The majority of gastrinomas are sporadic, but approximately 20% of these tumors are associated with MEN-1 syndrome [58,59,61,62]. There is conflicting data on preference for male or female sex, with some studies indicating a slight male preference [59,63,64]. The age range of sporadic tumors is typically 45–55 years, while MEN-1-associated gastrinomas occur in the 25–35-year-old age range [65]. Approximately 90% of all tumors are in the gastrinoma triangle, with a predominance for the duodenal bulb, while the remaining 10% of gastrinomas occur in the pancreatic tail [58,59,64,66,67].
3.1. Morbidity and Mortality
Unlike insulinoma, malignancy rates for gastrinoma are much higher and reported to be between 60 and 90% of all diagnosed gastrinomas, particularly in patients with concomitant MEN-1 syndrome [62,66]. Duodenal gastrinomas have less malignant potential than those occurring in the pancreas [58,68,69]. Survival for isolated gastrinomas without liver metastasis has been reported as up to 95% at 20 years after surgical excision; however, patients with metastatic spread to the liver have much worse outcomes with only 15% survival at 10 years [58,64,68]. Patients with MEN-1 syndrome present more frequently with distant metastasis, especially to the liver, and have worse outcomes than those with sporadic gastrinomas [66,70].
3.2. Clinical Presentation
Symptomatic gastrinomas lead to Zollinger–Ellison syndrome (ZES), which is characterized by diarrhea, GERD, and abdominal pain with recurrent peptic ulcer disease (PUD) [58,64,65,66]. In normal physiologic conditions, gastrin is released from G-cells located in the stomach and duodenum, which then stimulates the parietal cells of the stomach to release hydrochloric acid [59,64,71,72]. This acid then interacts with inactivated precursor molecules pepsinogen and chymotrypsin (from gastric chief cells), activating them to aid in the digestive process [73]. In the setting of gastrinoma, overproduction of gastrin leads to hypersecretion of gastric acid and results in severe PUD [58,59,64,66,74]. Clinical manifestations may be slightly blunted initially, as patients are frequently taking proton pump inhibitors (PPIs) and H2 receptor blockers prior to formal diagnosis [58,59,64,75].
3.3. Laboratory Diagnostic Factors
When a patient presents with recurrent PUD, serum gastrin levels should be measured. Gastrin values exceeding 1000 pg/mL are diagnostic of ZES/gastrinoma, while those with values > 100 pg/mL and associated clinical symptoms are highly suspicious for ZES/gastrinoma and warrant further workup with laboratory and imaging studies [58,64,72]. Gastric contents can be sampled during esophagogastroduodenoscopy and will show pH < 2 [59,66,67,76]. Hypercalcemia as well as elevated chromogranin A in the setting of PUD should also raise concern for ZES/gastrinoma [59,64,67]. Finally, a secretin stimulation test can be utilized for diagnosis—secretin normally inhibits gastrin production from G-cells; however, in the presence of a gastrinoma, secretin will paradoxically increase gastrin production >200 pg/mL over the course of 30–60 min and is diagnostic of the disease [58,64,74,75,76,77]. Importantly, patients on PPIs and H2 blockers must hold these medications for up to 2 weeks prior to laboratory evaluation for gastrinoma, as they will cause an underestimation of true serum gastrin levels [59,75].
3.4. Imaging Features
As with other F-PNENs, tumor localization with triphasic CT or MRI, as well as EUS with duodenal visualization, is crucial for surgical planning [67]. CT scans allow for the identification of both intra- and extra-pancreatic gastrinomas; MRI will reveal hypointense tumor characteristics on T-1 weighted imaging and hyperintense tumor characteristics on T-2 imaging [58,64,68]. Additional localization can be performed with somatostatin scintigraphy if CT or MRI fails to characterize the tumor well [67,70,78].
3.5. Medical Management
Medical management of gastrinomas is centered on symptom control of PUD. This is generally managed with PPIs and H2 blockers to suppress gastric acid production [58,79,80,81]. Long-term use and effects of these medications are still being studied to understand potential negative effects such as rebound hypersecretion of gastric acid if stopped, development of gastric tumors, and nutrient absorption impairment leading to vitamin deficiencies [80,82,83]. A somatostatin analog such as octreotide is also useful for decreasing gastrin secretion and has been shown to be an effective addition to acid suppression therapy [59,63,81,84]. This is particularly useful in patients with MEN-1 syndrome, who often present with advanced disease and may require multiple surgical interventions and multi-drug symptom control [84,85]. 177Lu-DOTATATE PRRT has shown efficacy in symptom control, tumor shrinkage, and decreased hormonal secretion [53,86]. Finally, chemotherapy is used in refractory cases as a second-line option, generally using the combination of doxorubicin and 5-FU [81,87,88]. While several retrospective studies support the activity of streptozocin-based therapy in combination with either 5-FU or doxorubicin, other studies have not confirmed significant efficacy of streptozocin with doxorubicin [89].
3.6. Surgical Management
The extent of surgical intervention is dependent upon the location of the gastrinoma. For tumors located within the gastrinoma triangle, enucleation is often attempted when feasible due to lower morbidity than pancreatic resection. This process can be aided by intraoperative EUS with transduodenal illumination for tumor demarcation during resection [58,59,64,90]. If the tumor involves the head of the pancreas with extension to the duodenum, or if enucleation is not safe, tumors in the gastrinoma triangle may require a Whipple procedure [58,85,90,91]. Finally, for the 10% of gastrinomas located in the pancreatic tail, a distal pancreatectomy is generally safe and effective for complete tumor resection [58,64]. If complete resection is not possible in a primary operation, patients can undergo staged resections with concurrent medical therapy for symptom control [85,90].
3.7. Recent Changes and Emerging Therapies
Although RFA has been described for other F-PNENs, it is not generally used for gastrinomas due to the availability of effective antihypersecretory medications [22]. Similarly, trans-arterial chemoembolization is not frequently performed when metastatic gastrinoma lesions are present in the liver [58,65,92]. However, in the setting of locally advanced gastrinomas with liver metastasis, liver transplant has grown in interest as a viable surgical option for management of liver lesions after primary tumor resection [58,64]. Finally, a recent study has shown that chemoradiotherapy for locally advanced unresectable disease is an effective method for managing tumor growth, even shrinking primary tumor size in several of their small patient cohort [93].
4. Glucagonoma
Glucagonomas represent approximately 2% of PNENs and present at an average age of 52.5 years [94,95,96]. A slight increase in prevalence for females has been reported [94,96]. Due to the rarity of the disease, there are limited data identifying racial correlations. Glucagonoma does have genetic associations, with between 1 and 5% of tumors occurring in patients with MEN-1 [44]. Thus, it is beneficial to evaluate for endocrine pathologies in a patient’s family history.
4.1. Morbidity and Mortality
Patients with localized and surgically resected glucagonoma have a very favorable prognosis, with a 10-year survival rate approaching 100% [94,96]. Approximately 35% of patients have clinically significant postoperative complications after laparoscopic or robotic surgery [97]. Unfortunately, 49.2% of patients are shown to have metastatic disease at the time of presentation [98]. In patients presenting with metastatic glucagonoma, the 10-year survival rate is reduced to approximately 50% [94,96]. The overall median survival time for glucagonoma is 7.7 years, although this is significantly shortened to 3.3 years in those with stage IV disease [69,94].
4.2. Clinical Presentation
Patients with glucagonoma may present with worsening of diabetes mellitus and significant weight loss [99]. Necrolytic migratory erythema (NME) is a characteristic dermatologic symptom in 80% of glucagonoma patients that consists of initial erythema of the skin and subsequent bullae, which ulcerate [99]. This forms a depressed lesion surrounded by brown pigment [94]. Typically, the rash is widespread, with involvement of the fingers, legs, feet, and perioral and perianal regions [100]. Additionally, oral findings may be present, with glossitis, cheilitis, and angular stomatitis being reported [99]. Furthermore, glucagonoma carries a risk of deep vein thrombosis, pulmonary embolism, normocytic anemia, hypo-aminoacidemia, and plasma zinc deficiency [94,101].
4.3. Laboratory Diagnostic Factors
The diagnosis of glucagonoma is aided by clinical indications in combination with elevated plasma glucagon levels. If the diagnosis of glucagonoma is suspected, blood glucose and glucagon testing should be performed [44]. A plasma glucagon level greater than 500 pg/mL is considered diagnostic for glucagonoma [94].
4.4. Imaging Features
For preoperative evaluation, multiphase MRI or CT may be performed [44]. The typical location is within the tail of the pancreas [44]. To evaluate metastatic involvement, PET/CT or PET/MRI is recommended with 68GA-DOTA-SST labeling due to the high somatostatin receptor expression in glucagonomas [44,94].
4.5. Medical Management
Supportive therapy may be indicated to correct nutrient abnormalities and control plasma glucose levels prior to surgical resection [102]. Additionally, SSA is indicated for symptomatic control prior to surgery. For localized glucagonomas, surgery is the only curative treatment, and patients should be placed on perioperative anticoagulation to minimize the risk of thromboembolic events [94].
In those patients with advanced unresectable or metastatic glucagonoma, SSA is first-line treatment [44]. These drugs have demonstrated improvements in PFS for both F- and NF-PNENs and reduce the NME rash for glucagonoma specifically [49,103]. Everolimus may be used as a second-line treatment, as it has shown reductions in plasma glucagon levels and a survival benefit, although this drug may contribute to the worsening of diabetic symptoms due to reductions in insulin secretion [104]. PRRT with 177Lu-DOTATATE has shown promise in the treatment of F-PNENs as a whole and specifically has a high symptomatic response rate, reduction in glucagon levels, and quality of life improvement for glucagonomas [53]. Similarly, sunitinib has demonstrated a survival benefit for F-PNENs, although results have not been reported for glucagonoma patients specifically [51]. A chemotherapeutic regimen of capecitabine and temozolomide has also demonstrated effectiveness in treating F-PNENs, although there is again no specific data for glucagonoma [54]. The chemotherapeutic combination of 5-flourouracil and streptozocin produced radiologic improvement in 50% of glucagonoma cases and may be indicated for progressive disease [105]. For liver-predominant metastatic disease, liver-directed therapy as discussed above may be indicated to reduce symptom burden [44].
4.6. Surgical Management
In the management of localized glucagonoma, surgical resection is the only curative treatment [94]. For tumors that occur in the distal pancreas, distal pancreatectomy in combination with peripancreatic lymphadenectomy and splenectomy is recommended [44]. For those rarer tumors that occur in the pancreatic head, pancreatoduodenectomy and peripancreatic lymphadenectomy are indicated [44].
An additional role of surgical management is in the cytoreduction of tumors that are metastatic or unresectable. In these cases, both the primary tumor and distant metastasis may be surgically excised to reduce tumor burden [44].
4.7. Recent Changes and Emerging Therapies
Recent changes to therapeutic directives include the updated guidance that temozolomide and capecitabine together are more efficacious in the treatment of advanced PNENs than temozolomide alone [54]. As with insulinoma, cytoreductive debulking surgery in cases of metastatic and advanced regional disease is now recommended when complete resection is not possible [44].
5. Somatostatinoma
Somatostatinomas constitute an exceedingly rare type of F-NEN of the GI tract, with most located in the pancreas and in the duodenum. Somatostatin, either in the 14- or 28-peptide form [106] and typically secreted from delta cells of the GI tract, is found in the stomach, small intestine, and pancreas [107,108]. Somatostatinomas of the GI tract have an annual incidence of 1 in 40,000,000 individuals [107,109,110], and specifically pancreatic somatostatinomas constitute approximately 1% to 4% of all PNENs [111,112]. Some studies have noted an equal incidence in both males and females, while others have found a 2:1 female predominance [109,111]. The average age at presentation with somatostatinomas is 50–55 years old, but studies have noted a range of cases in patients from 17 to 79 years old [111,113]. These tumors can be found throughout the pancreas, but a predilection for the pancreatic head has been observed in approximately 66% of cases [109,111,114].
5.1. Morbidity and Mortality
Survival rates of patients with pancreatic somatostatinomas are variable and limited throughout the literature [115]. Some studies have documented 5-year survival rates of 60–100% in patients with localized disease and 15–60% in those with disseminated disease [107]. Others have documented 5-year survival rates of 40% [116]. Approximately 60–75% of all pancreatic somatostatinomas are malignant and present with a high degree of local invasion and metastatic spread [109,116,117]. While a majority of somatostatinomas occur secondary to sporadic genetic mutations of sentinel tumor cells, there are associations of somatostatinoma development in patients with known genetic disorders, specifically MEN-1, the VHL syndromes, and NF-1 [107,116]. Patients with pathologic genetic variants of the hypoxia-inducible factor 2 alpha (HIF-2α) gene, also known as EPAS1, are also known to develop a triad of polycythemia, somatostatinomas, and pheochromocytomas/paragangliomas [118].
