
Analysis of Short-Range Ordering Effect on Tensile Deformation Behavior of Equiatomic High-Entropy Alloys TiNbZrV, TiNbZrTa and TiNbZrHf Based on Atomistic Simulations


 ,
, 
Abstract
1. Introduction
2. Modelling
2.1. Materials and MD Model
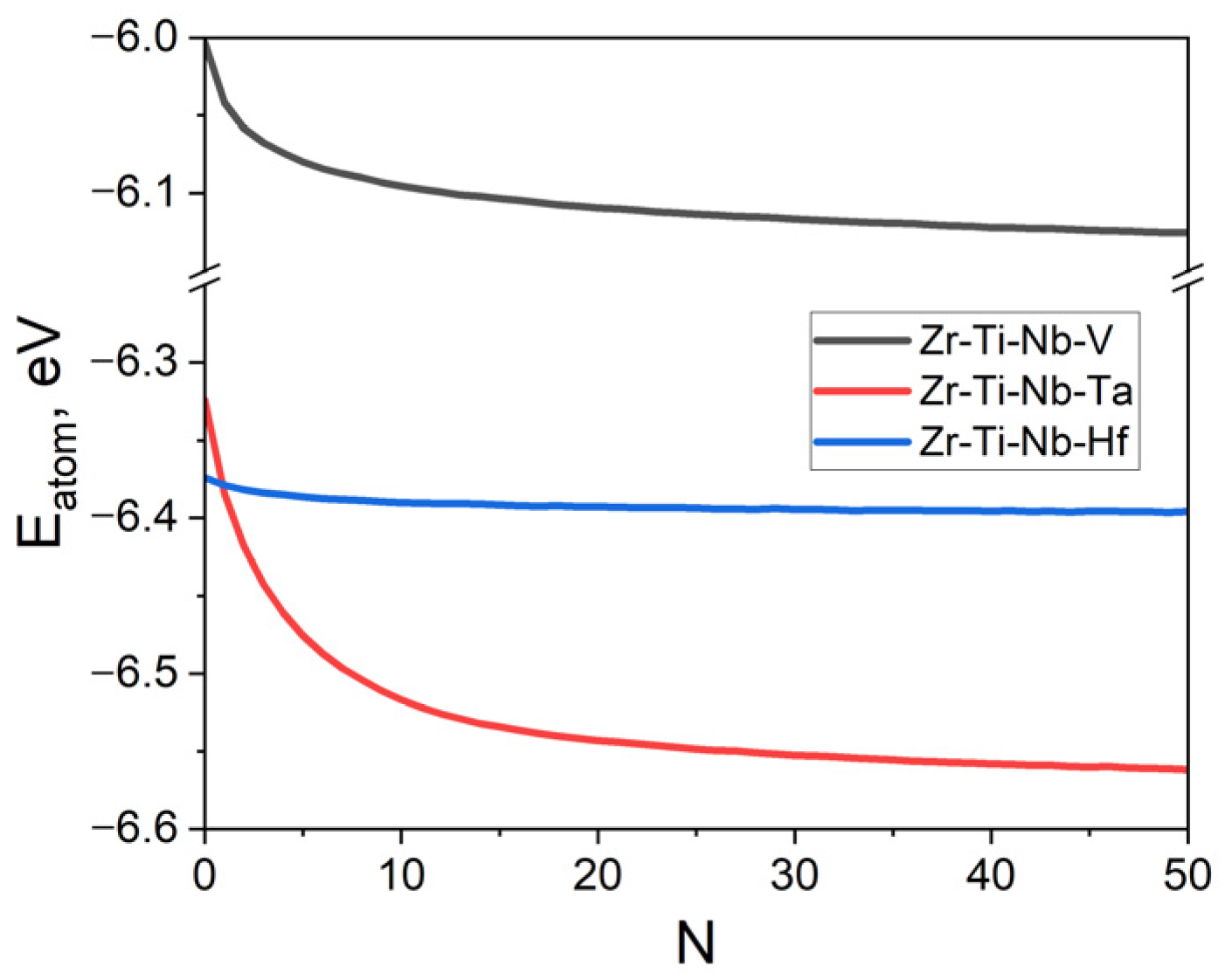
2.2. MD/MC Relaxation Procedure
2.3. Tensile Deformation
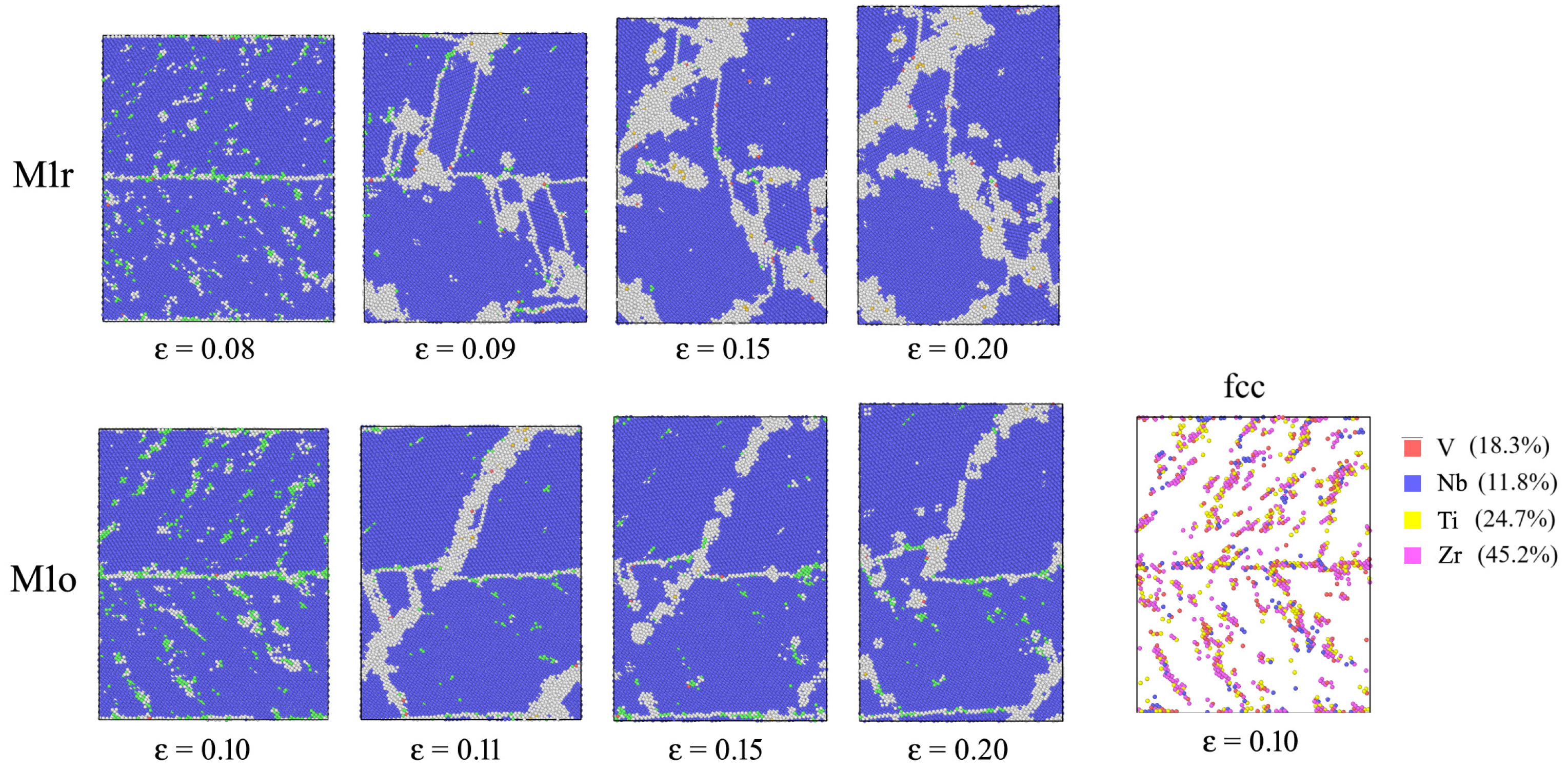
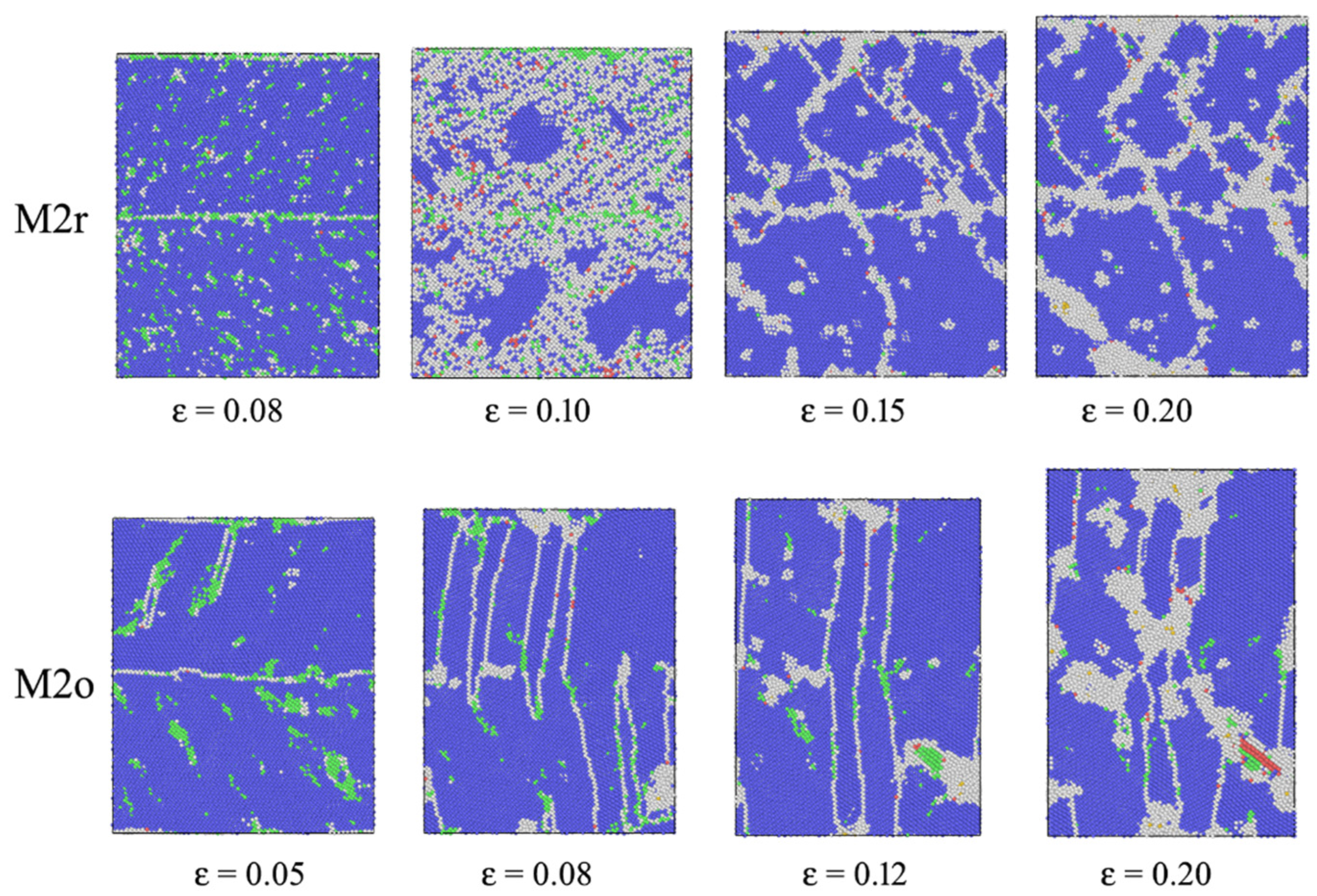
3. Results
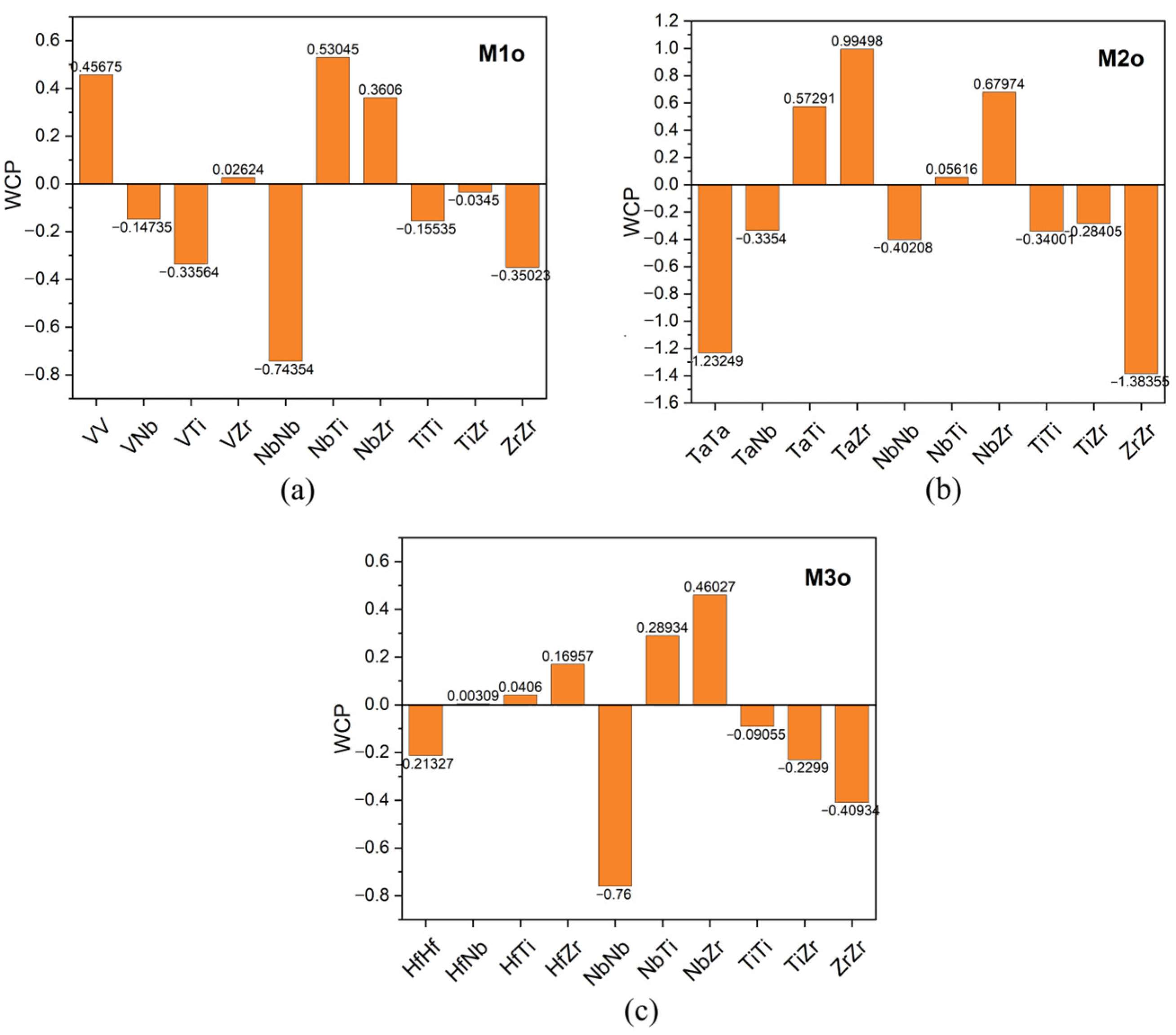
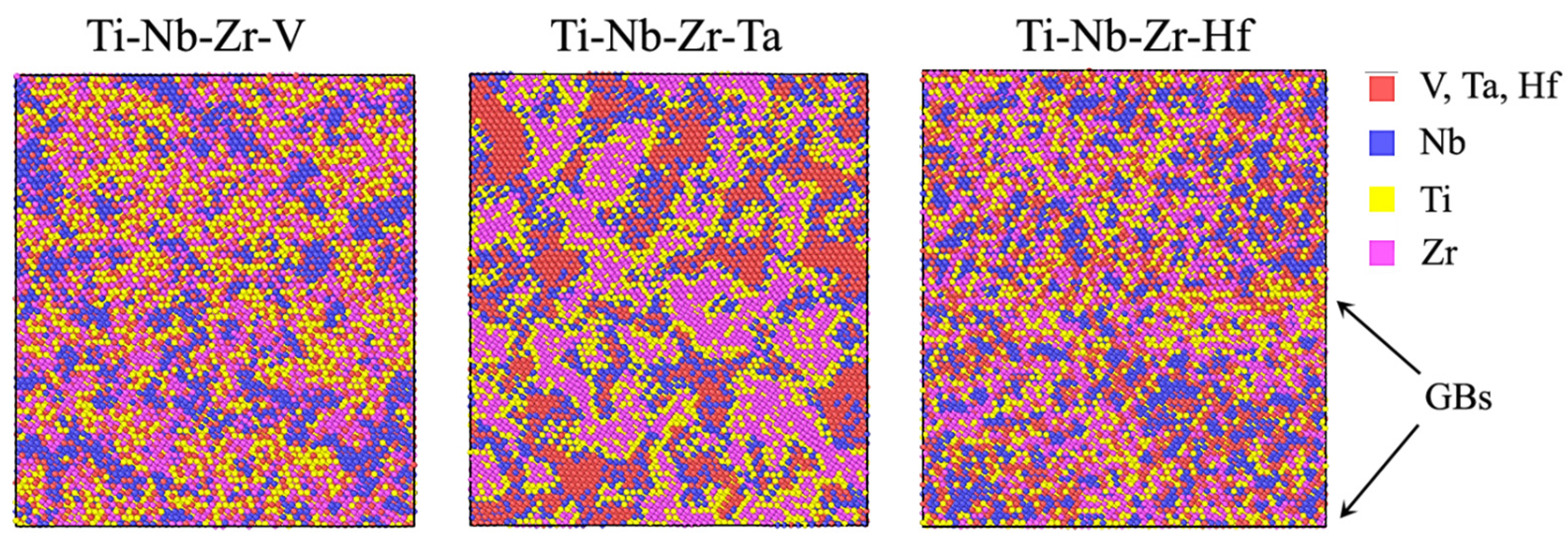
3.1. SRO
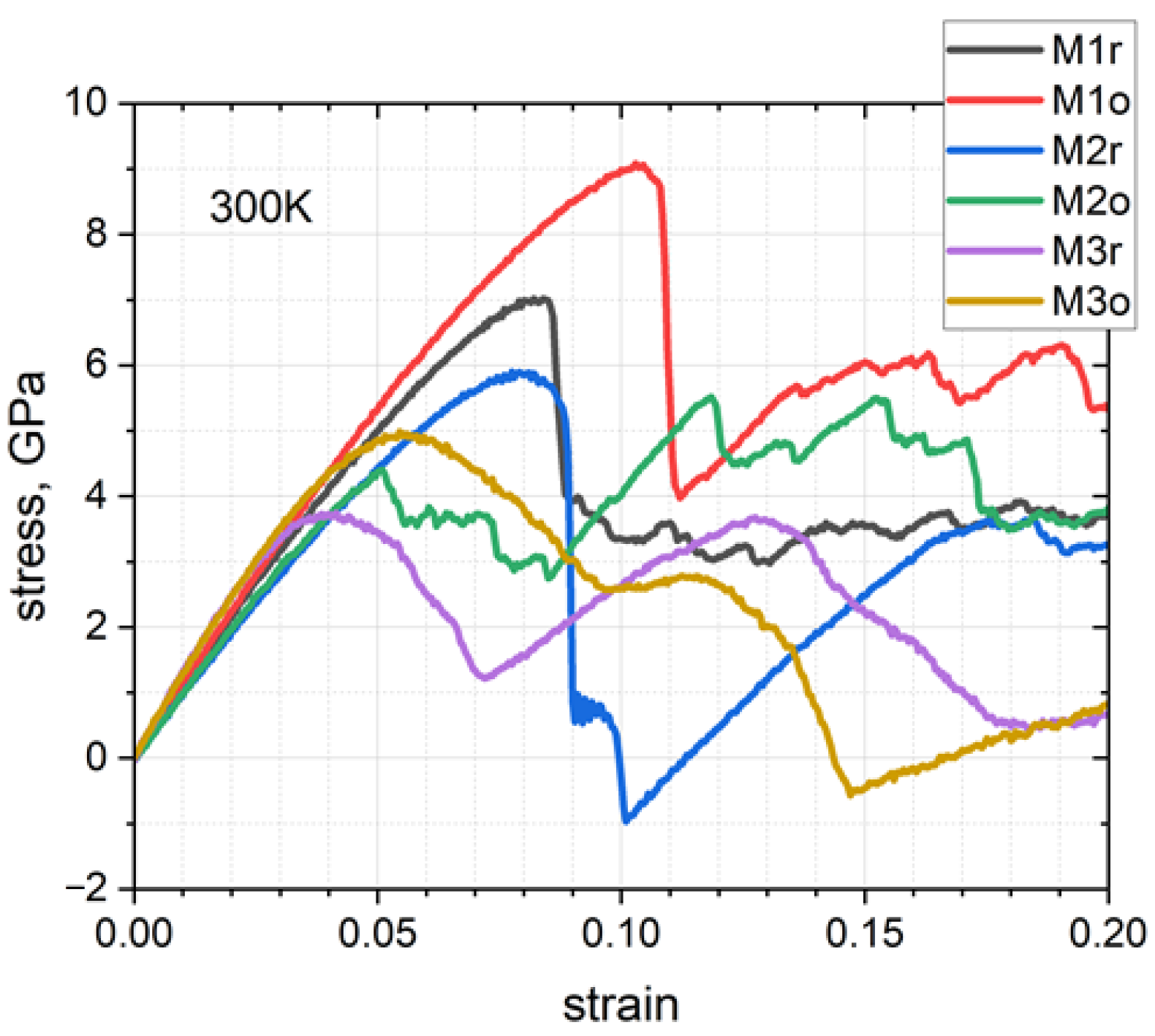
3.2. Stress–Strain Curves
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fan, X.J.; Qu, R.T.; Zhang, Z.F. Remarkably high fracture toughness of HfNbTaTiZr refractory high-entropy alloy. J. Mater. Sci. Technol. 2022, 123, 70–77. [Google Scholar] [CrossRef]
- Li, H.; Cao, F.; Li, T.; Tan, Y.; Chen, Y.; Wang, H.; Liaw, P.K.; Dai, L. Enhanced plasticity in refractory high-entropy alloy via multicomponent ceramic nanoparticle. J. Mater. Sci. Technol. 2024, 194, 51–62. [Google Scholar] [CrossRef]
- Zhang, Y.; Bu, Z.; Yao, T.; Yang, L.; Li, W.; Li, J. Novel BCC Ti-Al-Nb-Zr medium-entropy alloys with ultrahigh specific strength and ductility. J. Alloys Compd. 2023, 936, 168290. [Google Scholar] [CrossRef]
- Jia, Y.; Zhang, L.; Li, P.; Ma, X.; Xu, L.; Wu, S.; Jia, Y.; Wang, G. Microstructure and Mechanical Properties of Nb–Ti–V–Zr Refractory Medium-Entropy Alloys. Front. Mater. 2020, 7, 172. [Google Scholar] [CrossRef]
- Wang, S.; Wu, M.; Shu, D.; Zhu, G.; Wang, D.; Sun, B. Mechanical instability and tensile properties of TiZrHfNbTa high entropy alloy at cryogenic temperatures. Acta Mater. 2020, 201, 517–527. [Google Scholar] [CrossRef]
