
Assessment of Computational Tools for Predicting Supramolecular Synthons



Abstract
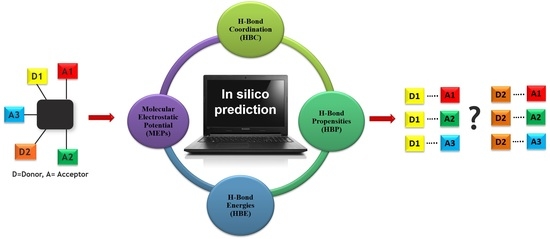
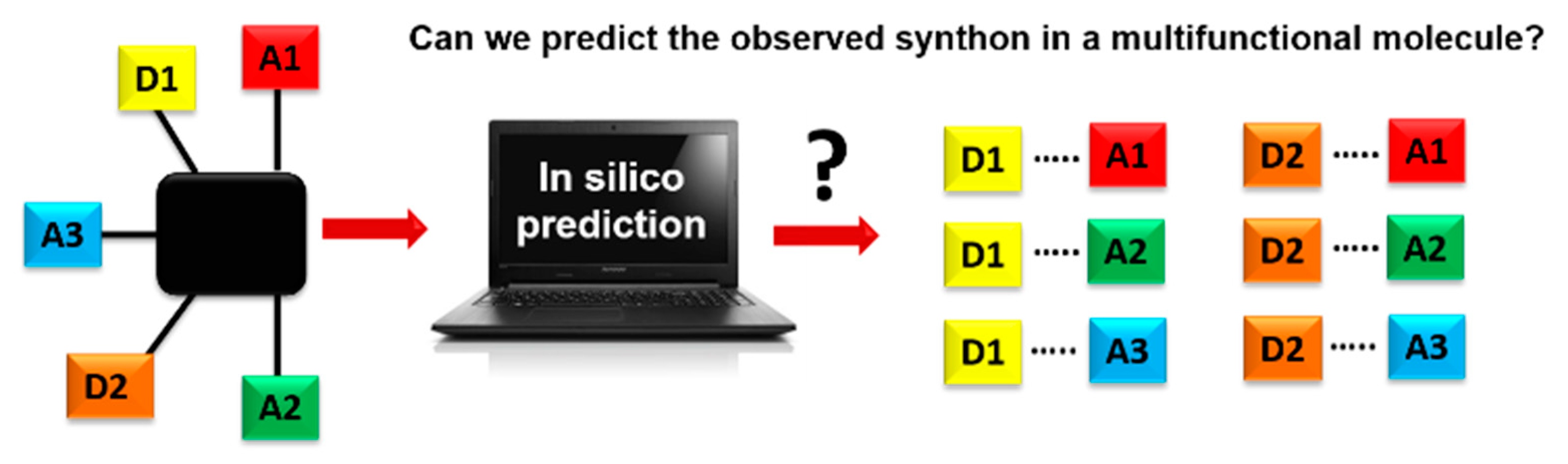
1. Introduction
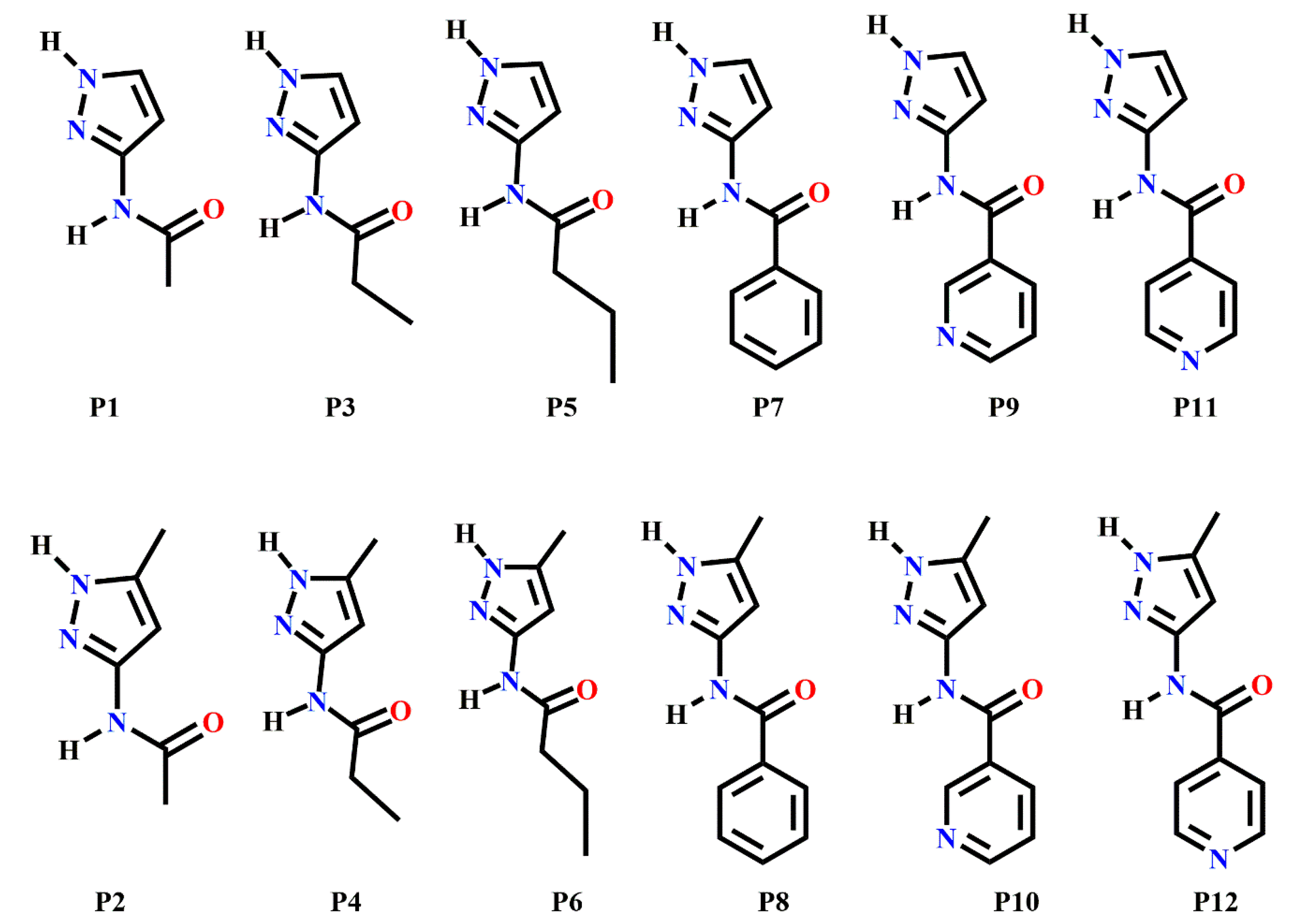
- Which method is more suitable for predicting the synthon outcome in the crystal structures of P1–P12?
- Is a combination of prediction methods better than individual methods?
- Which synthon is most optimal in group 1 (P1–P8) and how does adding an acceptor group affect the choice of synthon in P9–P12?
- Which molecules present the larger risk of synthon polymorphism, and which method is most suitable for predicting synthon polymorphism in this group of molecules?