5.2. Clinical Presentation
Most patients are asymptomatic and present with an incidental finding on imaging or have non-specific symptoms such as weight loss, abdominal pain, and/or painless jaundice secondary to obstruction of the biliary tract due to the size of the tumor [107]. Due to the non-specific symptomatology, patients typically present with significantly progressive disease, frequently with evidence of metastases [107,110,119]. The most common sites of metastases include the liver and lymph nodes, with less frequent metastases to the lung and brain [111,120].
5.3. Laboratory Diagnostic Factors
These tumors are characterized by cells that stain grossly positive on IHC for chromogranin A, synaptophysin, and somatostatin [111]. On histologic analysis these tumors have been found to demonstrate trabecular growth patterns, with some demonstrating pseudoglandular components and others with paraganglioma-like appearance and psammoma bodies [111]. On serological evaluation, patients demonstrate increased serum levels of somatostatin, three times the upper limit of normal (>25–30 pg/mL) [109], and patients with significantly elevated somatostatin levels may present with somatostatinoma syndrome [107,111] characterized by diabetes mellitus, diarrhea, cholelithiasis, and hypochlorhydria/achlorhydria [109,111,113]. While this constellation of symptoms is distinctive for this tumor type, less than 10% of patients present with somatostatin syndrome [109,111]. Other serological diagnostic biomarkers that are increasing in their utility include circulating tumor cells, miRNA, mRNA, and metabolomics [121].
5.4. Imaging Features
Initial imaging studies used to identify these tumors and disseminated disease include CT and MRI [107]. For more specific and sensitive imaging, radiolabeled scans, including 68GA-DOTA-SSA PET/CT, indium-111-pentetreotide scintigraphy, 18fluoride-FDG-PET/CT, or 18fluoride-DOPA PET/CT, are performed [121].
5.5. Medical and Surgical Management
Curative treatment for pancreatic somatostatinomas is surgical resection of the primary tumor as well as resection, or debulking, of any metastatic lesions [122]. In patients with unresectable metastases to the liver, adjunct treatments are recommended, including microwave ablation, bland embolization, cryoablation, RFA, TAE, and TACE [122,123]. Patients with large tumors (>3 cm), tumors with poor cellular differentiation, and evidence of lymphatic spread have a worse prognosis [107]. Additionally, those who undergo surgical resection of their tumor and resection of the majority or all metastases have significantly better outcomes [124]. One study demonstrated a 10-year survival rate of 72% in patients who underwent aggressive surgical intervention for somatostatinomas of the duodenum and pancreas [111].
5.6. Recent Changes and Emerging Therapies
Therapies for metastatic disease include PRRT, which more specifically targets tumor receptors, and systemic cytotoxic chemotherapy regimens using a combination of streptozotocin and either doxorubicin or 5-fluorouracil [53,89,122,125]. While several retrospective studies support the activity of streptozocin-based therapy in combination with either 5-FU or doxorubicin, other studies have not confirmed significant efficacy of streptozocin with doxorubicin [89]. Finally, medical therapy to slow tumor progression and increase survival includes the use of mTOR inhibitors and SSAs such as everolimus and sunitinib [89].
6. VIPoma
6.1. Incidence/Epidemiology
VIPomas represent approximately 5% of all F-PNENs [126] and have been reported to represent 0.01% [127] to 1.5% of all PNEN cases [128,129,130] emphasizing their rare incidence. Data collection is difficult due to rarity, with individual studies using as low as three cases to perform analysis. VIPomas are primarily sporadic, with only 5–15% associated with MEN-1. They present at a median age of 51–69 years, with both males and females equally affected [126,127,130,131,132].
6.2. Morbidity and Mortality
A single-center study found that VIPomas had significantly decreased median survival time compared to NF-PNENs, insulinomas, and gastrinomas [129]. A possible explanation could be the lack of specific hormone excess targets for VIPomas compared to insulinomas [129]. Another explanation could be that >50% and up to 80% of VIPomas present with distant metastasis at diagnosis, with the liver being the most common site [126,127,128]. Within the last decade, 5-year postoperative overall survival rates have been reported at 93.3% [132], 67.7% [133], 63.6% [126,127], and 50% [129]. This variation is likely due, in part, to relatively small sample sizes for primary investigations, ranging from 4 to 15 patients. The 67.7% figure was derived from a systematic review of 65 patients and may represent the most accurate overall survival rate [133]. As for all F-PNENs, chromogranin A levels are an important consideration for VIPoma prognosis, specifically when considering post-resection recurrence. VIPomas have a higher recurrence rate than other F-PNENs at 40% [129,133]. Additional useful prognostic factors for VIPoma are increased Ki-67 index and plasma vasoactive intestinal peptide (VIP) concentration, which have been shown to significantly decrease overall survival [127].
6.3. Clinical Presentation
VIPomas are characterized by excess secretion of vasoactive intestinal peptide (VIP), leading to a group of symptoms commonly presenting together. These include watery diarrhea, hyperkalemia, and achlorydia, collectively referred to as WDHA syndrome or Verner–Morrison syndrome [128]. A systematic review of VIPoma reported that out of 65 cases of VIPoma, 27 patients (42.4%) presented with all three symptoms [133]. Secretory diarrhea is the most common symptom of VIPoma, presenting in up to 54.5% of cases [133]. Thus, any patient presenting with diarrhea should be considered for VIPoma as part of their differential. Excess blood VIP levels can lead to the following biologic changes: increased cardiac output, peripheral vasodilation, impairment of renal blood flow, and abnormal glucose tolerance [130]. Resulting fluid-electrolyte imbalances can lead to dehydration, acute renal failure, and non-anion gap metabolic acidosis, often requiring hospitalization [127]. Additional less prominent symptoms seen with VIPoma are weight loss and facial flushing [132].
6.4. Laboratory Diagnostic Factors
Due to their rare incidence, there are no defined guidelines for VIPoma diagnosis. Most diagnostic algorithms are based on extrapolations for established guidelines for the more common F-PNENs. Commonly, clinical symptoms combined with elevated fasting VIP levels are sufficient for diagnosis. Median VIP levels in confirmed cases of VIPoma can be as high as 636 pg/mL, but any value >200 pg/mL is considered diagnostic for VIPoma [133]. During asymptomatic periods, a patient’s VIP levels may appear normal, making biochemical work-up during the time of symptom onset critical [131]. Other causes of elevated VIP that should be ruled out are congestive heart failure, renal insufficiency, small bowel ischemia, and other etiologies of diarrhea [131,133]. Hypokalemia, hypochlorhydria, and hyperglycemia can help to confirm VIPoma diagnosis.
6.5. Imaging Features
VIPomas tend to occur in the tail of the pancreas (>50%) [127,130,132]. Other common locations are the head (20–30%) and body (15–20%) of the pancreas [128,132,133]. VIPomas are typically larger than other F-PNENs, with median tumor sizes reported as 44–57 mm, making them easily detectable on CT scans [132,133]. First-line imaging consists of multi-phasic contrast-enhanced CT scans [23,132], while MRI is second-line in evaluating F-PNENs [23,132]. The MRI findings are often inconclusive when suspecting VIPoma, as these tend to overlap with image findings for other F-PNENs. SRS can be utilized to visualize tumors that are undetectable on CT or MRI. SRS can also be utilized to determine how responsive a VIPoma may be to somatostatin analogs or targeted therapy with radiolabeled somatostatin analogs [23]. More than 80% of the time, patients with VIPoma will have positive SRS [130]. Recently, the FDA approved 68-Ga DOTATATE with PET for the detection of F-PNENs, including VIPomas, as it demonstrated greater sensitivity than SRS [23]. EUS-guided biopsy, or EUS-FNA, is a common method of obtaining tissue samples for histological analysis. In the case of VIPoma, which often occurs in the tail of the pancreas, it is important to note that sonographic visualization of pancreatic tail masses can be difficult due to a limited ultrasonic window [23]. VIPomas will stain positive for VIP with IHC and are often well-differentiated grade 2 tumors with a mean Ki-67% labeling index of 7.2 [132]. A higher Ki-67 index has been reported to significantly decrease 5-year overall survival [127]. Overall, to make a diagnosis of VIPoma, imaging is typically paired with symptoms and high laboratory values of plasma VIP or positive IHC for VIP. Additional imaging, such as SRS or 68Ga DOTATE with PET, is useful in guiding surgical and medical decisions. Imaging also plays an important role in monitoring response to treatment.
6.6. Medical Management
In the acute setting, rapid management of electrolytes, fluids, and diarrhea is critical to prevent severe complications such as organ damage. Steroids, clonidine, loperamide, or opioid-based treatments have been utilized for the specific treatment of VIPoma-related diarrhea [134]. Currently, no defined protocol for long-term treatment and management of VIPomas exists due to their rarity. The primary goals of medical therapies can be divided into anti-secretory (for symptom control) or anti-tumor (for tumor growth restriction). Many therapies have both anti-secretory and anti-tumor action.
First-line medical therapy is typically SSA, such as octreotide, lanreotide, and pasireotide. SSA can be prescribed pre- or postoperatively. SSA has a high rate of anti-secretory symptom relief initially (66.7%), but a 1-year symptom-free rate has been reported as 0% for VIPoma [127]. For this reason, SSA is rarely prescribed alone and is often paired with other drugs such as chemotherapies and/or molecular direct targeted therapies for a multi-modal approach. Common chemotherapies utilized for the treatment of VIPomas are streptozotocin and 5-FU [134]. Chemotherapies are often applied in the situation of progressive or recurring VIPoma metastasis. These are often second-line medical therapies for VIPoma with an 83.3% rate of initial symptom relief (vs. SSA 66.7%), a 50% 1-year secretory-control rate, and a 47.3% 1-year PFS [127]. The median PFS for chemotherapy without surgery has been reported as 9.2–30 months compared to 7.6–24.5 months for SSA [127,132]. The major drawback of chemotherapy for VIPoma is the high risk of toxicity and adverse effects, specifically the worsening of diarrhea. A benefit of chemotherapy is that it can be effective for both high- and low-somatostatin receptor-expressing VIPomas.
Sunitinib and everolimus are two types of molecular direct-targeted therapies that target somatostatin receptors and mTOR, respectively, highlighting the importance of SRS or 68Ga DOTATE with PET imaging when making treatment decisions. These drugs are commonly used as second- or third-line therapies for the treatment of VIPoma [127,132]. Sunitinib is shown to have better anti-secretory control versus everolimus, which provides slightly better anti-tumor/progression control but poor symptom control. Median PFS for both targeted therapies without surgery is around 11 months [127,132].
PRRT with lutetium 177 DOTATE is the newest treatment for PNENs with little evidence of efficacy in VIPoma, indicating a need for more VIPoma treatment studies [127]. A recent study showed PRRT to have the second-best tumor response, with chemotherapy being the first, for VIPoma treatment, alluding to its promising efficacy [132]. In this same study, PRRT used as monotherapy led to a median PFS of 26.5 months with complete resolution of symptoms, highlighting its anti-tumor and anti-secretory effect [132].
6.7. Surgical Management
Surgical resection remains the only curative option for non-metastatic VIPoma, with a study showing a 1-year tumor progression-free rate of 80% and a median PFS of 15–20 months (higher than the above medical therapies) [127,132]. A 5-year recurrence-free survival rate post-curative-intent surgery is as high as 60%, which is poor considering there is a greater than 50% chance of tumor recurrence despite surgery [127]. In the situation of metastasis, which is how the majority of VIPomas present, curative-intent surgery has promising results if the tumor is completely resectable with R0 margins. Alternatively, cytoreductive debulking surgical procedures can be performed in the instance of unresectable VIPoma metastasis with palliative intent. Approximately 47% of metastatic VIPoma cases are surgically treated [127,134]. Patients who undergo cytoreductive surgeries often have recurrence of symptoms and are frequently followed by multiple medical treatments [132]. Although surgery provides initial symptom relief and prolongs survival, a study reported that after curative-intent or cytoreductive surgery, only 18% of patients remained disease-free after 9.5 years [132]. Still, surgically treated VIPoma patients had prolonged median overall survival by 10 months when compared to non-surgically treated individuals (44 vs. 33 months) [132]. The surgical procedure commonly performed is a distal pancreatomy with and without splenectomy [133]. Compared to other F-PNENs, VIPomas tend to have increased postoperative complications and require reoperation [126]. SSA therapy is often prescribed perioperatively to control symptoms and prevent complications during operations, such as hypotensive crisis due to excess blood VIP levels. Neoadjuvant chemotherapy has also been utilized perioperatively [127,134].
Approximately 5% of VIPoma cases are inoperable [133]. Medical management is the mainstay of treatment in the case of unresectable tumors or patients who are not eligible for surgery. Chemotherapy is often the first line if initially unresponsive to SSA therapy, followed by the addition of molecular targeted therapies such as sunitinib and everolimus if symptoms persist. TACE is an emerging procedure that can be used to help shrink unresectable tumors to an operable size or employed as a method of cytoreduction and symptom relief for patients who are not surgical candidates and have failed medical treatments [134]. TACE is a newer therapeutic option for locoregional cytoreduction of liver metastases and is often utilized in a multi-modal treatment plan along with SSAs or sunitinib for symptom control. TACE is scarcely utilized for VIPoma but has been reported to lead to a median PFS of 17 months and a 1-year progression-free survival rate of 42.8% [127,132]. Further research regarding the utilization of TACE for VIPoma is warranted.