- Hu, M.; Wang, L.; Li, G.; Huang, Q.; Liu, Y.; He, J.; Wu, H.; Song, M. Investigations on microstructure and properties of Ti-Nb-Zr medium-entropy alloys for metallic biomaterials. Intermetallics 2022, 145, 107568. [Google Scholar] [CrossRef]
- Wong, K.-K.; Hsu, H.-C.; Wu, S.-C.; Ho, W.-F. Structure and properties of Ti-rich Ti–Zr–Nb–Mo medium-entropy alloys. J. Alloys Compd. 2021, 868, 159137. [Google Scholar] [CrossRef]
- Sahlberg, M.; Karlsson, D.; Zlotea, C.; Jansson, U. Superior hydrogen storage in high entropy alloys. Sci. Rep. 2016, 6, 36770. [Google Scholar] [CrossRef]
- Nygård, M.M.; Ek, G.; Karlsson, D.; Sørby, M.H.; Sahlberg, M.; Hauback, B.C. Counting electrons–A new approach to tailor the hydrogen sorption properties of high-entropy alloys. Acta Mater. 2019, 175, 121–129. [Google Scholar] [CrossRef]
- Edalati, P.; Floriano, R.; Mohammadi, A.; Li, Y.; Zepon, G.; Li, H.-W.; Edalati, K. Reversible room temperature hydrogen storage in high-entropy alloy TiZrCrMnFeNi. Scr. Mater. 2020, 178, 387–390. [Google Scholar] [CrossRef]
- Couzinié, J.-P.; Lilensten, L.; Champion, Y.; Dirras, G.; Perrière, L.; Guillot, I. On the room temperature deformation mechanisms of a TiZrHfNbTa refractory high-entropy alloy. Mater. Sci. Eng. A 2015, 645, 255–263. [Google Scholar] [CrossRef]
- Lilensten, L.; Couzinié, J.-P.; Perrière, L.; Hocini, A.; Keller, C.; Dirras, G.; Guillot, I. Study of a bcc multi-principal element alloy: Tensile and simple shear properties and underlying deformation mechanisms. Acta Mater. 2018, 142, 131–141. [Google Scholar] [CrossRef]
- Dirras, G.; Lilensten, L.; Djemia, P.; Laurent-Brocq, M.; Tingaud, D.; Couzinié, J.-P.; Perrière, L.; Chauveau, T.; Guillot, I. Elastic and plastic properties of as-cast equimolar TiHfZrTaNb high-entropy alloy. Mater. Sci. Eng. A 2016, 654, 30–38. [Google Scholar] [CrossRef]