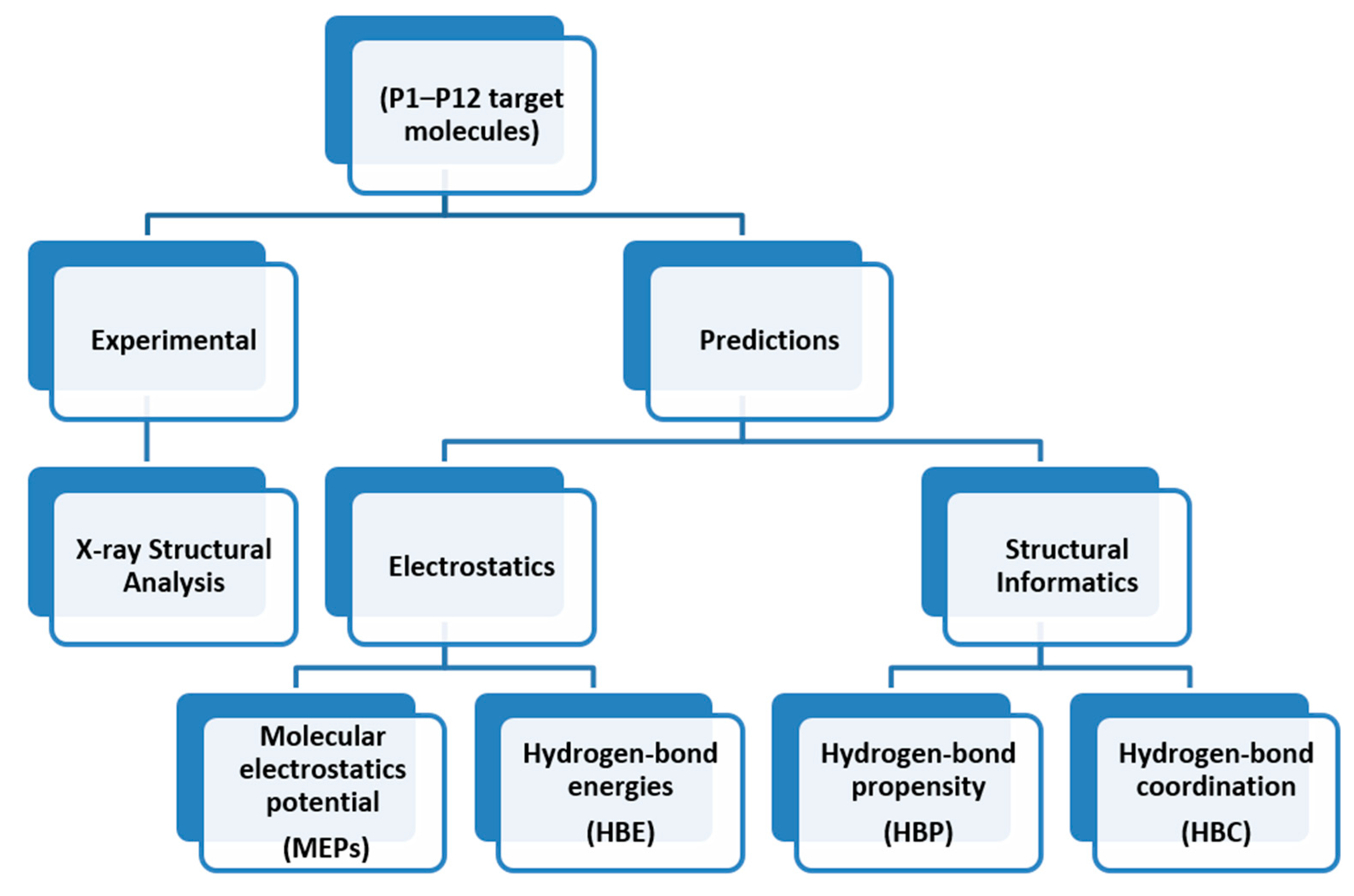
2. Materials and Methods
2.1. Molecular Electrostatic Potentials (MEPs)
2.2. Hydrogen-Bond Energies (HBE)
2.3. Hydrogen-Bond Propensity (HBP)
2.4. Hydrogen-Bond Coordination (HBC)
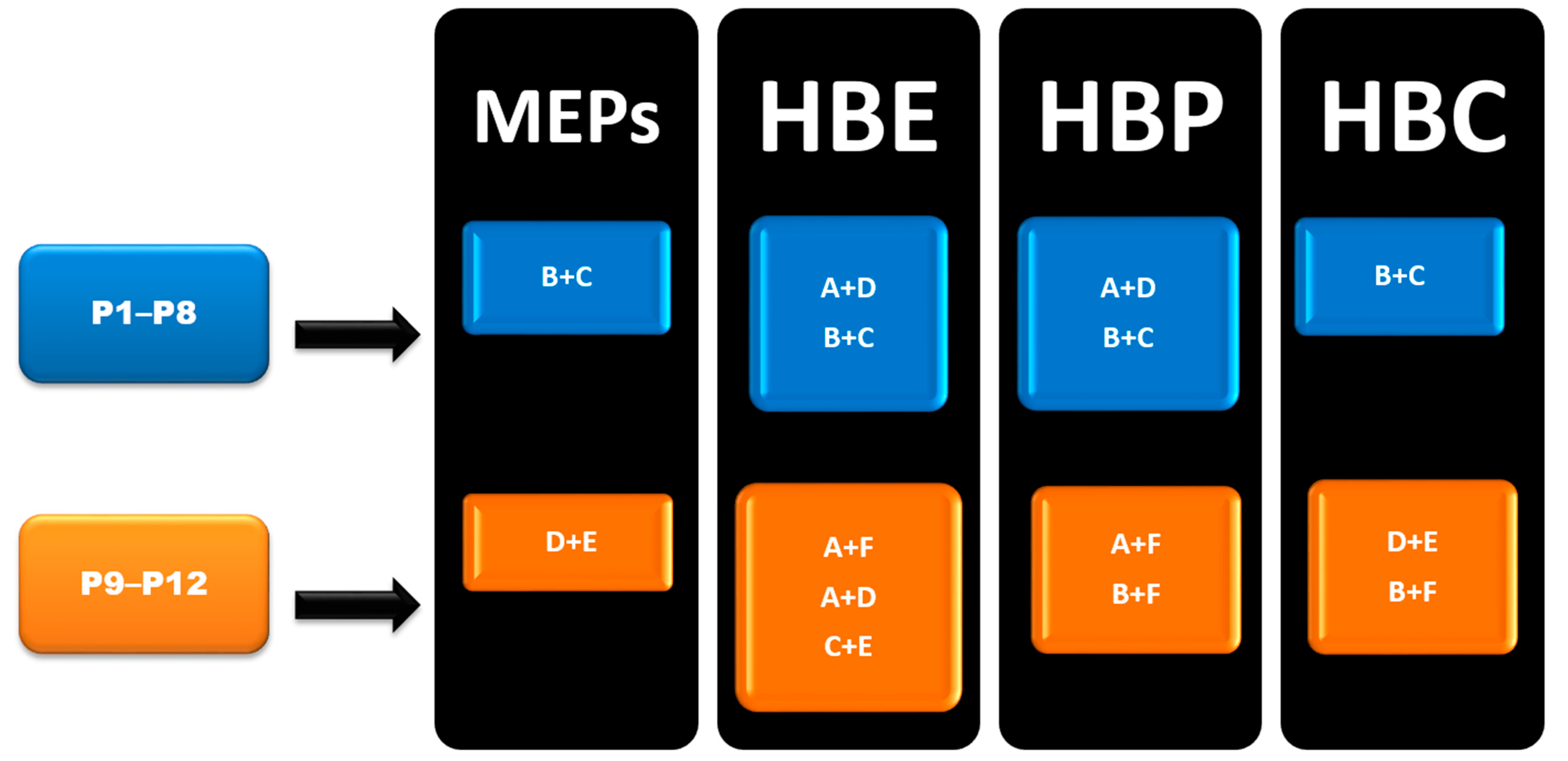
3. Results
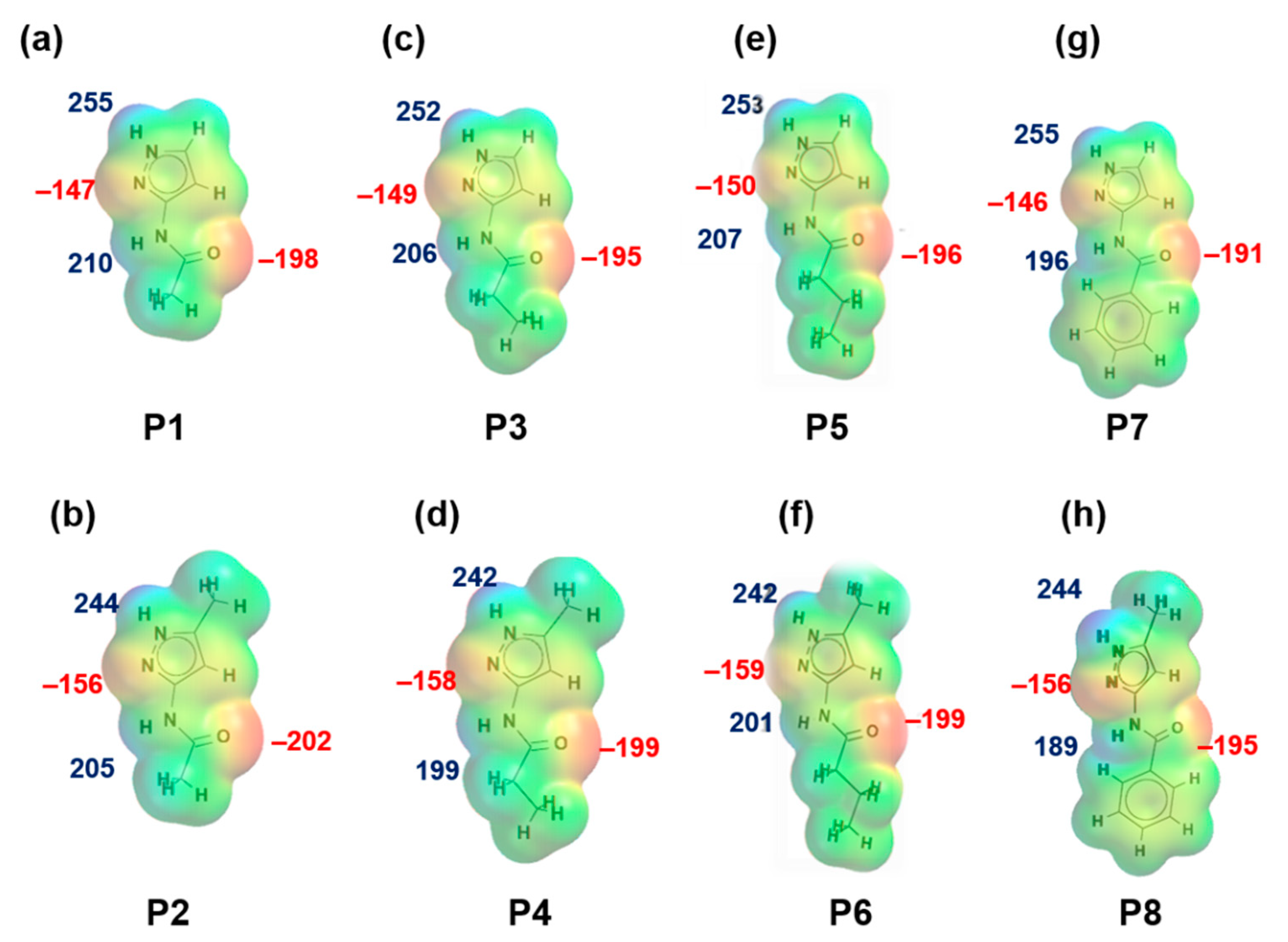
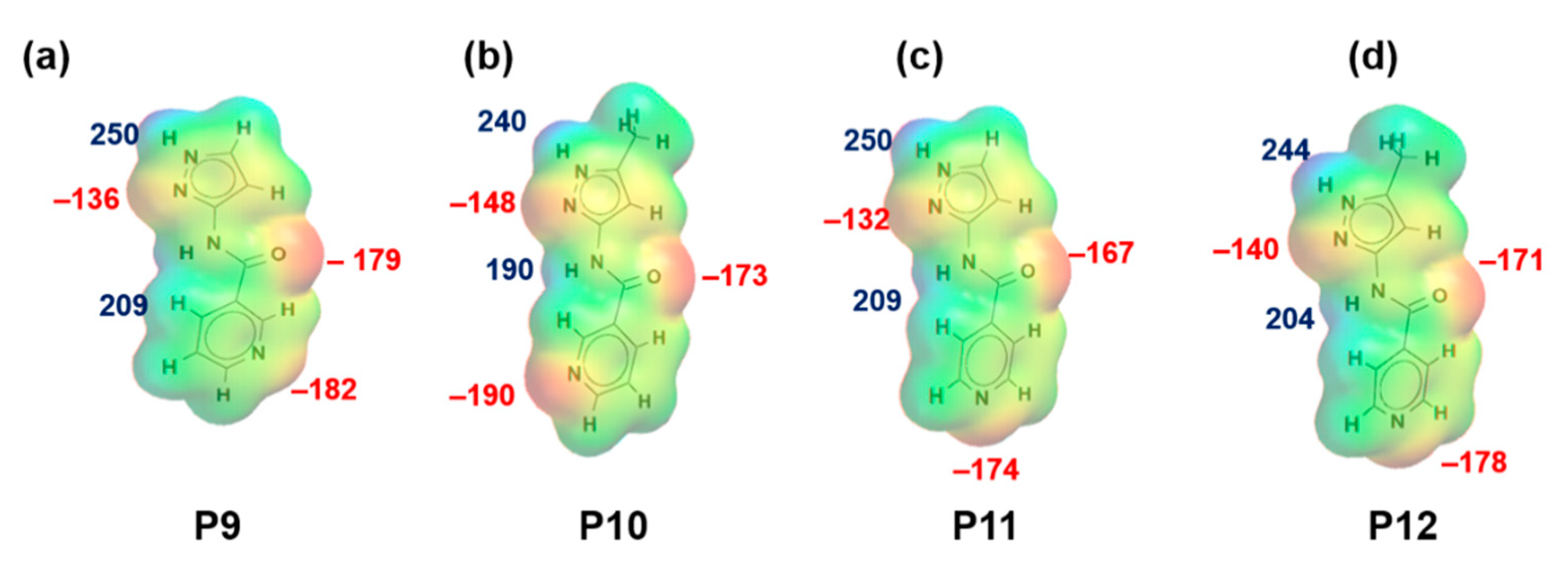
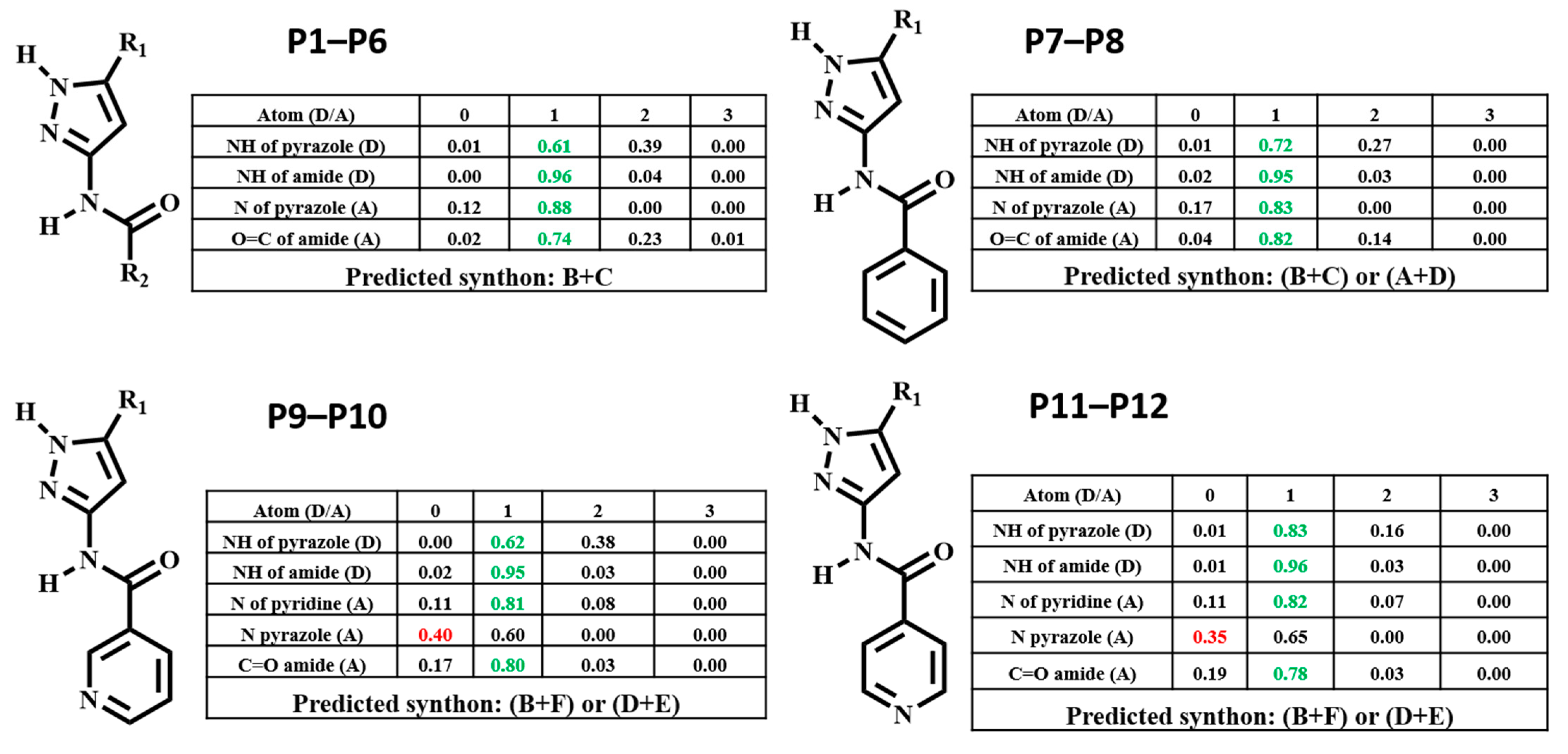
3.1. Molecular Electrostatics Potentials
3.2. Hydrogen-Bond Energies (HBE)
3.3. Hydrogen-Bond Propensities (HBP)
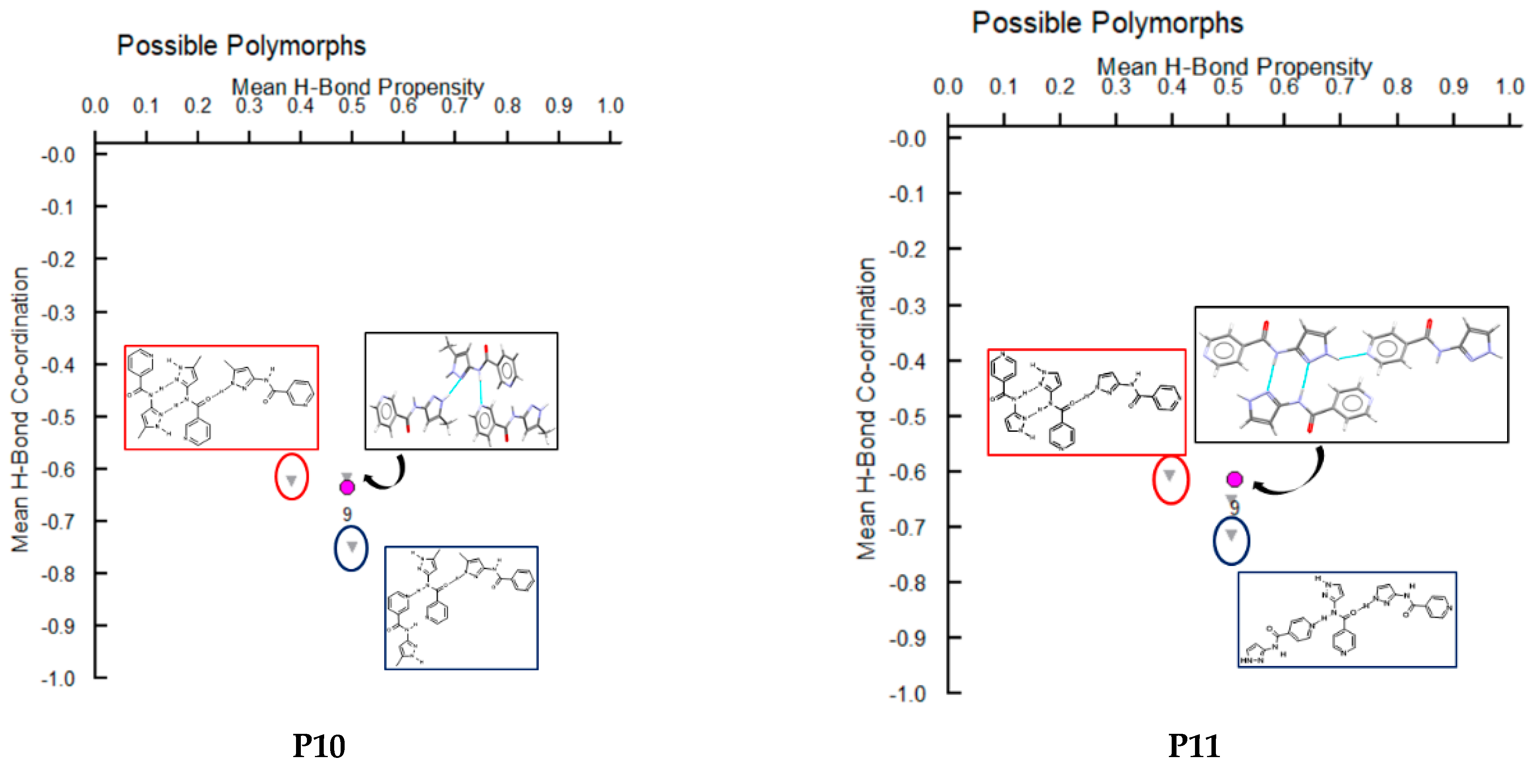
3.4. Hydrogen-Bond Coordination (HBC)
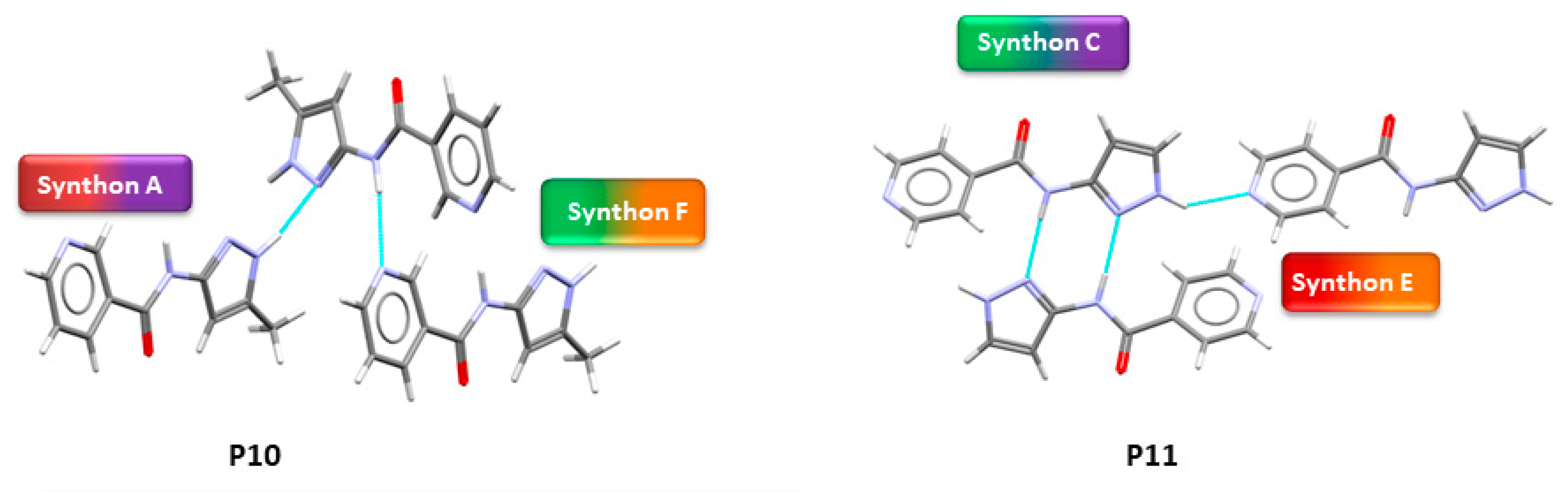
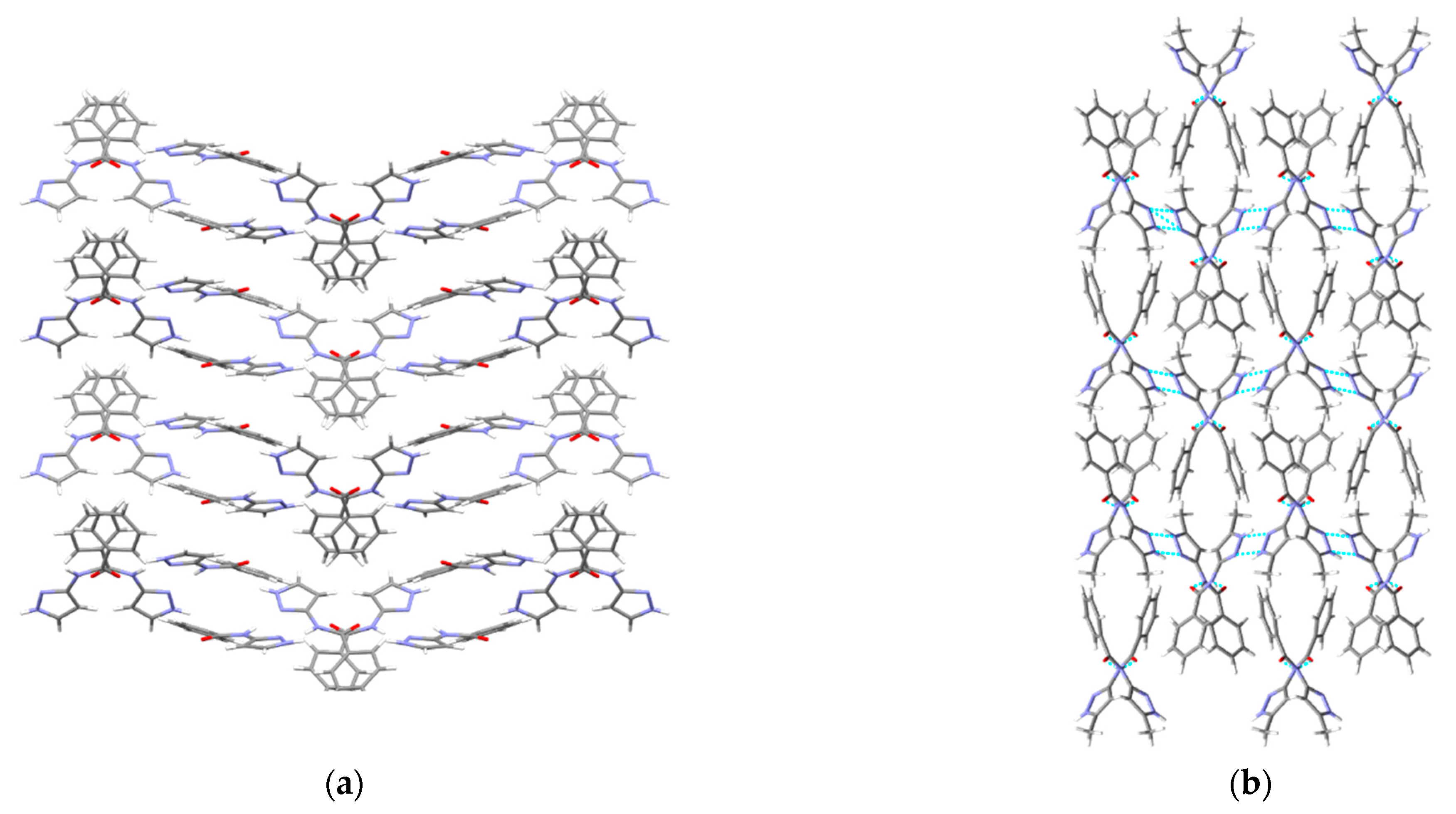
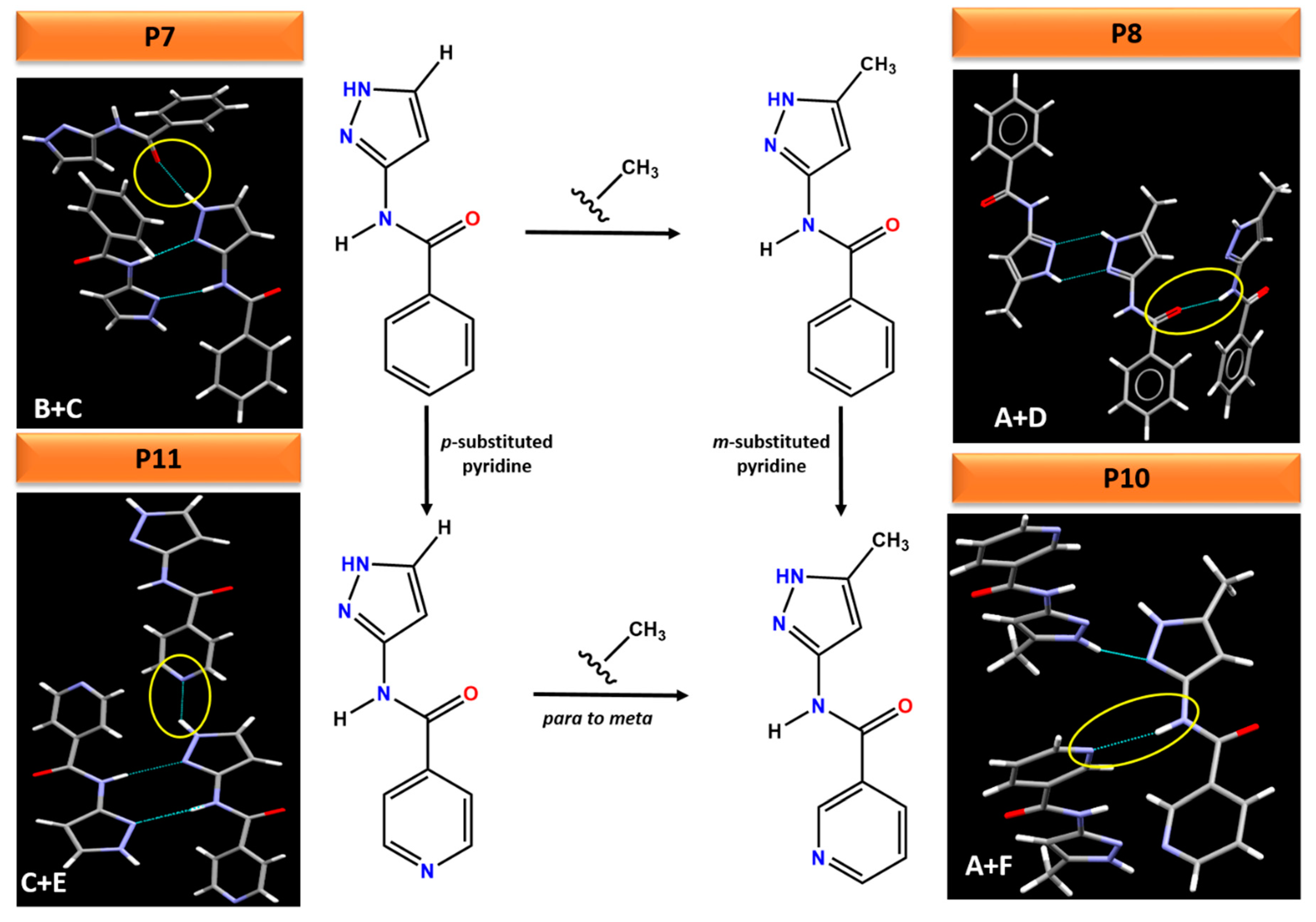
3.5. Experimentally Observed Crystal Structures
4. Discussion
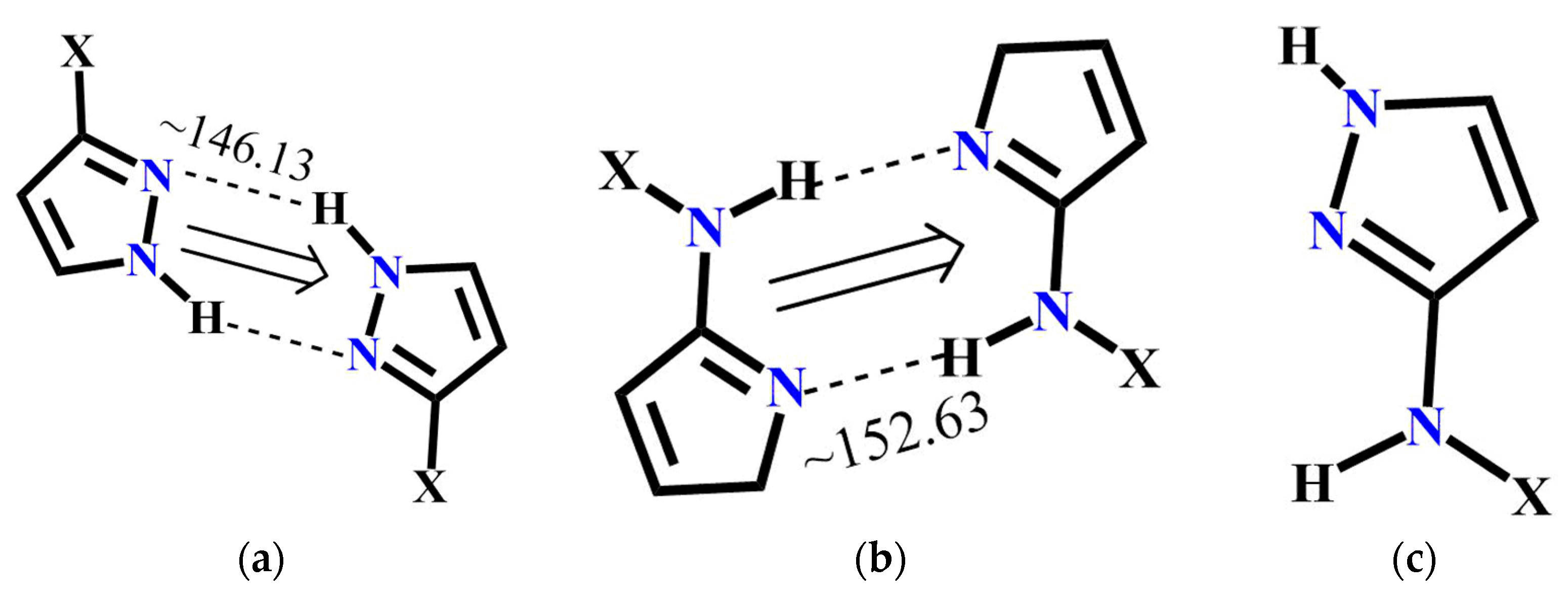
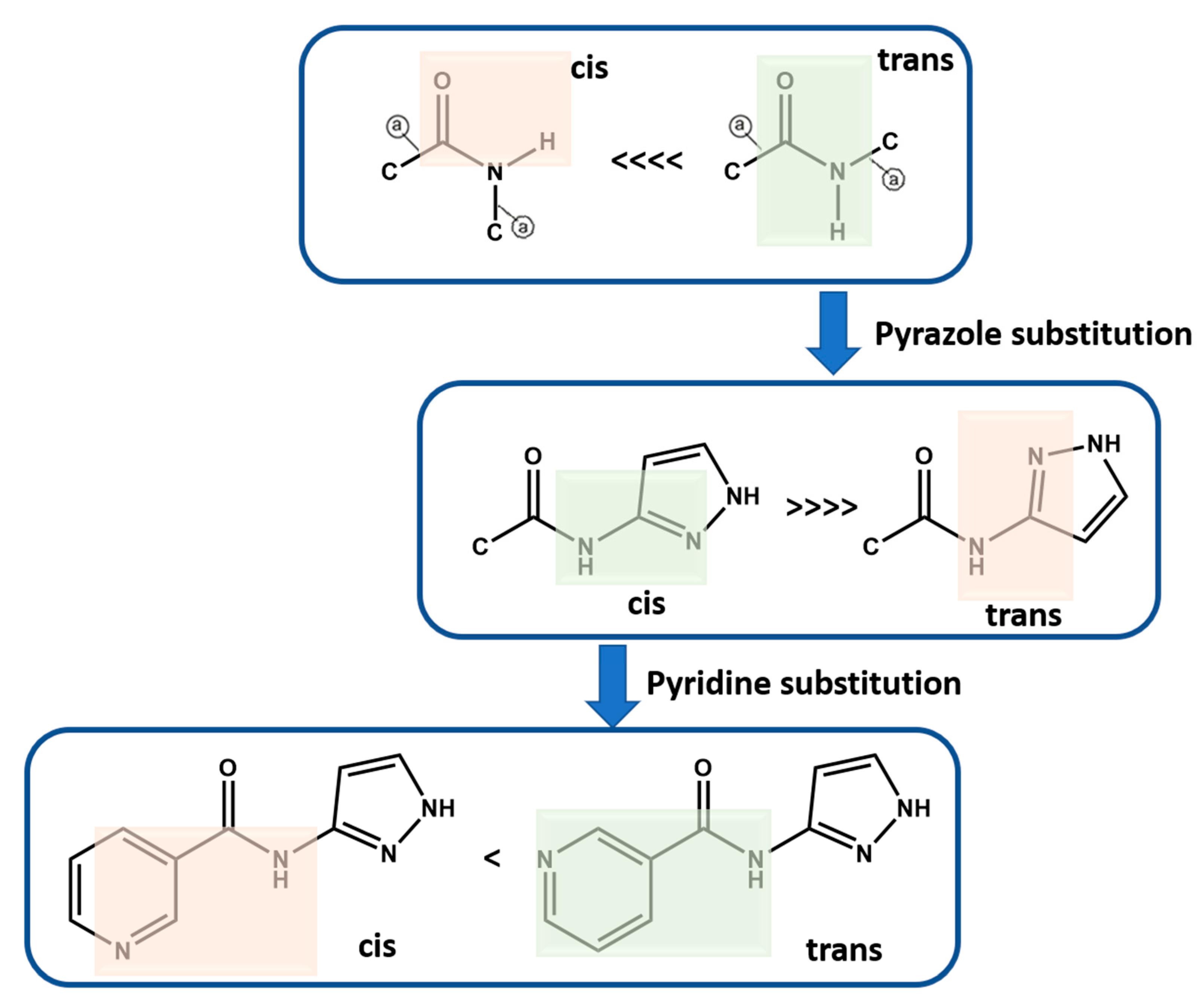
4.1. Molecular Conformational Analysis
4.2. Prediction Analysis
4.3. CSD Search-Molecular Geometric Complementarity
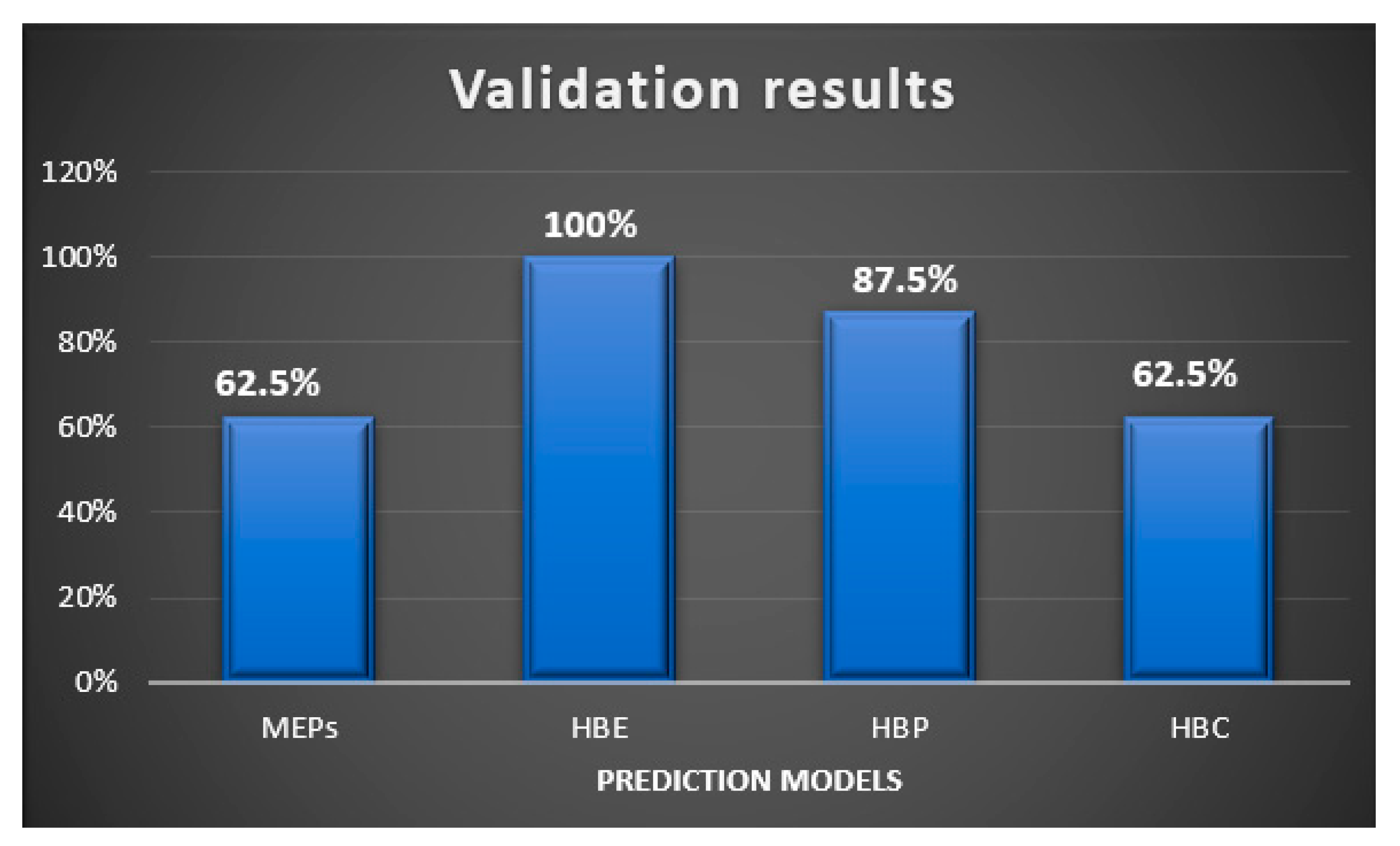
4.4. Validation Analysis
4.5. Polymorph Assessment of Experimental Structures
5. Conclusions
Supplementary Materials
 = bond is acyclic, and Hn = n bonded hydrogen atoms; Table S5. Hydrogen-bond propensities for each individual synthon possible in molecules P1-P12; Table S6. Hydrogen-bond propensities for combination synthons possible in molecules P1-P12. Combination synthon propensities are calculated by multiplying the individual synthon propensities; Table S7. Experimental details of crystals obtained in this study.
= bond is acyclic, and Hn = n bonded hydrogen atoms; Table S5. Hydrogen-bond propensities for each individual synthon possible in molecules P1-P12; Table S6. Hydrogen-bond propensities for combination synthons possible in molecules P1-P12. Combination synthon propensities are calculated by multiplying the individual synthon propensities; Table S7. Experimental details of crystals obtained in this study.Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Price, S.L. Predicting crystal structures of organic compounds. Chem. Soc. Rev. 2014, 43, 2098–2111. [Google Scholar] [CrossRef]
- Desiraju, G.R. Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis. Angew. Chem. Int. Ed. Engl. 1995, 34, 2311–2327. [Google Scholar] [CrossRef]
- Fedyanin, I.V.; Karnoukhova, V.A.; Lyssenko, K.A. Conformational analysis of a supramolecular synthon: A case study of 8-hydroxyquinoline. CrystEngComm 2018, 20, 652–660. [Google Scholar] [CrossRef]