6.8. Recent Changes and Emerging Therapies
Surveillance is extremely important not just for detecting recurrence but also due to reports of PNENs initially diagnosed as NF-PNEN becoming functional VIPomas during the course of a study [129]. Due to their extremely low incidence, utilization of multidisciplinary tumor boards is highly valuable and recommended when making treatment decisions [127]. The majority of VIPomas are detected with distant disease, thus limiting treatment options and worsening prognosis [127,128,135]. This highlights the need for research to identify novel tests that would allow detection of VIPomas at an earlier stage, thus decreasing the incidence of distant disease and improving prognosis.
7. ACTHoma
ACTHoma, alternatively known as ectopic Cushing’s syndrome, is an exceedingly rare disease, comprising less than 1.2% of all F-PNEN cases [136]. Women account for 66.4% of reported ACTHomas, and the mean age of diagnosis is 44.7 years, which is remarkably younger than other tumor types included in this review [137]. Approximately 1.5% of ACTHomas diagnosed are concurrent with heritable endocrine diseases, including MEN-1 and VHL [137].
7.1. Morbidity and Mortality
84.3% of patients with ACTHoma have metastatic disease at presentation, and the prognosis is relatively poor. In one study, only about 10% of patients with recorded follow-up survived without disease-related symptoms [137].
7.2. Clinical Presentation
The clinical features of pancreatic ACTHoma can mimic those of hypercortisolism, hyperaldosteronism, and excessive sex steroid production. Addisonian pigmentation, characterized by hyperpigmentation of the skin in sun-exposed areas, is a key early indicator of hypercortisolism [138]. Hypokalemia, diabetes mellitus, muscle weakness, moon facies, hirsutism, central obesity, and edema may also be present [22,137].
7.3. Laboratory Diagnostic Factors
Laboratory findings of ACTHoma may include elevated levels of adrenocorticotropic hormone (ACTH), hypercortisolism without diurnal variation, and elevated 24 h urine cortisol [139]. A dexamethasone suppression test will not show suppression of fasting blood cortisol or ACTH levels, which establishes an ectopic source of ACTH secretion [139]. Following these laboratory results, imaging is necessary to localize the ectopic source of ACTH to the pancreas.
7.4. Imaging Features
ACTHomas most commonly occur in the pancreatic tail (44.4%) and head (34.9%) [137]. Like other F-PNENs, multiphasic CT or MRI may be used for the localization or detection of ACTHomas [137]. Because of its high sensitivity, EUS may be useful in cases where other imaging modalities fail to detect or localize the tumor, or where tumor biopsy is needed [140]. Additionally, the most sensitive diagnostic and staging procedure is PET/CT or PET/MRI with 68DOTA SST radiolabeling [141,142].
7.5. Medical Management
Medical management of ACTHoma is indicated when surgical interventions are unable to control the tumor burden or symptoms [137]. The first-line medical management for pancreatic ACTHoma includes enzymatic inhibitors such as metyrapone, osilodrostat, ketoconazole, etomidate, and fluconazole [22,141]. Combination therapy of octreotide and cabergoline has also shown reductions in ACTH levels and symptomatic improvement [143]. Mifepristone may increase symptomatic response to octreotide therapy by upregulating somatostatin receptors [144]. In tumors that are refractory to medical management, 177Lu-DOTATATE PRRT therapy may be helpful in the management of symptomatic and tumor burden, although limited data exist for its usage in ACTHoma [53,141,145]. Patients undergoing medical or surgical therapy for ACTHoma may require replacement or supplementation of endogenous cortisol [143].
7.6. Surgical Management
As with other F-PNENs, surgical management remains the only curative treatment option for ACTHoma, and primary tumor resection may be indicated for symptomatic management in cases of metastatic disease [146]. For tumors that occur in the pancreatic head, enucleation or pancreaticoduodenectomy may be considered depending on the extent of involvement [146]. Tumors in the pancreatic tail may be resected with distal pancreatectomy, with or without concomitant splenectomy [146]. In patients with metastatic disease, cytoreductive surgery may assist in controlling hormonal symptoms [141]. When metastatic involvement is liver predominant, liver-directed therapy such as TAE, TACE, or RFA may be helpful [141].
7.7. Recent Changes and Emerging Therapies
Due to the relative rarity of ACTHoma, treatment updates specific to the tumor type occur infrequently. Despite this, PRRT therapy with 177Lu-DOTATATE is an emerging option in the treatment of ACTHoma and other PNENs refractory to enzymatic modulators and surgery. Although data are limited, reductions in plasma cortisol levels and tumor burden have been reported, offering a promising option for patients who have exhausted other therapies [145].
8. F-PNENs: Insights and Associated Challenges
PNENs represent a complex and fascinating group of tumors, intertwining both endocrine and oncologic elements. The presence of a functional syndrome makes the management of these uncommon types of cancers challenging, as clinicians must manage both tumor growth and hormonal hypersecretion [147]. Consequently, diagnosis and treatment require extensive experience [148]. Furthermore, NF-PNENs represent the majority of PNENs; therefore, the bulk of data for PNENs is skewed towards this subcategory. Thus, data describing PNENs overall may be less applicable when focusing on F-PNEN subtypes.
It has been noted that accurate diagnosis of F-PNENs is increasingly challenging [149]. A knowledge gap exists, where F-PNENs such as the insulinoma and gastrinoma are well characterized, while the rarer subtypes, owing to their clinical prevalence or available limited evidence, have a paucity in reference and discussion (as also noted in this review). As per the latest guidelines from ENETS and NANETS, F-PNENs have plasma elevations of specific neuropeptides/biomarkers and an appropriate constellation of associated symptoms presenting as syndromes or hormone excess-specific symptoms [22,89]. Insulin, gastrin, glucagon, VIP, somatostatin, and adrenocorticotropic hormone (ACTH) are the most common elevated hormones in F-PNENs. Once an F-PNEN is diagnosed, continued measurement of the specific hormone before and after therapeutic intervention, in conjunction with radiologic testing, can help determine tumor progression, recurrence, and response to therapy [89,150]. Before planning any invasive treatment (such as surgery, locoregional therapy, or PRRT), the clinician must consider the possibility of hormonal crisis induced by the procedure. The prevention of hormonal crises must be considered in all patients, especially those with insulinoma, VIPoma, and gastrinoma [147]. Furthermore, it has been appreciated that the clinical presentation of the more common gastrinoma/ZES is further nuanced due to the widespread use of PPIs [149]. As discussed within the NANETS guidelines, continuing PPI use can lead to chronically elevated gastrin levels, whereas stopping the PPI will reverse hypergastrinemia unless there is underlying atrophic gastritis [89]. However, PPI withdrawal is potentially risky in gastrinoma patients such that routine withdrawal for the purpose of gastrin measurement is not always advisable [89].
The majority of F-PNENs are indolent and slow-growing but still preserve malignant potential [151]. There is a high degree of heterogeneity in tumor behavior, ranging from nearly benign to extremely aggressive [152], with significant biological and clinical heterogeneity observed within the same subtype. For example, the vast majority (90%) of the insulinomas are benign and localized in the pancreas, but a small fraction (10%) can be malignant and aggressive [153]. This reflects the difficulty of developing experimental models that can capture the heterogeneous characteristics of PNENs. A large proportion of well-differentiated PNENs are diagnosed at more advanced stages and, if not detected early, grow and eventually spread to the liver, making this progression the most common cause of mortality [148].
Tumor heterogeneity contributes to errors in the choice of treatment strategy, especially in metastatic disease, because the expression of targets for therapy in the primary tumor may not necessarily warrant their expression in metastases [154]. PET/CT imaging overcomes these limitations by detecting heterogeneity in the primary tumor and metastases, as well as identifying target expression throughout the body, which is essential for clinical decision-making. In this context, advances in molecular imaging using dual tracer PET with [68Ga] Ga-DOTA-SSTR and [18F] F-FDG are emerging as a potential tool to investigate lesion differentiation, affecting patient management [155,156]. Imaging studies with PET tracers acting as fibroblast activation protein inhibitors have also shown promising results that could usefully complement [18F]-FDG in cancer imaging [157] and may be efficacious in the diagnostic workup for PNENs by providing relevant information regarding tumor behavior and aggressiveness [158,159].
9. F-PNEN Therapies: Clinical Synthesis and Confines
Recent excellent reviews have thoroughly consolidated information related to the status of SSAs, PRRT, and other associated therapies for NENs [24,156,160]. To further consolidate this information is beyond the scope of this review, but the fact remains: there is a lack of trials and directives focusing specifically on the management of F-PNENs.
Systemic therapies, including SSAs, PRRT, targeted therapies (everolimus and sunitinib), and chemotherapies, are some of the established treatments for recurrent or unresectable F-PNENs. SSAs have emerged as a foundational therapy in NEN treatment, exerting antisecretory and antiproliferative effects by acting on somatostatin receptors. Despite their proven efficacy, there are factors that limit their long-term effectiveness, including challenges associated with receptor downregulation and tumor heterogeneity [24]. That said, SSAs have shown evidence of immense synergistic potential in the treatment of gastroenteropancreatic (GEP) NENs when combined with PRRT (NETTER-2 trial) and everolimus (RADIANT-2 trial) [24,36,37]. Furthermore, novel SSA formulations such as CAM2029 subcutaneous octreotide are being evaluated for safety and efficacy in NENs [24].
For F-PNENs, if supportive or surgical measures are insufficient to control the hormonal symptoms, a trial of SSAs may reduce functioning activity [22]. SSA antisecretory effects can palliate hormonal syndromes such as gastrinoma, glucagonoma, and VIPoma. Therefore, for most patients with SSTR-positive tumors, an SSA is the first-line treatment of choice, particularly if the disease is relatively non-bulky and unaggressive [22].
A critical question that requires thorough evaluation is when the diagnostic studies should be performed in relation to when the pharmacologic control of the hormone hypersecretion has begun [32]. An unphysiological hormone secretion validates an F-PNEN diagnosis. This can be an issue with F-PNENs treated with SSAs, because they can decrease the ectopic hormone release, affecting plasma hormone levels and controlling the hormone-excess state, thus complicating diagnosis [161]. The same could occur with the use of PRRT or other medical treatments (diazoxide in insulinomas), which can decrease the hormone secretion rate [32].
SSAs are of limited use in non-metastatic insulinomas, and caution should be exercised when considering SSA therapy, as hypoglycemia may worsen due to a more profound suppression of counterregulatory hormones, such as glucagon, than tumor-produced insulin [162]. In metastatic insulinomas, mTOR inhibitors can reduce hypoglycemic episodes (e.g., the RADIANT-4 trial, where treatment with everolimus was associated with significant improvement in PFS in patients) [163]. In most patients with insulinomas, glucagonomas, somatostatinomas, and the other infrequent F-PNEN syndromes, emergent immediate treatment is not required, and time can be taken to establish the diagnosis prior to the start of pharmacologic therapy [32].
In contrast, immediate treatment of the hormone-hypersecretory state is needed in almost all patients with gastrinomas, who frequently have acid hypersecretion rates, as well as patients with VIPomas with high-volume diarrhea, hypokalemia, and dehydration [32]. In patients already on antisecretory drug treatments, the establishment of the diagnosis of gastrinoma is difficult. For gastrinoma, the gastric acid hypersecretion is treated first (use of PPIs). SSAs can also control acid hypersecretion in patients with gastrinomas and have a favorable outcome for PFS [162]. The combination of PPI and SSAs offers an advantageous additive effect on gastric acid suppression. For VIPoma, SSAs may reduce tumor VIP secretion by more than 50% and inhibit intestinal water and electrolyte secretion; these drugs control secretory diarrhea in more than 50% of patients [162].
PRRT is the newest modality with continually emerging data exploring its efficacy [30]. The NETTER-1 trial showed the safety and efficacy of PRRT as a second-line therapy for unresectable, metastatic, or locally advanced well-differentiated midgut NENs after disease progression in patients who initially received SSA treatment [164]. Interestingly, the NETTER-1 had no dedicated subgroup analysis for F-NENs, which led to FDA approval of PRRT treatment in advanced NENs [31]. NETTER-2 confirmed the relevant benefits in survival and tumor response of the SSA-PRRT combination therapy to SSA alone (control arm) as first-line treatment for advanced, higher-grade 2–3 GEP NENs. Data on the efficacy of PRRT in PNENs or related to the proper timing of PRRT initiation and its effectiveness in comparison with chemotherapy or targeted therapy for PNENs/F-PNENs are lacking [164]. The combination of PRRT with cytotoxic chemotherapy, capecitabine, and temozolomide (CAPTEM) has become clinically useful, especially in PNENs, demonstrating an acceptable safety profile [156]. As outlined in the ENETS, ESMO, and the ASCO guidelines [160], the specific role of PRRT varies according to the grade and type of GEP NETs. This reiterates the importance of performing trials and coalescing data to design therapeutic strategies specific to F-PNENs.