- Jarlöv, A.; Ji, W.; Babicheva, R.; Tian, Y.; Hu, Z.; Seet, H.L.; Tan, L.; Liu, F.; Liu, Y.; Nai, M.L.S.; et al. Tailoring short-range order and dislocation evolution in Cr–Co–Ni medium-entropy alloys: A molecular dynamics study. Mater. Des. 2024, 240, 112840. [Google Scholar] [CrossRef]
- Rao, S.I.; Varvenne, C.; Woodward, C.; Parthasarathy, T.A.; Miracle, D.; Senkov, O.N.; Curtin, W.A. Atomistic simulations of dislocations in a model BCC multicomponent concentrated solid solution alloy. Acta Mater. 2017, 125, 311–320. [Google Scholar] [CrossRef]
- Li, X. Phase stability and micromechanical properties of TiZrHf-based refractory high-entropy alloys: A first-principles study. Phys. Rev. Mater. 2023, 7, 113604. [Google Scholar] [CrossRef]
- Qu, N.; Chen, Y.; Lai, Z.; Liu, Y.; Zhu, J. The phase selection via machine learning in high entropy alloys. Procedia Manuf. 2019, 37, 299–305. [Google Scholar] [CrossRef]
- Huang, H.; Wu, Y.; He, J.; Wang, H.; Liu, X.; An, K.; Wu, W.; Lu, Z. Phase-Transformation Ductilization of Brittle High-Entropy Alloys via Metastability Engineering. Adv. Mater. 2017, 29, 1701678. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tang, Y.; Li, S.; Zhang, H.; Ye, Y.; Zhu, L.; Ai, Y.; Bai, S. Novel metastable engineering in single-phase high-entropy alloy. Mater. Des. 2019, 162, 256–262. [Google Scholar] [CrossRef]
- Lilensten, L.; Couzinié, J.-P.; Bourgon, J.; Perrière, L.; Dirras, G.; Prima, F.; Guillot, I. Design and tensile properties of a bcc Ti-rich high-entropy alloy with transformation-induced plasticity. Mater. Res. Lett. 2017, 5, 110–116. [Google Scholar] [CrossRef]
- Eleti, R.R.; Klimova, M.; Tikhonovsky, M.; Stepanov, N.; Zherebtsov, S. Exceptionally high strain-hardening and ductility due to transformation induced plasticity effect in Ti-rich high-entropy alloys. Sci. Rep. 2020, 10, 13293. [Google Scholar] [CrossRef] [PubMed]