- Thalladi, V.R.; Goud, B.S.; Hoy, V.J.; Allen, F.H.; Howard, J.A.K.; Desiraju, G.R. Supramolecular synthons in crystal engineering. Structure simplification, synthon robustness and supramolecular retrosynthesis. Chem. Commun. 1996, 3, 401–402. [Google Scholar] [CrossRef]
- Nangia, A.; Desiraju, G.R. Supramolecular Synthons and Pattern Recognition. In Design of Organic Solids; Weber, E., Aoyama, Y., Caira, M.R., Desiraju, G.R., Glusker, J.P., Hamilton, A.D., Meléndez, R.E., Nangia, A., Eds.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 57–95. [Google Scholar] [CrossRef]
- Hofmann, D.W.M.; Kuleshova, L.N.; Antipin, M.Y. Supramolecular Synthons and Crystal Structure Prediction of Organic Compounds. Cryst. Growth Des. 2004, 4, 1395–1402. [Google Scholar] [CrossRef]
- Mukherjee, A.; Dixit, K.; Sarma, S.P.; Desiraju, G.R. Aniline-phenol recognition: From solution through supramolecular synthons to cocrystals. IUCrJ 2014, 1, 228–239. [Google Scholar] [CrossRef]
- Reddy, D.S.; Ovchinnikov, Y.E.; Shishkin, O.V.; Struchkov, Y.T.; Desiraju, G.R. Supramolecular Synthons in Crystal Engineering. 3. Solid State Architecture and Synthon Robustness in Some 2,3-Dicyano-5,6-dichloro-1,4-dialkoxybenzenes1. J. Am. Chem. Soc. 1996, 118, 4085–4089. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: A Holistic View. Angew. Chem. Int. Ed. 2007, 46, 8342–8356. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R. Designer crystals: Intermolecular interactions, network structures and supramolecular synthons. Chem. Commun. 1997, 16, 1475–1482. [Google Scholar] [CrossRef]
- Merz, K.; Vasylyeva, V. Development and boundaries in the field of supramolecular synthons. CrystEngComm 2010, 12, 3989–4002. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal engineering: Solid state supramolecular synthesis. Curr. Opin. Solid State Mater. Sci. 1997, 2, 451–454. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular Chemistry and Chemical Synthesis. In Chemical Synthesis: Gnosis to Prognosis; Chatgilialoglu, C., Snieckus, V., Eds.; Springer: Dordrecht, The Netherlands, 1996; pp. 511–524. [Google Scholar] [CrossRef]
- Desiraju, G.R. Crystal Engineering: From Molecule to Crystal. J. Am. Chem. Soc. 2013, 135, 9952–9967. [Google Scholar] [CrossRef] [PubMed]