Prospective randomized trials involving direct comparisons are currently underway, including the COMPOSE and the COMPETE trials that evaluate PRRT (with 177Lu-Edotreotide) to targeted molecular therapy with everolimus in patients with inoperable, progressive GEP NENs [165,166]. Everolimus is an oral inhibitor of mTOR and registered for advanced NEN patients based on the RADIANT-3 and RADIANT-4 trials [52,163]. Sunitinib, a multi-targeted tyrosine kinase inhibitor registered for grade 1–2 advanced, progressive PNEN patients, was shown to have improved median PFS in a randomized phase 3 clinical trial [51]. A direct comparison between PRRT and sunitinib was recently conducted in the OCLURANDOM trial, a multicenter randomized phase 2 trial, indicating PRRT as a more effective second-line treatment with less toxicity than sunitinib [167]. The efficacy of cabozantinib, another multi-targeted tyrosine kinase inhibitor, was identified in the latest phase 3 CABINET trial in patients with advanced PNEN after progression on PRRT or targeted therapy [168]; however, a direct comparison between PRRT and cabozantinib still remains unavailable. Cabozantinib has been recommended as a next-line treatment for patients with progressive grade 1–3 PNEN previously treated with PRRT [160]; however, its effectiveness as a targeted therapy for patients within the F-PNEN subcategory is still largely unknown.
A multicenter, retrospective cohort study including 508 patients has directly evaluated second-line therapies PRRT, targeted therapy, and chemotherapy in patients with advanced GEP NENs [164]. Data from this study revealed that treatment with upfront PRRT in patients with GEP NENs who had experienced disease progression with SSA treatment was associated with significantly improved survival outcomes compared with upfront chemotherapy or targeted therapy [164]. There seems to be a potential beneficial role of neoadjuvant PRRT in unresectable and metastatic GEP NENs [169], but prospective studies are required from multiple center collaborations to obtain sufficient high-quality evidence. Two clinical studies are currently recruiting patients with either GEP-NENs or PNENs receiving neoadjuvant PRRT followed by surgery [156].
A revised therapeutic algorithm for PRRT has been proposed based on recent findings from the NETTER-2 trial, long-term outcomes from the NETTER-1 trial, and the OCLURANDOM trial [160]. Such algorithms are critical and foundational for tailoring targeted therapies specific to F-PNENs. Data from the COMPOSE and COMPETE trials are anticipated to provide further insights into the role of PRRT as a first-line or second-line treatment for PNENs. Neoadjuvant PRRT prior to surgery, PRRT combinations of intravenous and intraarterial routes of application, combinations of PRRT with differently radiolabeled SSTR-targeting agonists and antagonists, inhibitors of immune checkpoints (ICIs), and other key molecular targets are currently being investigated in ongoing clinical trials [156]. Despite the emergence of several novel radiopharmaceuticals for NENs, there is still a significant subset of patients where imaging-based biomarkers fall short in disease assessment, prognostication, and improving outcomes [170].
10. Other Limitations and Future Perspectives
As discussed in the earlier sections, diagnostic subtleties to distinguish F-PNENs from NF-PNENs require skill and extensive clinical experience from within two distinct clinical disciplines [148]. Although the medical therapeutic implications may not be significantly different, personalized patient care requires a nuanced understanding of the disease. However, the current knowledge gap in the field hinders early detection and appropriate management of F-PNENs [149].
A decreased trend to evaluate circulating markers has been observed in recent years [149], and this, unfortunately, has stimulated the notion that all markers, including the specific ones, are optional, thereby adding additional complexities to underdiagnosing F-PNENs [149,171]. Apart from the fact that they are cost-effective and minimally invasive [172], the importance of testing specific circulating markers as critical and mandatory has been heavily emphasized for precise diagnosis and effective management of F-PNENs [149,150].
Conducting translational work in a rare disease such as PNEN, particularly owing to its inherent heterogeneity, is challenging. Additional limiting factors include a lack of access to the tumor tissue and complex treatment paradigms without a consensus pathway through the therapeutic options. Novel molecular strategies for targeted therapies, i.e., the development of chimeric antigen receptor (CAR) T cells targeting CDH17 to suppress PNENs [173] and a systems biology approach to define mechanisms, phenotypes, and drivers in PNENs [151], have been evaluated.
International consortiums, collaborations, and the establishment of several high-quality prospective bio-banks with matched clinical data hold promise to streamline existing and novel information for F-PNENs. For example, the development of ENETS centers of excellence across Europe over the past 15 years has supported a more standardized approach with minimum datasets and continues to facilitate international partnerships, which are mandatory for studying this rare disease [174]. The relatively uncommon nature of F-PNENs is another critical factor affecting the design and execution of randomized trials at single and even multicenter levels. In this context, the recommendation of NANETS, ENETS, and other governing bodies to broadly extend eligible criteria to admit patients into clinical trials whenever feasible has been very positively received. Such clauses help overcome barriers to participation and move patient care forward.
11. Conclusions
F-PNENs are an interesting and rare group of neoplasms. This review presents a consolidated clinical overview of some of the more well-characterized F-PNENs, with medical therapies and surgical management drawn from established clinical guidelines (Table 2). Surgical resection remains the gold standard for treatment, with distal pancreatectomy and pancreaticoduodenectomy serving as common procedures. Unfortunately, many of these tumors are detected late in their course, with metastasis having already occurred. For advanced or metastatic disease, there is increased hope of symptomatic control or PFS with medical therapy such as SSAs, chemotherapies, and PRRT. Advancements in minimally invasive treatment options, such as RFA, have further broadened the therapeutic landscape in managing advanced F-PNENs. Improved understanding of these rare groups of pancreatic tumors at the genetic, molecular, and biochemical levels has resulted in the development of novel therapies. Yet, their relative rarity has hampered efforts to optimize their diagnostic markers and therapeutic management as well as characterize the molecular environment that leads to their development. In-depth basic science and clinical translational investigations are warranted to identify novel molecular therapeutic targets and biomarkers to enhance diagnosis, and large multinational cohort studies could provide the sample size necessary to further improve disease management and augment clinical outcomes.
Table 2.
A summary of the specific laboratory findings, medical and surgical management and recent advances for each F-PNEN.
Author Contributions
Conceptualization, E.A.M., B.P.D., C.D.W., T.H.J. and S.N.; data curation, E.A.M., B.P.D., C.D.W. and T.H.J.; writing—original draft and figure preparation, E.A.M., B.P.D., C.D.W. and T.H.J.; writing—review and editing, E.A.M., B.P.D., C.D.W., T.H.J., S.N. and G.E.B. All authors have read and agreed to the published version of the manuscript.
Funding
This work has not received any funding.
Data Availability Statement
No new data were created or analyzed in this study.
Conflicts of Interest
This research received no external funding.
References
- Mukkala, A.N.; Ray, S.; Bevacqua, D.; McGilvray, I.; Sapisochin, G.; Moulton, C.-A.; Gallinger, S.; Cleary, S.P.; Shwaartz, C.; Wei, A.C.; et al. Disease-Free Survival after Pancreatectomy for Pancreatic Neuroendocrine Tumors: A 17-Year Single-Center Experience of 223 Patients. J. Gastrointest. Surg. 2024, 28, 1485–1492. [Google Scholar] [CrossRef]
- Bader, A.; Landau, S.; Hwang, J.; Passman, J.; Lee, M.K.; Fraker, D.; Vollmer, C.; Wachtel, H. Recurrence and Treatment Trends of Pancreatic Neuroendocrine Tumors. Surgery 2025, 177, 108835. [Google Scholar] [CrossRef]
- Brooks, J.C.; Shavelle, R.M.; Vavra-Musser, K.N. Life Expectancy in Pancreatic Neuroendocrine Cancer. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Ro, C.; Chai, W.; Yu, V.E.; Yu, R. Pancreatic Neuroendocrine Tumors: Biology, Diagnosis, and Treatment. Chin. J. Cancer 2013, 32, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Alshareefy, Y.; Cummins, S.; Mazzoleni, A.; Sharma, V.; Guggilapu, S.; Leong, A.W.Y.; Wireko, A.A. A Review of Functional Pancreatic Neuroendocrine Tumors: Exploring the Molecular Pathogenesis, Diagnosis and Treatment. Medicine 2023, 102, e36094. [Google Scholar] [CrossRef] [PubMed]
- Cloyd, J.M.; Poultsides, G.A. Non-Functional Neuroendocrine Tumors of the Pancreas: Advances in Diagnosis and Management. World J. Gastroenterol. 2015, 21, 9512–9525. [Google Scholar] [CrossRef]
- Park, Y.S. Less Common Types of Pancreatic Neuroendocrine Tumors. In Neuroendocrine Tumours: Diagnosis and Management; Yalcin, S., Öberg, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 271–274. [Google Scholar]
- Jensen, R.T.; Berna, M.J.; Bingham, D.B.; Norton, J.A. Inherited Pancreatic Endocrine Tumor Syndromes: Advances in Molecular Pathogenesis, Diagnosis, Management, and Controversies. Cancer 2008, 113, 1807–1843. [Google Scholar] [CrossRef]
- Alexakis, N.; Connor, S.; Ghaneh, P.; Lombard, M.; Smart, H.L.; Evans, J.; Hughes, M.; Garvey, C.J.; Vora, J.; Vinjamuri, S.; et al. Hereditary Pancreatic Endocrine Tumours. Pancreatology 2004, 4, 417–435. [Google Scholar] [CrossRef]
- Scarpa, A.; Chang, D.K.; Nones, K.; Corbo, V.; Patch, A.-M.; Bailey, P.; Lawlor, R.T.; Johns, A.L.; Miller, D.K.; Mafficini, A.; et al. Whole-Genome Landscape of Pancreatic Neuroendocrine Tumours. Nature 2017, 543, 65–71. [Google Scholar] [CrossRef]
- Helbing, A.; Menon, G.; Tuma, F. Pancreatic Neuroendocrine Tumors; Statpearls: Treasure Island, FL, USA, 2025. [Google Scholar]