- Babicheva, R.; Jarlöv, A.; Zheng, H.; Dmitriev, S.; Korznikova, E.; Ling Sharon Nai, M.; Ramamurty, U.; Zhou, K. Effect of short-range ordering and grain boundary segregation on shear deformation of CoCrFeNi high-entropy alloys with Al addition. Comput. Mater. Sci. 2022, 215, 111762. [Google Scholar] [CrossRef]
- Wang, L.; Zhao, Y.; Dou, Y.; He, X.; Zhang, Z.; Chen, M.; Deng, H.; Yang, W. Atomistic Study on Defect–Grain Boundary Interactions in TiVTa Concentrated Solid–Solution Alloys. Crystals 2024, 14, 166. [Google Scholar] [CrossRef]
- Li, Y.; Qiang, W. Defect properties of a body-centered cubic equiatomic TiVZrTa high-entropy alloy from atomistic simulations. J. Phys. Condens. Matter 2023, 35, 345701. [Google Scholar] [CrossRef]
- Cao, P. Maximum strength and dislocation patterning in multi–principal element alloys. Sci. Adv. 2022, 8, eabq7433. [Google Scholar] [CrossRef]
- Jing, W.; Wu, M.S. Inhibiting the inverse Hall-Petch behavior in CoCuFeNiPd high-entropy alloys with short-range ordering and grain boundary segregation. Scr. Mater. 2022, 221, 114950. [Google Scholar] [CrossRef]
- Jarlöv, A.; Ji, W.; Zhu, Z.; Tian, Y.; Babicheva, R.; An, R.; Seet, H.L.; Nai, M.L.S.; Zhou, K. Molecular dynamics study on the strengthening mechanisms of Cr–Fe–Co–Ni high-entropy alloys based on the generalized stacking fault energy. J. Alloys Compd. 2022, 905, 164137. [Google Scholar] [CrossRef]
- Chen, S.; Aitken, Z.H.; Pattamatta, S.; Wu, Z.; Yu, Z.G.; Srolovitz, D.J.; Liaw, P.K. Simultaneously enhancing the ultimate strength and ductility of high-entropy alloys via short-range ordering. Nat. Commun. 2021, 12, 4953. [Google Scholar] [CrossRef]