- Lehn, J.-M. Supramolecular Chemistry—Scope and Perspectives Molecules, Supermolecules, and Molecular Devices (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1988, 27, 89–112. [Google Scholar] [CrossRef]
- Dunitz, J.D. Introduction. In X-ray Analysis and the Structure of Organic Molecules; © Verlag Helvetica Chimica Acta: Zürich, Switzerland, 1995; pp. 17–21. [Google Scholar] [CrossRef]
- Dunitz, J.D. Diffraction of X-Rays by Crystals. In X-ray Analysis and the Structure of Organic Molecules; © Verlag Helvetica Chimica Acta: Zürich, Switzerland, 1995; pp. 23–72. [Google Scholar] [CrossRef]
- Dunitz, J.D. Internal Symmetry of Crystals. In X-ray Analysis and the Structure of Organic Molecules; © Verlag Helvetica Chimica Acta: Zürich, Switzerland, 1995; pp. 73–111. [Google Scholar] [CrossRef]
- Dey, A.; Pati, N.N.; Desiraju, G.R. Crystal structure prediction with the supramolecular synthon approach: Experimental structures of 2-amino-4-ethylphenol and 3-amino-2-naphthol and comparison with prediction. CrystEngComm 2006, 8, 751–755. [Google Scholar] [CrossRef]
- Sarma, J.A.R.P.; Desiraju, G.R. The Supramolecular Synthon Approach to Crystal Structure Prediction. Cryst. Growth Des. 2002, 2, 93–100. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Wijethunga, T.K.; Desper, J. Molecular electrostatic potential dependent selectivity of hydrogen bonding. New J. Chem. 2015, 39, 822–828. [Google Scholar] [CrossRef]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Sandhu, B.; McLean, A.; Sinha, A.S.; Desper, J.; Sarjeant, A.A.; Vyas, S.; Reutzel-Edens, S.M.; Aakeröy, C.B. Evaluating Competing Intermolecular Interactions through Molecular Electrostatic Potentials and Hydrogen-Bond Propensities. Cryst. Growth Des. 2018, 18, 466–478. [Google Scholar] [CrossRef]
- Singh, M.K. Predicting lattice energy and structure of molecular crystals by first-principles method: Role of dispersive interactions. J. Cryst. Growth 2014, 396, 14–23. [Google Scholar] [CrossRef]
- Atahan-Evrenk, S.; Aspuru-Guzik, A. Prediction and Calculation of Crystal Structures: Methods and Applications; Springer: Berlin/Heidelberg, Germany, 2014. [Google Scholar]
- Gharagheizi, F.; Sattari, M.; Tirandazi, B. Prediction of Crystal Lattice Energy Using Enthalpy of Sublimation: A Group Contribution-Based Model. Ind. Eng. Chem. Res. 2011, 50, 2482–2486. [Google Scholar] [CrossRef]