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. Daxx/Atrx, Men1, and Mtor Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science 2011, 331, 1199–1203. [Google Scholar] [CrossRef]
- Yousaf, M.N.; Chaudhary, F.S.; Ehsan, A.; Suarez, A.L.; Muniraj, T.; Jamidar, P.; Aslanian, H.R.; Farrell, J.J. Endoscopic Ultrasound (Eus) and the Management of Pancreatic Cancer. BMJ Open Gastroenterol. 2020, 7, e000408. [Google Scholar] [CrossRef] [PubMed]
- Mullen, B.; Sy, A.L.; Dias Goncalves, P.; Zhang, M.L. Navigating the Diagnostic Gray Zone: A Challenging Case of Pancreatic High-Grade Neuroendocrine Neoplasm. Diagn. Pathol. 2024, 19, 123. [Google Scholar] [CrossRef]
- Katsuda, H.; Kobayashi, M.; Ito, G.; Kawamoto, A.; Krimura, S.; Sato, H.; Hirakawa, A.; Akahoshi, K.; Kudo, A.; Ohtsuka, K.; et al. Evaluating Endoscopic Ultrasound-Guided Tissue Acquisition for Diagnosis of Small Pancreatic Neuroendocrine Neoplasms. Endosc. Int. Open 2024, 12, E1379–E1385. [Google Scholar] [CrossRef] [PubMed]
- Tacelli, M.; Bina, N.; Crinò, S.F.; Facciorusso, A.; Celsa, C.; Vanni, A.S.; Fantin, A.; Antonini, F.; Falconi, M.; Monica, F.; et al. Reliability of Grading Preoperative Pancreatic Neuroendocrine Tumors on Eus Specimens: A Systematic Review with Meta-Analysis of Aggregate and Individual Data. Gastrointest. Endosc. 2022, 96, 898–908.e23. [Google Scholar] [CrossRef] [PubMed]
- Inzani, F.; Petrone, G.; Fadda, G.; Rindi, G. Cyto-Histology in Net: What Is Necessary Today and What Is the Future? Rev. Endocr. Metab. Disord. 2017, 18, 381–391. [Google Scholar] [CrossRef]
- Rindi, G.; Mete, O.; Uccella, S.; Basturk, O.; La Rosa, S.; Brosens, L.A.A.; Ezzat, S.; de Herder, W.W.; Klimstra, D.S.; Papotti, M.; et al. Overview of the 2022 Who Classification of Neuroendocrine Neoplasms. Endocr. Pathol. 2022, 33, 115–154. [Google Scholar] [CrossRef]
- Sonnen, A.F.P.; Verschuur, A.V.D.; Brosens, L.A.A. Diagnostic and Prognostic Biomarkers for Pancreatic Neuroendocrine Neoplasms. Die Pathol. 2024, 45, 74–82. [Google Scholar] [CrossRef]
- Bellizzi, A.M. Immunohistochemistry in the Diagnosis and Classification of Neuroendocrine Neoplasms: What Can Brown Do for You? Hum. Pathol. 2020, 96, 8–33. [Google Scholar] [CrossRef]
- Koo, J.; Mertens, R.B.; Mirocha, J.M.; Wang, H.L.; Dhall, D. Value of Islet 1 and Pax8 in Identifying Metastatic Neuroendocrine Tumors of Pancreatic Origin. Mod. Pathol. 2012, 25, 893–901. [Google Scholar] [CrossRef]
- Hofland, J.; Falconi, M.; Christ, E.; Castano, J.P.; Faggiano, A.; Lamarca, A.; Perren, A.; Petrucci, S.; Prasad, V.; Ruszniewski, P.; et al. European Neuroendocrine Tumor Society 2023 Guidance Paper for Functioning Pancreatic Neuroendocrine Tumour Syndromes. J. Neuroendocrinol. 2023, 35, e13318. [Google Scholar] [CrossRef]
- Lee, D.W.; Kim, M.K.; Kim, H.G. Diagnosis of Pancreatic Neuroendocrine Tumors. Clin. Endosc. 2017, 50, 537–545. [Google Scholar] [CrossRef]
- Massironi, S.; Albertelli, M.; Hasballa, I.; Paravani, P.; Ferone, D.; Faggiano, A.; Danese, S. “Cold” Somatostatin Analogs in Neuroendocrine Neoplasms: Decoding Mechanisms, Overcoming Resistance, and Shaping the Future of Therapy. Cells 2025, 14, 245. [Google Scholar] [CrossRef] [PubMed]
- Rabei, R.; Fidelman, N. Liver-Directed Therapy for Neuroendocrine Tumor Metastases in the Era of Peptide Receptor Radionuclide Therapy. Curr. Treat. Options Oncol. 2023, 24, 1994–2004. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.J.; Naguib, N.N.; Zangos, S.; Eichler, K.; Hedayati, A.; Nour-Eldin, N.E. Liver Metastases of Neuroendocrine Carcinomas: Interventional Treatment Via Transarterial Embolization, Chemoembolization and Thermal Ablation. Eur. J. Radiol. 2009, 72, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Ostapenko, A.; Stroever, S.; Eyasu, L.; Kim, M.; Aploks, K.; Dong, X.D.; Seshadri, R. Role of Ablation Therapy in Conjunction with Surgical Resection for Neuroendocrine Tumors Involving the Liver. World J. Gastrointest. Surg. 2024, 16, 768–776. [Google Scholar] [CrossRef]
- Lehrman, E.D.; Fidelman, N. Liver-Directed Therapy for Neuroendocrine Tumor Liver Metastases in the Era of Peptide Receptor Radionuclide Therapy. Semin. Intervent Radiol. 2020, 37, 499–507. [Google Scholar] [CrossRef]
- Pérez-Saborido, B.; Bailón-Cuadrado, M.; Tejero-Pintor, F.J.; Choolani-Bhojwani, E.; Marcos-Santos, P.; Pacheco-Sánchez, D. Pancreatic Neuroendocrine Tumors: Diagnosis, Management, and Intraoperative Techniques. In Recent Innovations in Surgical Procedures of Pancreatic Neoplasms; Bellido Luque, J., Muñoz, A.N., Eds.; Springer International Publishing: Cham, Switzerland, 2023; pp. 35–53. [Google Scholar]
- Partelli, S.; Landoni, L.; Bartolomei, M.; Zerbi, A.; Grana, C.M.; Boggi, U.; Butturini, G.; Casadei, R.; Bassi, C.; Falconi, M. 1186mo a Prospective Phase Ii Single-Arm Trial on Neoadjuvant Peptide Receptor Radionuclide Therapy (Prrt) with 177lu-Dotatate Followed by Surgery for Pancreatic Neuroendocrine Tumors (Neolupanet). Ann. Oncol. 2023, 34, S703. [Google Scholar] [CrossRef]
- Strosberg, J.; El-Haddad, G.; Wolin, E.; Hendifar, A.; Yao, J.; Chasen, B.; Mittra, E.; Kunz, P.L.; Kulke, M.H.; Jacene, H.; et al. Phase 3 Trial of (177)Lu-Dotatate for Midgut Neuroendocrine Tumors. N. Engl. J. Med. 2017, 376, 125–135. [Google Scholar] [CrossRef]
- Ito, T.; Jensen, R.T. Perspectives on the Current Pharmacotherapeutic Strategies for Management of Functional Neuroendocrine Tumor Syndromes. Expert. Opin. Pharmacother. 2021, 22, 685–693. [Google Scholar] [CrossRef]
- Veltroni, A.; Cosaro, E.; Spada, F.; Fazio, N.; Faggiano, A.; Colao, A.; Pusceddu, S.; Zatelli, M.C.; Campana, D.; Piovesan, A.; et al. Clinico-Pathological Features, Treatments and Survival of Malignant Insulinomas: A Multicenter Study. Eur. J. Endocrinol. 2020, 182, 439–446. [Google Scholar] [CrossRef]
- Kwekkeboom, D.J.; de Herder, W.W.; Kam, B.L.; van Eijck, C.H.; van Essen, M.; Kooij, P.P.; Feelders, R.A.; van Aken, M.O.; Krenning, E.P. Treatment with the Radiolabeled Somatostatin Analog [177 Lu-Dota 0,Tyr3]Octreotate: Toxicity, Efficacy, and Survival. J. Clin. Oncol. 2008, 26, 2124–2130. [Google Scholar] [CrossRef]
- Del Olmo-Garcia, M.I.; Muros, M.A.; Lopez-de-la-Torre, M.; Agudelo, M.; Bello, P.; Soriano, J.M.; Merino-Torres, J.F. Prevention and Management of Hormonal Crisis During Theragnosis with Lu-Dota-Tate in Neuroendocrine Tumors. A Systematic Review and Approach Proposal. J. Clin. Med. 2020, 9, 2203. [Google Scholar] [CrossRef]
- Singh, S.; Halperin, D.; Myrehaug, S.; Herrmann, K.; Pavel, M.; Kunz, P.L.; Chasen, B.; Tafuto, S.; Lastoria, S.; Capdevila, J.; et al. [(177)Lu]Lu-Dota-Tate Plus Long-Acting Octreotide Versus High-Dose Long-Acting Octreotide for the Treatment of Newly Diagnosed, Advanced Grade 2-3, Well-Differentiated, Gastroenteropancreatic Neuroendocrine Tumours (Netter-2): An Open-Label, Randomised, Phase 3 Study. Lancet 2024, 403, 2807–2817. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.E.; Hainsworth, J.D.; Baudin, E.; Peeters, M.; Horsch, D.; Winkler, R.E.; Klimovsky, J.; Lebwohl, D.; Jehl, V.; Wolin, E.M.; et al. Everolimus Plus Octreotide Long-Acting Repeatable for the Treatment of Advanced Neuroendocrine Tumours Associated with Carcinoid Syndrome (Radiant-2): A Randomised, Placebo-Controlled, Phase 3 Study. Lancet 2011, 378, 2005–2012. [Google Scholar] [CrossRef] [PubMed]
- Mehrabi, A.; Fischer, L.; Hafezi, M.; Dirlewanger, A.; Grenacher, L.; Diener, M.K.; Fonouni, H.; Golriz, M.; Garoussi, C.; Fard, N.; et al. A Systematic Review of Localization, Surgical Treatment Options, and Outcome of Insulinoma. Pancreas 2014, 43, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Kurakawa, K.I.; Okada, A.; Manaka, K.; Konishi, T.; Jo, T.; Ono, S.; Uda, K.; Michihata, N.; Matsui, H.; Fushimi, K.; et al. Clinical Characteristics and Incidences of Benign and Malignant Insulinoma Using a National Inpatient Database in Japan. J. Clin. Endocrinol. Metab. 2021, 106, 3477–3486. [Google Scholar] [CrossRef]
- Hofland, J.; Refardt, J.C.; Feelders, R.A.; Christ, E.; de Herder, W.W. Approach to the Patient: Insulinoma. J. Clin. Endocrinol. Metab. 2023, 109, 1109–1118. [Google Scholar] [CrossRef]
- Whipple, A.O.; Frantz, V.K. Adenoma of Islet Cells with Hyperinsulixism. Ann. Surg. 1935, 101, 1299–1365. [Google Scholar] [CrossRef]
- Service, F.J. Hypoglycemic Disorders. N. Engl. J. Med. 1995, 332, 1144–1152. [Google Scholar] [CrossRef]
- Valente, L.G.; Antwi, K.; Nicolas, G.P.; Wild, D.; Christ, E. Clinical Presentation of 54 Patients with Endogenous Hyperinsulinaemic Hypoglycaemia: A Neurological Chameleon (Observational Study). Swiss Med. Wkly. 2018, 148, w14682. [Google Scholar] [CrossRef]
- Nccn Guidelines Neuroendocrine and Adrenal Tumors (Version 2.2025). Available online: https://www.nccn.org/professionals/physician_gls/pdf/neuroendocrine.pdf (accessed on 28 May 2025).
- Gouya, H.; Vignaux, O.; Augui, J.; Dousset, B.; Palazzo, L.; Louvel, A.; Chaussade, S.; Legmann, P. Ct, Endoscopic Sonography, and a Combined Protocol for Preoperative Evaluation of Pancreatic Insulinomas. Am. J. Roentgenol. 2003, 181, 987–992. [Google Scholar] [CrossRef]
- Antwi, K.; Fani, M.; Heye, T.; Nicolas, G.; Rottenburger, C.; Kaul, F.; Merkle, E.; Zech, C.J.; Boll, D.; Vogt, D.R.; et al. Comparison of Glucagon-Like Peptide-1 Receptor (Glp-1r) Pet/Ct, Spect/Ct and 3t Mri for the Localisation of Occult Insulinomas: Evaluation of Diagnostic Accuracy in a Prospective Crossover Imaging Study. Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 2318–2327. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, A.; Boursier, C.; Sahakian, N.; Ouvrard, E.; Chevalier, E.; Sebag, F.; Addeo, P.; Taïeb, D. Value of 68ga-Dotatoc and Carbidopa-Assisted 18f-Dopa Pet/Ct for Insulinoma Localization. J. Nucl. Med. 2022, 63, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Ba, Y.; Xing, Q.; Cai, R.-C. Diagnostic Value of Asvs for Insulinoma Localization: A Systematic Review and Meta-Analysis. PLoS ONE 2019, 14, e0224928. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; Ćwikła, J.B.; Lombard-Bohas, C.; Borbath, I.; Shah, T.; Pape, U.F.; Capdevila, J.; Panzuto, F.; Truong Thanh, X.-M.; Houchard, A.; et al. Efficacy and Safety of High-Dose Lanreotide Autogel in Patients with Progressive Pancreatic or Midgut Neuroendocrine Tumours: Clarinet Forte Phase 2 Study Results. Eur. J. Cancer 2021, 157, 403–414. [Google Scholar] [CrossRef]