- Kazakov, A.M.; Yakhin, A.V.; Karimov, E.Z.; Babicheva, R.I.; Kistanov, A.A.; Korznikova, E.A. Effect of Segregation on Deformation Behaviour of Nanoscale CoCrCuFeNi High-Entropy Alloy. Appl. Sci. 2023, 13, 4013. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, F.; Yuan, X.; Huang, H.; Wen, X.; Wang, Y.; Zhang, M.; Wu, H.; Liu, X.; Wang, H.; et al. Short-range ordering and its effects on mechanical properties of high-entropy alloys. J. Mater. Sci. Technol. 2021, 62, 214–220. [Google Scholar] [CrossRef]
- Mei, Z.-G.; Liang, L.; Kim, Y.S.; Wiencek, T.; O’Hare, E.; Yacout, A.M.; Hofman, G.; Anitescu, M. Grain growth in U–7Mo alloy: A combined first-principles and phase field study. J. Nucl. Mater. 2016, 473, 300–308. [Google Scholar] [CrossRef]
- Scheiber, D.; Renk, O.; Popov, M.; Romaner, L. Temperature dependence of surface and grain boundary energies from first principles. Phys. Rev. B 2020, 101, 174103. [Google Scholar] [CrossRef]
- Metropolis, N.; Rosenbluth, A.W.; Rosenbluth, M.N.; Teller, A.H.; Teller, E. Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1953, 21, 1087–1092. [Google Scholar] [CrossRef]
- Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Babicheva, R.I.; Evazzade, I.; Korznikova, E.A.; Shepelev, I.A.; Zhou, K.; Dmitriev, S.V. Low-energy channel for mass transfer in Pt crystal initiated by molecule impact. Comput. Mater. Sci. 2019, 163, 248–255. [Google Scholar] [CrossRef]
- Shepelev, I.A.; Dmitriev, S.V.; Kudreyko, A.A.; Velarde, M.G.; Korznikova, E.A. Supersonic voidions in 2D Morse lattice. Chaos Solitons Fractals 2020, 140, 110217. [Google Scholar] [CrossRef]
- Shelepev, I.A.; Kolesnikov, I.D. 2022 Excitation and propagation of 1-crowdion in bcc niobium lattice. Mater. Technol. Des. 2022, 4, 5–10. [Google Scholar] [CrossRef]