- Day, G.M. Current approaches to predicting molecular organic crystal structures. Crystallogr. Rev. 2011, 17, 3–52. [Google Scholar] [CrossRef]
- Musumeci, D.; Hunter, C.A.; Prohens, R.; Scuderi, S.; McCabe, J.F. Virtual cocrystal screening. Chem. Sci. 2011, 2, 883–890. [Google Scholar] [CrossRef]
- Hunter, C.A. Quantifying Intermolecular Interactions: Guidelines for the Molecular Recognition Toolbox. Angew. Chem. Int. Ed. 2004, 43, 5310–5324. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Comparison of Bifurcated Halogen with Hydrogen Bonds. Molecules 2021, 26, 350. [Google Scholar] [CrossRef]
- Hou, L.; Gao, L.; Zhang, W.; Yang, X.-J.; Wu, B. Quaternary Cocrystals Based on Halide-Binding Foldamers through Both Hydrogen and Halogen Bonding. Cryst. Growth Design 2021. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Chopade, P.D.; Desper, J. Avoiding “Synthon Crossover” in Crystal Engineering with Halogen Bonds and Hydrogen Bonds. Cryst. Growth Des. 2011, 11, 5333–5336. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Fasulo, M.; Schultheiss, N.; Desper, J.; Moore, C. Structural Competition between Hydrogen Bonds and Halogen Bonds. J. Am. Chem. Soc. 2007, 129, 13772–13773. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, B.; Ji, W.-J.; Guo, C.-Y.; Hong, M.; Qi, M.-H.; Ren, G.-B. Insight into the Formation of Cocrystals of Flavonoids and 4,4′-Vinylenedipyridine: Heteromolecular Hydrogen Bonds, Molar Ratio, and Structural Analysis. Cryst. Growth Design 2021. [Google Scholar] [CrossRef]
- Aakeroy, C.B.; Spartz, C.L.; Dembowski, S.; Dwyre, S.; Desper, J. A systematic structural study of halogen bonding versus hydrogen bonding within competitive supramolecular systems. IUCrJ 2015, 2, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.D.; Desper, J.; Sinha, A.S.; Aakeröy, C.B. Impact and importance of electrostatic potential calculations for predicting structural patterns of hydrogen and halogen bonding. CrystEngComm 2016, 18, 8631–8636. [Google Scholar] [CrossRef]
- Zheng, Q.; Unruh, D.K.; Hutchins, K.M. Cocrystallization of Trimethoprim and Solubility Enhancement via Salt Formation. Cryst. Growth Design 2021, 21, 1507–1517. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Wijethunga, T.K.; Desper, J.; Đaković, M. Electrostatic Potential Differences and Halogen-Bond Selectivity. Cryst. Growth Des. 2016, 16, 2662–2670. [Google Scholar] [CrossRef]