- Oziel-Taieb, S.; Maniry-Quellier, J.; Chanez, B.; Poizat, F.; Ewald, J.; Niccoli, P. Pasireotide for Refractory Hypoglycemia in Malignant Insulinoma- Case Report and Review of the Literature. Front. Endocrinol. 2022, 13, 860614. [Google Scholar] [CrossRef]
- Raymond, E.; Dahan, L.; Raoul, J.-L.; Bang, Y.-J.; Borbath, I.; Lombard-Bohas, C.; Valle, J.; Metrakos, P.; Smith, D.; Vinik, A.; et al. Sunitinib Malate for the Treatment of Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 501–513. [Google Scholar] [CrossRef]
- Yao, J.C.; Shah, M.H.; Ito, T.; Bohas, C.L.; Wolin, E.M.; Cutsem, E.V.; Hobday, T.J.; Okusaka, T.; Capdevila, J.; Vries, E.G.E.d.; et al. Everolimus for Advanced Pancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2011, 364, 514–523. [Google Scholar] [CrossRef]
- Zandee, W.T.; Brabander, T.; Blažević, A.; Kam, B.L.R.; Teunissen, J.J.M.; Feelders, R.A.; Hofland, J.; de Herder, W.W. Symptomatic and Radiological Response to 177lu-Dotatate for the Treatment of Functioning Pancreatic Neuroendocrine Tumors. J. Clin. Endocrinol. Metab. 2019, 104, 1336–1344. [Google Scholar] [CrossRef]
- Kunz, P.L.; Graham, N.T.; Catalano, P.J.; Nimeiri, H.S.; Fisher, G.A.; Longacre, T.A.; Suarez, C.J.; Martin, B.A.; Yao, J.C.; Kulke, M.H.; et al. Randomized Study of Temozolomide or Temozolomide and Capecitabine in Patients with Advanced Pancreatic Neuroendocrine Tumors (Ecog-Acrin E2211). J. Clin. Oncol. 2023, 41, 1359–1369. [Google Scholar] [CrossRef]
- de Carbonnières, A.; Challine, A.; Cottereau, A.S.; Coriat, R.; Soyer, P.; Abou Ali, E.; Prat, F.; Terris, B.; Bertherat, J.; Dousset, B.; et al. Surgical Management of Insulinoma over Three Decades. Hpb 2021, 23, 1799–1806. [Google Scholar] [CrossRef]
- Crinò, S.F.; Napoleon, B.; Facciorusso, A.; Lakhtakia, S.; Borbath, I.; Caillol, F.; Do-Cong Pham, K.; Rizzatti, G.; Forti, E.; Palazzo, L.; et al. Endoscopic Ultrasound-Guided Radiofrequency Ablation Versus Surgical Resection for Treatment of Pancreatic Insulinoma. Clin. Gastroenterol. Hepatol. 2023, 21, 2834–2843.e2. [Google Scholar] [CrossRef]
- Habibollahi, P.; Bai, H.X.; Sanampudi, S.; Soulen, M.C.; Dagli, M. Effectiveness of Liver-Directed Therapy for the Management of Intractable Hypoglycemia in Metastatic Insulinoma. Pancreas 2020, 49, 763–767. [Google Scholar] [CrossRef]
- Chatzipanagiotou, O.; Schizas, D.; Vailas, M.; Tsoli, M.; Sakarellos, P.; Sotiropoulou, M.; Papalambros, A.; Felekouras, E. All You Need to Know About Gastrinoma Today | Gastrinoma and Zollinger-Ellison Syndrome: A Thorough Update. J. Neuroendocrinol. 2023, 35, e13267. [Google Scholar] [CrossRef] [PubMed]
- Rossi, R.E.; Elvevi, A.; Citterio, D.; Coppa, J.; Invernizzi, P.; Mazzaferro, V.; Massironi, S. Gastrinoma and Zollinger Ellison Syndrome: A Roadmap for the Management between New and Old Therapies. World J. Gastroenterol. 2021, 27, 5890–5907. [Google Scholar] [CrossRef] [PubMed]
- Anlauf, M.; Garbrecht, N.; Henopp, T.; Schmitt, A.; Schlenger, R.; Raffel, A.; Krausch, M.; Gimm, O.; Eisenberger, C.F.; Knoefel, W.T.; et al. Sporadic Versus Hereditary Gastrinomas of the Duodenum and Pancreas: Distinct Clinico-Pathological and Epidemiological Features. World J. Gastroenterol. 2006, 12, 5440–5446. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Igarashi, H.; Jensen, R.T. Zollinger-Ellison Syndrome: Recent Advances and Controversies. Curr. Opin. Gastroenterol. 2013, 29, 650–661. [Google Scholar] [CrossRef]
- Camera, L.; Boccadifuoco, F.; Modica, R.; Messerini, L.; Faggiano, A.; Romeo, V.; Gaudieri, V.; Colao, A.; Maurea, S.; Brunetti, A. Gastrinomas and Non-Functioning Pancreatic Endocrine Tumors in Multiple Endocrine Neoplasia Syndrome Type-1 (Men-1). Endocrine 2023, 81, 459–463. [Google Scholar] [CrossRef]
- Guarnotta, V.; Martini, C.; Davì, M.V.; Pizza, G.; Colao, A.; Faggiano, A. The Zollinger-Ellison Syndrome: Is There a Role for Somatostatin Analogues in the Treatment of the Gastrinoma? Endocrine 2018, 60, 15–27. [Google Scholar] [CrossRef]
- Cingam, S.R.; Botejue, M.; Hoilat, G.J.; Karanchi, H. Gastrinoma; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Falconi, M.; Eriksson, B.; Kaltsas, G.; Bartsch, D.K.; Capdevila, J.; Caplin, M.; Kos-Kudla, B.; Kwekkeboom, D.; Rindi, G.; Klöppel, G.; et al. Enets Consensus Guidelines Update for the Management of Patients with Functional Pancreatic Neuroendocrine Tumors and Non-Functional Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2016, 103, 153–171. [Google Scholar] [CrossRef]
- Norton, J.A.; Foster, D.S.; Ito, T.; Jensen, R.T. Gastrinomas: Medical or Surgical Treatment. Endocrinol. Metab. Clin. N. Am. 2018, 47, 577–601. [Google Scholar] [CrossRef]
- Pratap, T.; Jalal, M.J.A.; Jacob, D.; Mahadevan, P.; Nair, S.S.; Raja, S. Radiological Features of Zollinger-Ellison Syndrome: A Report of Two Cases. Indian. J. Radiol. Imaging 2022, 32, 395–402. [Google Scholar] [CrossRef]
- Krampitz, G.W.; Norton, J.A. Current Management of the Zollinger-Ellison Syndrome. Adv. Surg. 2013, 47, 59–79. [Google Scholar] [CrossRef]
- Keutgen, X.M.; Nilubol, N.; Kebebew, E. Malignant-Functioning Neuroendocrine Tumors of the Pancreas: A Survival Analysis. Surgery 2016, 159, 1382–1389. [Google Scholar] [CrossRef]
- Jadhav, A.; Ayyat, M.; Serafini, F.M. Presentation, Diagnosis, and Treatment of a Sporadic Gastrinoma with Liver Metastases. Cureus 2024, 16, e61426. [Google Scholar] [CrossRef] [PubMed]
- Suissa, Y.; Magenheim, J.; Stolovich-Rain, M.; Hija, A.; Collombat, P.; Mansouri, A.; Sussel, L.; Sosa-Pineda, B.; McCracken, K.; Wells, J.M.; et al. Gastrin: A Distinct Fate of Neurogenin3 Positive Progenitor Cells in the Embryonic Pancreas. PLoS ONE 2013, 8, e70397. [Google Scholar] [CrossRef] [PubMed]
- Prosapio, J.G.; Sankar, P.; Jialal, I. Physiology, Gastrin; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Hsu, M.; Safadi, A.O.; Lui, F. Physiology, Stomach; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Shah, P.; Singh, M.H.; Yang, Y.X.; Metz, D.C. Hypochlorhydria and Achlorhydria Are Associated with False-Positive Secretin Stimulation Testing for Zollinger-Ellison Syndrome. Pancreas 2013, 42, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Goldman, J.A.; Blanton, W.P.; Hay, D.W.; Wolfe, M.M. False-Positive Secretin Stimulation Test for Gastrinoma Associated with the Use of Proton Pump Inhibitor Therapy. Clin. Gastroenterol. Hepatol. 2009, 7, 600–602. [Google Scholar] [CrossRef]
- Phan, J.; Benhammou, J.N.; Pisegna, J.R. Gastric Hypersecretory States: Investigation and Management. Curr. Treat. Options Gastroenterol. 2015, 13, 386–397. [Google Scholar] [CrossRef]
- DiGregorio, N.; Sharma, S. Physiology, Secretin; StatPearls: Treasure Island, FL, USA, 2023. [Google Scholar]
- Zhang, J.M.; Zheng, C.W.; Li, X.W.; Fang, Z.Y.; Yu, M.X.; Shen, H.Y.; Ji, X. Typical Zollinger-Ellison Syndrome-Atypical Location of Gastrinoma and Absence of Hypergastrinemia: A Case Report and Review of Literature. World J. Clin. Cases 2023, 11, 6223–6230. [Google Scholar] [CrossRef]
- Ito, T.; Ramos-Alvarez, I.; Jensen, R.T. Successful Lifetime/Long-Term Medical Treatment of Acid Hypersecretion in Zollinger-Ellison Syndrome (Zes): Myth or Fact? Insights from an Analysis of Results of Nih Long-Term Prospective Studies of Zes. Cancers 2023, 15, 1377. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.T. Consequences of Long-Term Proton Pump Blockade: Insights from Studies of Patients with Gastrinomas. Basic. Clin. Pharmacol. Toxicol. 2006, 98, 4–19. [Google Scholar] [CrossRef] [PubMed]
- Auernhammer, C.J.; Göke, B. Medical Treatment of Gastrinomas. Wien. Klin. Wochenschr. 2007, 119, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ramos-Alvarez, I.; Jensen, R.T. Long-Term Proton Pump Inhibitor-Acid Suppressive Treatment Can Cause Vitamin B(12) Deficiency in Zollinger-Ellison Syndrome (Zes) Patients. Int. J. Mol. Sci. 2024, 25, 7286. [Google Scholar] [CrossRef]
- Lee, L.; Ramos-Alvarez, I.; Ito, T.; Jensen, R.T. Insights into Effects/Risks of Chronic Hypergastrinemia and Lifelong Ppi Treatment in Man Based on Studies of Patients with Zollinger-Ellison Syndrome. Int. J. Mol. Sci. 2019, 20, 5128. [Google Scholar] [CrossRef]
- Massironi, S.; Cavalcoli, F.; Elvevi, A.; Quatrini, M.; Invernizzi, P. Somatostatin Analogs in Patients with Zollinger Ellison Syndrome (Zes): An Observational Study. Endocrine 2022, 75, 942–948. [Google Scholar] [CrossRef]
- Daniels, L.M.; Khalili, M.; Morano, W.F.; Simoncini, M.; Mapow, B.C.; Leaf, A.; Bowne, W.B. Case Report: Optimal Tumor Cytoreduction and Octreotide with Durable Disease Control in a Patient with Men-1 and Zollinger-Ellison Syndrome—Over a Decade of Follow-Up. World J. Surg. Oncol. 2019, 17, 213. [Google Scholar] [CrossRef]
- Mathew, D.; Sunny, S.S.; Benjamin, J.; John, J.R.; Jebasingh, F.K.; Georgy, J.T.; Singh, A.; Oommen, R. Outcome of (177)Lu-Dotatate Peptide Receptor Radionuclide Therapy in Progressive Metastatic Neuroendocrine Tumors from a Tertiary Care Center. Indian J. Endocrinol. Metab. 2024, 28, 601–610. [Google Scholar] [CrossRef]
- Ito, T.; Igarashi, H.; Uehara, H.; Jensen, R.T. Pharmacotherapy of Zollinger-Ellison Syndrome. Expert. Opin. Pharmacother. 2013, 14, 307–321. [Google Scholar] [CrossRef]
- Rogers, J.E.; Lam, M.; Halperin, D.M.; Dagohoy, C.G.; Yao, J.C.; Dasari, A. Fluorouracil, Doxorubicin with Streptozocin and Subsequent Therapies in Pancreatic Neuroendocrine Tumors. Neuroendocrinology 2022, 112, 34–42. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Strosberg, J.R.; Tang, L.; Bellizzi, A.M.; Bergsland, E.K.; O’Dorisio, T.M.; Halperin, D.M.; Fishbein, L.; Eads, J.; Hope, T.A.; et al. The North American Neuroendocrine Tumor Society Consensus Guidelines for Surveillance and Medical Management of Pancreatic Neuroendocrine Tumors. Pancreas 2020, 49, 863–881. [Google Scholar] [CrossRef]
- Robin, L.; Sauvanet, A.; Walter, T.; Najah, H.; Falconi, M.; Pattou, F.; Gaujoux, S. Recurrence after Surgical Resection of Nonmetastatic Sporadic Gastrinoma: Which Prognostic Factors and Surgical Procedure? Surgery 2023, 173, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Imamura, M.; Komoto, I.; Taki, Y. How to Treat Gastrinomas in Patients with Multiple Endocrine Neoplasia Type1: Surgery or Long-Term Proton Pump Inhibitors? Surg. Today 2023, 53, 1325–1334. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.; Öberg, K.; Falconi, M.; Krenning, E.P.; Sundin, A.; Perren, A.; Berruti, A. Gastroenteropancreatic Neuroendocrine Neoplasms: Esmo Clinical Practice Guidelines for Diagnosis, Treatment and Follow-Up. Ann. Oncol. 2020, 31, 844–860. [Google Scholar] [CrossRef] [PubMed]