- Babicheva, R.I.; Dahanayaka, M.; Liu, B.; Korznikova, E.A.; Dmitriev, S.V.; Wu, M.S.; Zhou, K. Characterization of two carbon allotropes, cyclicgraphene and graphenylene, as semi-permeable materials for membranes. Mater. Sci. Eng. B 2020, 259, 114569. [Google Scholar] [CrossRef]
- Yankovskaya, U.I.; Zakharov, P.V. Heat resistance of a Pt crystal reinforced with CNT’s. Mater. Technol. Des. 2021, 3, 64–67. [Google Scholar] [CrossRef]
- Yankovskaya, U.I.; Sharapova, Y.R.; Zakharov, P.V. Effect of carbon nanotubes on the mechanical properties of Ni3Al polycrystal. Mater. Technol. Des. 2022, 4, 76–81. [Google Scholar] [CrossRef]
- Rysaeva, L.K.; Bachurin, D.V.; Murzaev, R.T.; Abdullina, D.U.; Korznikova, E.A.; Mulyukov, R.R.; Dmitriev, S.V. Evolution Of The Carbon Nanotube Bundle Structure Under Biaxial And Shear Strains. FU Mech. Eng. 2020, 18, 525–536. [Google Scholar] [CrossRef]
- Nitol, M.S.; Echeverria, M.J.; Dang, K.; Baskes, M.I.; Fensin, S.J. New modified embedded-atom method interatomic potential to understand deformation behavior in VNbTaTiZr refractory high entropy alloy. Comput. Mater. Sci. 2024, 237, 112886. [Google Scholar] [CrossRef]
- Huang, X.; Liu, L.; Duan, X.; Liao, W.; Huang, J.; Sun, H.; Yu, C. Atomistic simulation of chemical short-range order in HfNbTaZr high entropy alloy based on a newly-developed interatomic potential. Mater. Des. 2021, 202, 109560. [Google Scholar] [CrossRef]
- Dmitriev, S.V.; Kistanov, A.A.; Kosarev, I.V.; Scherbinin, S.A.; Shapeev, A.V. Construction of machine learning interatomic potentials for metals. Russ. Phys. J. 2024, 67, 1408–1413. [Google Scholar] [CrossRef]
- Wang, F.; Wu, H.; Dong, L.; Pan, G.; Zhou, X.; Wang, S.; Guo, R.; Wu, G.; Gao, J.; Dai, F.; et al. Atomic-scale simulations in multi-component alloys and compounds: A review on advances in interatomic potential. J. Mater. Sci. Technol. 2023, 165, 49–65. [Google Scholar] [CrossRef]