- Wang, L.; Liu, S.; Chen, J.-m.; Wang, Y.-x.; Sun, C.C. Novel Salt-Cocrystals of Berberine Hydrochloride with Aliphatic Dicarboxylic Acids: Odd–Even Alternation in Physicochemical Properties. Mol. Pharm. 2021, 18, 1758–1767. [Google Scholar] [CrossRef]
- Gunawardana, C.A.; Desper, J.; Sinha, A.S.; Ðaković, M.; Aakeröy, C.B. Competition and selectivity in supramolecular synthesis: Structural landscape around 1-(pyridylmethyl)-2,2′-biimidazoles. Faraday Discuss. 2017, 203, 371–388. [Google Scholar] [CrossRef] [PubMed]
- Aakeröy, C.B.; Beatty, A.M.; Helfrich, B.A. “Total Synthesis” Supramolecular Style: Design and Hydrogen-Bond-Directed Assembly of Ternary Supermolecules. Angew. Chem. Int. Ed. 2001, 40, 3240–3242. [Google Scholar] [CrossRef]
- Bennion, J.C.; Matzger, A.J. Development and Evolution of Energetic Cocrystals. Acc. Chem. Res. 2021, 54, 1699–1710. [Google Scholar] [CrossRef] [PubMed]
- Groom, C.R.; Allen, F.H. The Cambridge Structural Database in Retrospect and Prospect. Angew. Chem. Int. Ed. 2014, 53, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.A.; Feeder, N.; Furlow, M.; Galek, P.T.A.; Groom, C.R.; Pidcock, E. Knowledge-based approaches to co-crystal design. CrystEngComm 2014, 16, 5839–5848. [Google Scholar] [CrossRef]
- Galek, P.T.A.; Chisholm, J.A.; Pidcock, E.; Wood, P.A. Hydrogen-bond coordination in organic crystal structures: Statistics, predictions and applications. Acta Crystallogr. Sect. B 2014, 70, 91–105. [Google Scholar] [CrossRef] [PubMed]
- Nauha, E.; Bernstein, J. “Predicting” Crystal Forms of Pharmaceuticals Using Hydrogen Bond Propensities: Two Test Cases. Cryst. Growth Des. 2014, 14, 4364–4370. [Google Scholar] [CrossRef]
- Sarkar, N.; Aakeröy, C.B. Evaluating hydrogen-bond propensity, hydrogen-bond coordination and hydrogen-bond energy as tools for predicting the outcome of attempted co-crystallisations. Supramol. Chem. 2020, 32, 81–90. [Google Scholar] [CrossRef]
- Sarkar, N.; Sinha, A.S.; Aakeröy, C.B. Systematic investigation of hydrogen-bond propensities for informing co-crystal design and assembly. CrystEngComm 2019, 21, 6048–6055. [Google Scholar] [CrossRef]
- Naim, M.; Alam, O.; Nawaz, F.; Alam, M.; Alam, P. Current status of pyrazole and its biological activities. J. Pharm. Bioallied Sci. 2016, 8, 2–17. [Google Scholar] [CrossRef]