- Strosberg, J.; Hoffe, S.; Gardner, N.; Choi, J.; Kvols, L. Effective Treatment of Locally Advanced Endocrine Tumors of the Pancreas with Chemoradiotherapy. Neuroendocrinology 2007, 85, 216–220. [Google Scholar] [CrossRef]
- de Herder, W.W.; Hofland, J. Glucagon & Glucagonoma Syndrome. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., Hofland, J., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Yao, J.C.; Eisner, M.P.; Leary, C.; Dagohoy, C.; Phan, A.; Rashid, A.; Hassan, M.; Evans, D.B. Population-Based Study of Islet Cell Carcinoma. Ann. Surg. Oncol. 2007, 14, 3492–3500. [Google Scholar] [CrossRef]
- Soga, J.; Yakuwa, Y. Glucagonomas/Diabetico-Dermatogenic Syndrome (Dds): A Statistical Evaluation of 407 Reported Cases. J. Hepato-Biliary-Pancreat. Surg. 1998, 5, 312–319. [Google Scholar] [CrossRef]
- Najafi, N.; Mintziras, I.; Wiese, D.; Albers, M.B.; Maurer, E.; Bartsch, D.K. A Retrospective Comparison of Robotic Versus Laparoscopic Distal Resection and Enucleation for Potentially Benign Pancreatic Neoplasms. Surg. Today 2020, 50, 872–880. [Google Scholar] [CrossRef]
- Song, X.; Zheng, S.; Yang, G.; Xiong, G.; Cao, Z.; Feng, M.; Zhang, T.; Zhao, Y. Glucagonoma and the Glucagonoma Syndrome. Oncol. Lett. 2018, 15, 2749–2755. [Google Scholar] [CrossRef]
- Povoski, S.P.; Zaman, S.A.; Ducatman, B.S.; McFadden, D.W. Dermatitis, Glossitis, Stomatitis, Cheilitis, Anemia and Weight Loss: A Classic Presentation of Pancreatic Glucagonoma. West. Va. Med. J. 2002, 98, 12–14. [Google Scholar]
- Wermers, R.A.; Fatourechi, V.; Wynne, A.G.; Kvols, L.K.; Lloyd, R.V. The Glucagonoma Syndrome Clinical and Pathologic Features in 21 Patients. Medicine 1996, 75, 53–63. [Google Scholar] [CrossRef]
- Delli Colli, M.; Alamri, B.N.; Palma, L.; Rivera, J. A Glucagonoma Presenting as Cerebral Vein Thrombosis and Diabetes. Case Rep. Endocrinol. 2022, 2022, 7659341. [Google Scholar] [CrossRef] [PubMed]
- Jin, X.-F.; Spampatti, M.P.; Spitzweg, C.; Auernhammer, C.J. Supportive Therapy in Gastroenteropancreatic Neuroendocrine Tumors: Often Forgotten but Important. Rev. Endocr. Metab. Disord. 2018, 19, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Caplin, M.E.; Pavel, M.; Ćwikła, J.B.; Phan, A.T.; Raderer, M.; Sedláčková, E.; Cadiot, G.; Wolin, E.M.; Capdevila, J.; Wall, L.; et al. Lanreotide in Metastatic Enteropancreatic Neuroendocrine Tumors. N. Engl. J. Med. 2014, 371, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Pavel, M.E.; Chen, D.; He, W.; Cushman, S.; Voi, M.; de Vries, E.G.E.; Baudin, E.; Yao, J.C. Everolimus Effect on Gastrin and Glucagon in Pancreatic Neuroendocrine Tumors. Pancreas 2017, 46, 751–757. [Google Scholar] [CrossRef][Green Version]
- Kindmark, H.; Sundin, A.; Granberg, D.; Dunder, K.; Skogseid, B.; Janson, E.T.; Welin, S.; Öberg, K.; Eriksson, B. Endocrine Pancreatic Tumors with Glucagon Hypersecretion: A Retrospective Study of 23 Cases During 20 Years. Med. Oncol. 2007, 24, 330–337. [Google Scholar] [CrossRef]
- Hofland, J.; Kaltsas, G.; de Herder, W.W. Advances in the Diagnosis and Management of Well-Differentiated Neuroendocrine Neoplasms. Endocr. Rev. 2020, 41, 371–403. [Google Scholar] [CrossRef]
- Williamson, J.; Thorn, C.; Spalding, D.; Williamson, R. Pancreatic and Peripancreatic Somatostatinomas. Ann. R. Coll. Surg. Engl. 2011, 93, 356–360. [Google Scholar] [CrossRef]
- Schubert, M.L.; Rehfeld, J.F. Gastric Peptides-Gastrin and Somatostatin. Compr. Physiol. 2019, 10, 197–228. [Google Scholar] [CrossRef]
- Mastoraki, A.; Schizas, D.; Papoutsi, E.; Ntella, V.; Kanavidis, P.; Sioulas, A.; Tsoli, M.; Charalampopoulos, G.; Vailas, M.; Felekouras, E. Clinicopathological Data and Treatment Modalities for Pancreatic Somatostatinomas. In Vivo 2020, 34, 3573–3582. [Google Scholar] [CrossRef]
- Zakaria, A.; Hammad, N.; Vakhariya, C.; Raphael, M. Somatostatinoma Presented as Double-Duct Sign. Case Rep. Gastrointest. Med. 2019, 2019, 9506405. [Google Scholar] [CrossRef]
- Garbrecht, N.; Anlauf, M.; Schmitt, A.; Henopp, T.; Sipos, B.; Raffel, A.; Eisenberger, C.F.; Knoefel, W.T.; Pavel, M.; Fottner, C.; et al. Somatostatin-Producing Neuroendocrine Tumors of the Duodenum and Pancreas: Incidence, Types, Biological Behavior, Association with Inherited Syndromes, and Functional Activity. Endocr. Relat. Cancer 2008, 15, 229–241. [Google Scholar] [CrossRef]
- Elangovan, A.; Mathias, P.M.; Zulfiqar, H. Somatostatinoma. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2024. [Google Scholar]
- Nesi, G.; Marcucci, T.; Rubio, C.A.; Brandi, M.L.; Tonelli, F. Somatostatinoma: Clinico-Pathological Features of Three Cases and Literature Reviewed. J. Gastroenterol. Hepatol. 2008, 23, 521–526. [Google Scholar] [CrossRef]
- Bevere, M.; Gkountakos, A.; Martelli, F.M.; Scarpa, A.; Luchini, C.; Simbolo, M. An Insight on Functioning Pancreatic Neuroendocrine Neoplasms. Biomedicines 2023, 11, 303. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gou, S.; Liu, Z.; Ye, Z.; Wang, C. Assessment of the American Joint Commission on Cancer 8th Edition Staging System for Patients with Pancreatic Neuroendocrine Tumors: A Surveillance, Epidemiology, and End Results Analysis. Cancer Med. 2018, 7, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Mansour, J.C.; Chen, H. Pancreatic Endocrine Tumors. J. Surg. Res. 2004, 120, 139–161. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.M. Rare Endocrine Tumours of the Gi Tract. Best. Pract. Res. Clin. Gastroenterol. 2005, 19, 807–817. [Google Scholar] [CrossRef]
- Alzahrani, A.S.; Alswailem, M.; Buffet, A.; Alghamdi, B.; Alobaid, L.; Alsagheir, O.; Al-Hindi, H.; Pacak, K. Epas1-Related Pheochromocytoma/Paraganglioma. Endocr. Relat. Cancer 2024, 31, e230303. [Google Scholar] [CrossRef]
- Shibata, C.; Funayama, Y.; Sasaki, I. Pancreatic Endocrine Tumors: Epidemiology, Pathology, Pathophysiology, and Diagnosis. In Diseases of the Pancreas: Current Surgical Therapy; Beger, H.G., Matsuno, S., Cameron, J.L., Rau, B.M., Sunamura, M., Schulick, R.D., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; pp. 707–713. [Google Scholar]
- Abe, T.; Oshida, K.; Matsumoto, K.; Iida, M.; Sanno, N. Brain Metastasis from Malignant Pancreatic Somatostatinoma: Case Report. J. Neurosurg. 1996, 85, 681–684. [Google Scholar] [CrossRef]
- Oberg, K.; Krenning, E.; Sundin, A.; Bodei, L.; Kidd, M.; Tesselaar, M.; Ambrosini, V.; Baum, R.P.; Kulke, M.; Pavel, M.; et al. A Delphic Consensus Assessment: Imaging and Biomarkers in Gastroenteropancreatic Neuroendocrine Tumor Disease Management. Endocr. Connect. 2016, 5, 174–187. [Google Scholar] [CrossRef]
- Brandl, A.; Lundon, D.; Siriwardena, A.K.; Sochorova, D.; Ceelen, W.; Besselink, M.; Soreide, K.; Stättner, S. Surgical Management of Pancreatic Neuroendocrine Tumors—An Eysac and E-Ahpba International Survey of Current Practice. Eur. J. Surg. Oncol. 2024, 50, 108544. [Google Scholar] [CrossRef]
- Frilling, A.; Al-Nahhas, A.; Clift, A.K. Transplantation and Debulking Procedures for Neuroendocrine Tumors. Front. Horm. Res. 2015, 44, 164–176. [Google Scholar] [CrossRef]
- Fendrich, V.; Langer, P.; Celik, I.; Bartsch, D.K.; Zielke, A.; Ramaswamy, A.; Rothmund, M. An Aggressive Surgical Approach Leads to Long-Term Survival in Patients with Pancreatic Endocrine Tumors. Ann. Surg. 2006, 244, 845–851; discussion 852–843. [Google Scholar] [CrossRef] [PubMed]
- Sitani, K.; Parghane, R.V.; Talole, S.; Basu, S. Long-Term Outcome of Indigenous (177)Lu-Dotatate Prrt in Patients with Metastatic Advanced Neuroendocrine Tumours: A Single Institutional Observation in a Large Tertiary Care Setting. Br J. Radiol. 2021, 94, 20201041. [Google Scholar] [CrossRef] [PubMed]
- Albers, M.B.; Sevcik, M.; Wiese, D.; Manoharan, J.; Rinke, A.; Jesinghaus, M.; Bartsch, D.K. Characteristics, Therapy, and Outcome of Rare Functioning Pancreatic Neuroendocrine Neoplasms. Sci. Rep. 2024, 14, 18507. [Google Scholar] [CrossRef] [PubMed]
- Brugel, M.; Walter, T.; Goichot, B.; Smith, D.; Lepage, C.; Do Cao, C.; Hautefeuille, V.; Rebours, V.; Cadiot, G.; de Mestier, L. Efficacy of Treatments for Vipoma: A Gte Multicentric Series. Pancreatology 2021, 21, 1531–1539. [Google Scholar] [CrossRef]
- Lam, A.K.; Ishida, H. Pancreatic Neuroendocrine Neoplasms: Clinicopathological Features and Pathological Staging. Histol. Histopathol. 2021, 36, 367–382. [Google Scholar] [CrossRef]
- Murakami, M.; Fujimori, N.; Matsumoto, K.; Ohno, A.; Teramatsu, K.; Takamatsu, Y.; Takeno, A.; Ueda, K.; Oono, T.; Ito, T.; et al. A Clinical Analysis on Functioning Pancreatic Neuroendocrine Tumors (Focusing on Vipomas): A Single-Center Experience. Endocr. J. 2022, 69, 1201–1209. [Google Scholar] [CrossRef]
- Siddappa, P.K.; Vege, S.S. Vasoactive Intestinal Peptide–Secreting Tumors: A Review. Pancreas 2019, 48, 1119–1125. [Google Scholar] [CrossRef]
- Kos-Kudła, B.; Castaño, J.P.; Denecke, T.; Grande, E.; Kjaer, A.; Koumarianou, A.; de Mestier, L.; Partelli, S.; Perren, A.; Stättner, S.; et al. European Neuroendocrine Tumour Society (Enets) 2023 Guidance Paper for Nonfunctioning Pancreatic Neuroendocrine Tumours. J. Neuroendocrinol. 2023, 35, e13343. [Google Scholar] [CrossRef]
- Angelousi, A.; Koffas, A.; Grozinsky-Glasberg, S.; Gertner, J.; Kassi, E.; Alexandraki, K.; Caplin, M.E.; Kaltsas, G.; Toumpanakis, C. Diagnostic and Management Challenges in Vasoactive Intestinal Peptide Secreting Tumors: A Series of 15 Patients. Pancreas 2019, 48, 934–942. [Google Scholar] [CrossRef]
- Schizas, D.; Mastoraki, A.; Bagias, G.; Patras, R.; Moris, D.; Lazaridis, I.I.; Arkadopoulos, N.; Felekouras, E. Clinicopathological Data and Treatment Modalities for Pancreatic Vipomas: A Systematic Review. J. BUON 2019, 24, 415–423. [Google Scholar]
- Azizian, A.; König, A.; Ghadimi, M. Treatment Options of Metastatic and Nonmetastatic Vipoma: A Review. Langenbecks Arch. Surg. 2022, 407, 2629–2636. [Google Scholar] [CrossRef]