- Semenova, M.N.; Bebikhov, Y.V.; Yakushev, I.A.; Semenov, A.S. Development of a program for selecting the interaction potential of particles in the crystal lattice of metals. Mater. Technol. Des. 2024, 6, 99–108. [Google Scholar] [CrossRef]
- Stukowski, A. Visualization and analysis of atomistic simulation data with OVITO–the Open Visualization Tool. Model. Simul. Mater. Sc. 2009, 18, 015012. [Google Scholar] [CrossRef]
- Stukowski, A. Structure identification methods for atomistic simulations of crystalline materials. Model. Simul. Mater. Sci. Eng. 2012, 20, 045021. [Google Scholar] [CrossRef]
- Stukowski, A.; Bulatov, V.V.; Arsenlis, A. Automated identification and indexing of dislocations in crystal interfaces. Model. Simul. Mater. Sci. Eng. 2012, 20, 085007. [Google Scholar] [CrossRef]
- Cowley, J.M. X-Ray Measurement of Order in Single Crystals of Cu3Au. J. Appl. Phys. 1950, 21, 24–30. [Google Scholar] [CrossRef]
- Aitken, Z.H.; Zhang, Y.W. Revealing the deformation twinning nucleation mechanism of CC HEAs. MRS Commun. 2019, 9, 406–412. [Google Scholar] [CrossRef]
- Han, Z.D.; Chen, N.; Zhao, S.F.; Fan, L.W.; Yang, G.N.; Shao, Y.; Yao, K.F. Effect of Ti additions on mechanical properties of NbMoTaW and VNbMoTaW refractory high entropy alloys. Intermetallics 2017, 84, 153–157. [Google Scholar] [CrossRef]
- Balokhonov, R.; Utyaganova, V.; Gatiyatullina, D.; Zemlianov, A.; Romanova, V. Interlayer effect on deformation and fracture of dendritic structure formed during wire-feed electron-beam additive manufacturing of Al-Si alloy. Facta Univ. Ser. Mech. Eng. 2024, 515–527. [Google Scholar] [CrossRef]