- Ansari, A.; Ali, A.; Asif, M.; Shamsuzzaman. Review: Biologically active pyrazole derivatives. New J. Chem. 2017, 41, 16–41. [Google Scholar] [CrossRef]
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M.h. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tan, D.-J.; Wang, T.; Jing, S.-S.; Kang, Y.; Zhang, Z.-T. Synthesis, crystal structure, characterization and antifungal activity of 3,4-diaryl-1H-Pyrazoles derivatives. J. Mol. Struct. 2017, 1149, 235. [Google Scholar] [CrossRef]
- El Shehry, M.F.; Ghorab, M.M.; Abbas, S.Y.; Fayed, E.A.; Shedid, S.A.; Ammar, Y.A. Quinoline derivatives bearing pyrazole moiety: Synthesis and biological evaluation as possible antibacterial and antifungal agents. Eur. J. Med. Chem. 2018, 143, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Du, S.; Tian, Z.; Yang, D.; Li, X.; Li, H.; Jia, C.; Che, C.; Wang, M.; Qin, Z. Synthesis, Antifungal Activity and Structure-Activity Relationships of Novel 3-(Difluoromethyl)-1-methyl-1H-pyrazole-4-carboxylic Acid Amides. Molecules 2015, 20, 8395–8408. [Google Scholar] [CrossRef]
- Meta, E.; Brullo, C.; Tonelli, M.; Franzblau, S.G.; Wang, Y.; Ma, R.; Baojie, W.; Orena, B.S.; Pasca, M.R.; Bruno, O. Pyrazole and imidazo[1,2-b]pyrazole Derivatives as New Potential Antituberculosis Agents. Med. Chem. 2019, 15, 17–27. [Google Scholar] [CrossRef]
- Sun, J.; Zhou, Y. Synthesis and antifungal activity of the derivatives of novel pyrazole carboxamide and isoxazolol pyrazole carboxylate. Molecules 2015, 20, 4383–4394. [Google Scholar] [CrossRef]
- Liu, J.J.; Zhao, M.Y.; Zhang, X.; Zhao, X.; Zhu, H.L. Pyrazole derivatives as antitumor, anti-inflammatory and antibacterial agents. Mini Rev. Med. Chem. 2013, 13, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, J.; Hu, D.; He, M.; Jin, L.; Song, B. Synthesis and antifungal activity of novel pyrazolecarboxamide derivatives containing a hydrazone moiety. Chem. Cent. J. 2012, 6, 51. [Google Scholar] [CrossRef]
- Taylor, R.; Macrae, C.F. Rules governing the crystal packing of mono- and dialcohols. Acta Crystallogr. Sect. B 2001, 57, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Bruno, I.J.; Cole, J.C.; Edgington, P.R.; Kessler, M.; Macrae, C.F.; McCabe, P.; Pearson, J.; Taylor, R. New software for searching the Cambridge Structural Database and visualizing crystal structures. Acta Crystallogr. Sect. B 2002, 58, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and analysis of crystal structures. J. Appl. Crystallogr. 