- Lakhtakia, S.; Mehrotra, K.; Sekaran, A.; Itha, S.; Duvvur, N.R. Endoscopic Ultrasound-Guided Radiofrequency Ablation of Metastatic Pancreatic Vipoma: A Novel Treatment. Cureus 2024, 16, e67114. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Tanaka, M.; Sasano, H.; Osamura, Y.R.; Sasaki, I.; Kimura, W.; Takano, K.; Obara, T.; Ishibashi, M.; Nakao, K.; et al. Preliminary Results of a Japanese Nationwide Survey of Neuroendocrine Gastrointestinal Tumors. J. Gastroenterol. 2007, 42, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Xiong, G.; Zhang, H.; Wang, M.; Zhu, F.; Qin, R. Adrenocorticotropic Hormone-Producing Pancreatic Neuroendocrine Neoplasms: A Systematic Review. Endocr. Pract. 2021, 27, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Sarkar, R.; Mendiratta, V.; Srivastava, A. Addisonian Pigmentation—The Great Mimicker—A Review. Indian. J. Dermatol. 2024, 69, 422. [Google Scholar] [CrossRef]
- Yoshihara, A.; Nishihama, K.; Inoue, C.; Okano, Y.; Eguchi, K.; Tanaka, S.; Maki, K.; Fridman D’Alessandro, V.; Takeshita, A.; Yasuma, T.; et al. Adrenocorticotropic Hormone-Secreting Pancreatic Neuroendocrine Carcinoma with Multiple Organ Infections and Widespread Thrombosis: A Case Report. World J. Clin. Cases 2022, 10, 5723–5731. [Google Scholar] [CrossRef]
- Fottner, C.; Ferrata, M.; Weber, M.M. Hormone Secreting Gastro-Entero-Pancreatic Neuroendocrine Neoplasias (Gep-Nen): When to Consider, How to Diagnose? Rev. Endocr. Metab. Disord. 2017, 18, 393–410. [Google Scholar] [CrossRef]
- Öberg, K. Management of Functional Neuroendocrine Tumors of the Pancreas. Gland. Surgery 2017, 7, 20–27. [Google Scholar] [CrossRef]
- Sundin, A.; Arnold, R.; Baudin, E.; Cwikla, J.B.; Eriksson, B.; Fanti, S.; Fazio, N.; Giammarile, F.; Hicks, R.J.; Kjaer, A.; et al. Enets Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: Radiological, Nuclear Medicine and Hybrid Imaging. Neuroendocrinology 2017, 105, 212–244. [Google Scholar] [CrossRef]
- Bruno, O.D.; Danilowicz, K.; Manavela, M.; Mana, D.; Rossi, M.A. Long-Term Management with Octreotide or Cabergoline in Ectopic Corticotropin Hypersecretion: Case Report and Literature Review. Endocr. Pract. 2010, 16, 829–834. [Google Scholar] [CrossRef]
- Moraitis, A.G.; Auchus, R.J. Mifepristone Improves Octreotide Efficacy in Resistant Ectopic Cushing’s Syndrome. Case Rep. Endocrinol. 2016, 2016, 8453801. [Google Scholar] [CrossRef][Green Version]
- Makis, W.; McCann, K.; Riauka, T.A.; McEwan, A.J.B. Ectopic Corticotropin-Producing Neuroendocrine Tumor of the Pancreas Treated with 177lu Dotatate Induction and Maintenance Peptide Receptor Radionuclide Therapy. Clin. Nucl. Med. 2016, 41, 50–52. [Google Scholar] [CrossRef]
- Akirov, A.; Larouche, V.; Alshehri, S.; Asa, S.L.; Ezzat, S. Treatment Options for Pancreatic Neuroendocrine Tumors. Cancers 2019, 11, 828. [Google Scholar] [CrossRef]
- Magi, L.; Marasco, M.; Rinzivillo, M.; Faggiano, A.; Panzuto, F. Management of Functional Pancreatic Neuroendocrine Neoplasms. Curr. Treat. Options Oncol. 2023, 24, 725–741. [Google Scholar] [CrossRef]
- Maxwell, J.E.; Sherman, S.K.; Howe, J.R. Translational Diagnostics and Therapeutics in Pancreatic Neuroendocrine Tumors. Clin. Cancer Res. 2016, 22, 5022–5029. [Google Scholar] [CrossRef] [PubMed]
- Massironi, S. The Diagnostic Challenges of Functioning Neuroendocrine Tumors: Balancing Accuracy, Availability, and Personalized Care. Expert. Rev. Endocrinol. Metab. 2024, 19, 99–101. [Google Scholar] [CrossRef] [PubMed]
- Koffas, A.; Giakoustidis, A.; Papaefthymiou, A.; Bangeas, P.; Giakoustidis, D.; Papadopoulos, V.N.; Toumpanakis, C. Diagnostic Work-up and Advancement in the Diagnosis of Gastroenteropancreatic Neuroendocrine Neoplasms. Front. Surg. 2023, 10, 1064145. [Google Scholar] [CrossRef] [PubMed]
- Werle, S.D.; Ikonomi, N.; Lausser, L.; Kestler, A.; Weidner, F.M.; Schwab, J.D.; Maier, J.; Buchholz, M.; Gress, T.M.; Kestler, A.M.R.; et al. A Systems Biology Approach to Define Mechanisms, Phenotypes, and Drivers in Pannets with a Personalized Perspective. NPJ Syst. Biol. Appl. 2023, 9, 22. [Google Scholar] [CrossRef]
- Buicko, J.L.; Finnerty, B.M.; Zhang, T.; Kim, B.J.; Fahey, T.J., 3rd; Nancy Du, Y.C. Insights into the Biology and Treatment Strategies of Pancreatic Neuroendocrine Tumors. Ann. Pancreat. Cancer 2019, 2, 12. [Google Scholar] [CrossRef]
- Masharani, U.; Lindsay, S.; Moon, F.; Paciorek, A.; Bergsland, E. Metastatic Insulinoma-Outcomes in the Current Era. Oncologist 2025, 30, oyae275. [Google Scholar] [CrossRef] [PubMed]
- Ryzhkova, D.; Mitrofanova, L.; Tsoy, U.; Grineva, E.; Schlyakhto, E. Dual-Tracer Pet/Ct Imaging to Determine Tumor Heterogeneity in a Patient with Metastatic Acth-Secreting Neuroendocrine Neoplasm: A Case Report and Literature Review. Front. Endocrinol. 2022, 13, 958442. [Google Scholar] [CrossRef] [PubMed]
- Nilica, B.; Waitz, D.; Stevanovic, V.; Uprimny, C.; Kendler, D.; Buxbaum, S.; Warwitz, B.; Gerardo, L.; Henninger, B.; Virgolini, I.; et al. Direct Comparison of (68)Ga-Dota-Toc and (18)F-Fdg Pet/Ct in the Follow-up of Patients with Neuroendocrine Tumour Treated with the First Full Peptide Receptor Radionuclide Therapy Cycle. Eur. J. Nucl. Med. Mol. Imaging 2016, 43, 1585–1592. [Google Scholar] [CrossRef]
- di Santo, G.; Santo, G.; Sviridenko, A.; Virgolini, I. Peptide Receptor Radionuclide Therapy Combinations for Neuroendocrine Tumours in Ongoing Clinical Trials: Status 2023. Theranostics 2024, 14, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Roth, K.S.; Voltin, C.A.; van Heek, L.; Wegen, S.; Schomacker, K.; Fischer, T.; Marnitz, S.; Drzezga, A.; Kobe, C. Dual-Tracer Pet/Ct Protocol with [(18)F]-Fdg and [(68)Ga]Ga-Fapi-46 for Cancer Imaging: A Proof of Concept. J. Nucl. Med. 2022, 63, 1683–1686. [Google Scholar] [CrossRef]
- Mapelli, P.; Partelli, S.; Salgarello, M.; Doraku, J.; Muffatti, F.; Schiavo Lena, M.; Pasetto, S.; Bezzi, C.; Bettinardi, V.; Andreasi, V.; et al. Dual Tracer 68ga-Dotatoc and 18f-Fdg Pet Improve Preoperative Evaluation of Aggressiveness in Resectable Pancreatic Neuroendocrine Neoplasms. Diagnostics 2021, 11, 192. [Google Scholar] [CrossRef]
- Chan, D.L.; Hayes, A.R.; Karfis, I.; Conner, A.; Furtado O’Mahony, L.; Mileva, M.; Bernard, E.; Roach, P.; Marin, G.; Pavlakis, N.; et al. Dual [(68)Ga]Dotatate and [(18)F]Fdg Pet/Ct in Patients with Metastatic Gastroenteropancreatic Neuroendocrine Neoplasms: A Multicentre Validation of the Netpet Score. Br. J. Cancer 2023, 128, 549–555. [Google Scholar] [CrossRef]
- Kuiper, J.; Zoetelief, E.; Brabander, T.; de Herder, W.W.; Hofland, J. Current Status of Peptide Receptor Radionuclide Therapy in Grade 1 and 2 Gastroenteropancreatic Neuroendocrine Tumours. J. Neuroendocrinol. 2025, 37, e13469. [Google Scholar] [CrossRef]
- Stueven, A.K.; Kayser, A.; Wetz, C.; Amthauer, H.; Wree, A.; Tacke, F.; Wiedenmann, B.; Roderburg, C.; Jann, H. Somatostatin Analogues in the Treatment of Neuroendocrine Tumors: Past, Present and Future. Int. J. Mol. Sci. 2019, 20, 3049. [Google Scholar] [CrossRef]
- Gut, P.; Waligorska-Stachura, J.; Czarnywojtek, A.; Sawicka-Gutaj, N.; Baczyk, M.; Ziemnicka, K.; Fischbach, J.; Wolinski, K.; Kaznowski, J.; Wrotkowska, E.; et al. Management of the Hormonal Syndrome of Neuroendocrine Tumors. Arch. Med. Sci. 2017, 13, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.C.; Fazio, N.; Singh, S.; Buzzoni, R.; Carnaghi, C.; Wolin, E.; Tomasek, J.; Raderer, M.; Lahner, H.; Voi, M.; et al. Everolimus for the Treatment of Advanced, Non-Functional Neuroendocrine Tumours of the Lung or Gastrointestinal Tract (Radiant-4): A Randomised, Placebo-Controlled, Phase 3 Study. Lancet 2016, 387, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Pusceddu, S.; Prinzi, N.; Tafuto, S.; Ibrahim, T.; Filice, A.; Brizzi, M.P.; Panzuto, F.; Baldari, S.; Grana, C.M.; Campana, D.; et al. Association of Upfront Peptide Receptor Radionuclide Therapy with Progression-Free Survival among Patients with Enteropancreatic Neuroendocrine Tumors. JAMA Netw. Open 2022, 5, e220290. [Google Scholar] [CrossRef] [PubMed]
- ClinicalTrialsUSA. Lutetium 177lu-Edotreotide Versus Best Standard of Care in Well-Differentiated Aggressive Grade-2 and Grade-3 Gastroenteropancreatic Neuroendocrine Tumors (Gep-Nets)—Compose (Compose). Available online: https://clinicaltrials.gov/study/NCT04919226 (accessed on 20 June 2025).
- ClinicalTrialsUSA. Efficacy and Safety of 177lu-Edotreotide Prrt in Gep-Net Patients (Compete). Available online: https://clinicaltrials.gov/study/NCT03049189 (accessed on 20 June 2025).
- Baudin, E.; Walter, T.; Beron, A.; Smith, D.; Hadoux, J.; Lachachi, C.; Taieb, D.; Ansquer, C.; Dierickx, L.; Du Bourg, L.d.M. 887o First Multicentric Randomized Phase Ii Trial Investigating the Antitumor Efficacy of Peptide Receptor Radionucleide Therapy with 177lutetium-Octreotate (Oclu) in Unresectable Progressive Neuroendocrine Pancreatic Tumor: Results of the Oclurandom Trial. Ann. Oncol. 2022, 33, S954. [Google Scholar] [CrossRef]
- Chan, J.A.; Geyer, S.; Zemla, T.; Knopp, M.V.; Behr, S.; Pulsipher, S.; Ou, F.-S.; Dueck, A.C.; Acoba, J.; Shergill, A. Phase 3 Trial of Cabozantinib in Advanced Neuroendocrine Tumors. N. Engl. J. Med. 2024, 392, 653. [Google Scholar] [CrossRef]
- Kashyap, R.; Raja, S.; Adusumilli, A.; Gopireddy, M.M.R.; Loveday, B.P.T.; Alipour, R.; Kong, G. Role of Neoadjuvant Peptide Receptor Radionuclide Therapy in Unresectable and Metastatic Gastro-Entero-Pancreatic Neuroendocrine Neoplasms: A Scoping Review. J. Neuroendocrinol. 2025, 37, e13425. [Google Scholar] [CrossRef]
- Puranik, A.D.; Dev, I.D.; Prasad, V. Frontiers in Radiopharmaceuticals for Neuroendocrine Tumors. J. Neuroendocrinol. 2025, 37, e70006. [Google Scholar] [CrossRef]
- Tsoli, M.; Koumarianou, A.; Angelousi, A.; Kaltsas, G. Established and Novel Circulating Neuroendocrine Tumor Biomarkers for Diagnostic, Predictive and Prognostic Use. Best Pract. Res. Clin. Endocrinol. Metab. 2023, 37, 101785. [Google Scholar] [CrossRef]
- Marien, L.; Islam, O.; Van Mileghem, L.; Lybaert, W.; Peeters, M.; Vandamme, T. Pathophysiology and Treatment of Pancreatic Neuroendocrine Neoplasms (Pnens): New Developments. In Endotext; Feingold, K.R., Ahmed, S.F., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Dhatariya, K., Dungan, K., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Feng, Z.; He, X.; Zhang, X.; Wu, Y.; Xing, B.; Knowles, A.; Shan, Q.; Miller, S.; Hojnacki, T.; Ma, J.; et al. Potent Suppression of Neuroendocrine Tumors and Gastrointestinal Cancers by Cdh17car T Cells without Toxicity to Normal Tissues. Nat. Cancer 2022, 3, 581–594. [Google Scholar] [CrossRef]
- Young, K.; Starling, N.; Sadanandam, A. The Molecular Biology of Pancreatic Neuroendocrine Neoplasms: Challenges and Translational Opportunities. Semin. Cancer Biol. 2020, 61, 132–138. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).