- Fisher, J.C. On the strength of solid solution alloys. Acta Metall. 1954, 2, 9–10. [Google Scholar] [CrossRef]
- Rajput, A.; Paul, S.K. Cyclic Plastic Deformation Response of Nanocrystalline BCC Iron. Met. Mater. Int. 2021, 27, 825–837. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HEA | M1r | M1o | M2r | M2o | M3r | M3o |
|---|---|---|---|---|---|---|
| εy | 0.084 | 0.103 | 0.078 | 0.051 | 0.033 | 0.051 |
| σy, GPa | 7.019 | 9.081 | 5.902 | 4.399 | 3.561 | 4.927 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Babicheva, R.I.; Semenov, A.S.; Izosimov, A.A.; Korznikova, E.A. Analysis of Short-Range Ordering Effect on Tensile Deformation Behavior of Equiatomic High-Entropy Alloys TiNbZrV, TiNbZrTa and TiNbZrHf Based on Atomistic Simulations. Modelling 2024, 5, 1853-1864. https://doi.org/10.3390/modelling5040096
Babicheva RI, Semenov AS, Izosimov AA, Korznikova EA. Analysis of Short-Range Ordering Effect on Tensile Deformation Behavior of Equiatomic High-Entropy Alloys TiNbZrV, TiNbZrTa and TiNbZrHf Based on Atomistic Simulations. Modelling. 2024; 5(4):1853-1864. https://doi.org/10.3390/modelling5040096
Chicago/Turabian StyleBabicheva, Rita I., Aleksander S. Semenov, Artem A. Izosimov, and Elena A. Korznikova. 2024. "Analysis of Short-Range Ordering Effect on Tensile Deformation Behavior of Equiatomic High-Entropy Alloys TiNbZrV, TiNbZrTa and TiNbZrHf Based on Atomistic Simulations" Modelling 5, no. 4: 1853-1864. https://doi.org/10.3390/modelling5040096
APA StyleBabicheva, R. I., Semenov, A. S., Izosimov, A. A., & Korznikova, E. A. (2024). Analysis of Short-Range Ordering Effect on Tensile Deformation Behavior of Equiatomic High-Entropy Alloys TiNbZrV, TiNbZrTa and TiNbZrHf Based on Atomistic Simulations. Modelling, 5(4), 1853-1864. https://doi.org/10.3390/modelling5040096