2006, 39, 453–457. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Shao, Y.; Molnar, L.F.; Jung, Y.; Kussmann, J.; Ochsenfeld, C.; Brown, S.T.; Gilbert, A.T.B.; Slipchenko, L.V.; Levchenko, S.V.; O’Neill, D.P.; et al. Advances in methods and algorithms in a modern quantum chemistry program package. Phys. Chem. Chem. Phys. 2006, 8, 3172–3191. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Kraatz, H.B. Metal complexes of 3-acetamido-5-methylpyrazole. Inorg. Chim. Acta 2004, 357, 159–166. [Google Scholar] [CrossRef]
- Daidone, G.; Maggio, B.; Raimondi, M.V.; Bombieri, G.; Marchini, N.; Artali, R. Comparative Structural Studies of 4-Diazopyrazole Derivatives by X-Ray Diffraction and Theoretical Investigation. Heterocycles 2005, 65, 2753. [Google Scholar] [CrossRef]









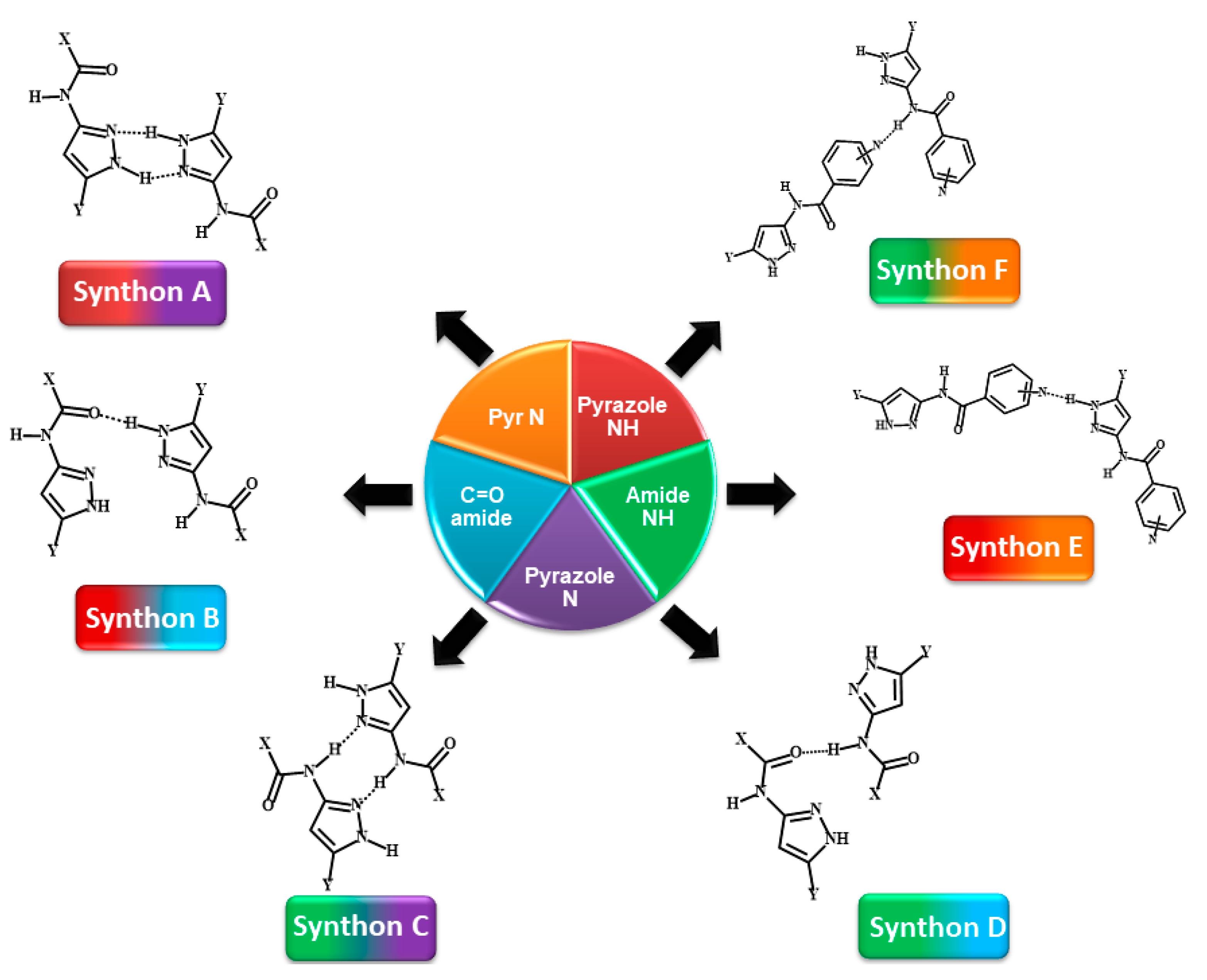
 indicates acyclic moiety.
indicates acyclic moiety.
indicates acyclic moiety.
indicates acyclic moiety.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A +() F) | (A + D) | (C + E) | (B + C) | (D + E) | (B + F) | |
|---|---|---|---|---|---|---|
| AVG (P1–P8) | N/A | −52 ± 2 | N/A | −50 ± 2 | N/A | N/A |
| AVG (P9–P12) | −44 ± 2 | −43 ± 1 | −43 ± 2 | −41 ± 1 | −37 ± 2 | −37 ± 2 |
| Synthon (A + F) | Synthon (A + D) | Synthon (C + E) | Synthon (B + C) | Synthon (D + E) | Synthon (B + F) | |
|---|---|---|---|---|---|---|
| P1 | N/A | 0.35 | N/A | 0.34 | N/A | N/A |
| P2 | N/A | 0.35 | N/A | 0.30 | N/A | N/A |
| P3 | N/A | 0.39 | N/A | 0.36 | N/A | N/A |
| P4 | N/A | 0.39 | N/A | 0.37 | N/A | N/A |
| P5 | N/A | 0.33 | N/A | 0.31 | N/A | N/A |
| P6 | N/A | 0.35 | N/A | 0.32 | N/A | N/A |
| P7 | N/A | 0.18 | N/A | 0.19 | N/A | N/A |
| P8 | N/A | 0.16 | N/A | 0.17 | N/A | N/A |
| P9 | 0.23 | 0.13 | 0.19 | 0.13 | 0.19 | 0.23 |
| P10 | 0.24 | 0.12 | 0.19 | 0.13 | 0.18 | 0.23 |
| P11 | 0.21 | 0.12 | 0.17 | 0.12 | 0.19 | 0.23 |
| P12 | 0.22 | 0.12 | 0.18 | 0.12 | 0.19 | 0.23 |
| Experimental | MEPs | HBE | HBP | HBC | |
|---|---|---|---|---|---|
| Group 1 | |||||
| P1 | Synthon (B + C) | Yes | Yes | Yes | Yes |
| P2 | Synthon (B + C) | Yes | Yes | Yes | Yes |
| P3 | Synthon (B + C) | Yes | Yes | Yes | Yes |
| P4 | Synthon (B + C) | Yes | Yes | Yes | Yes |
| P7 | Synthon (B + C) | Yes | Yes | Yes | Yes |
| P8 | Synthon (A + D) | No | Yes | Yes | No |
| Group 2 | |||||
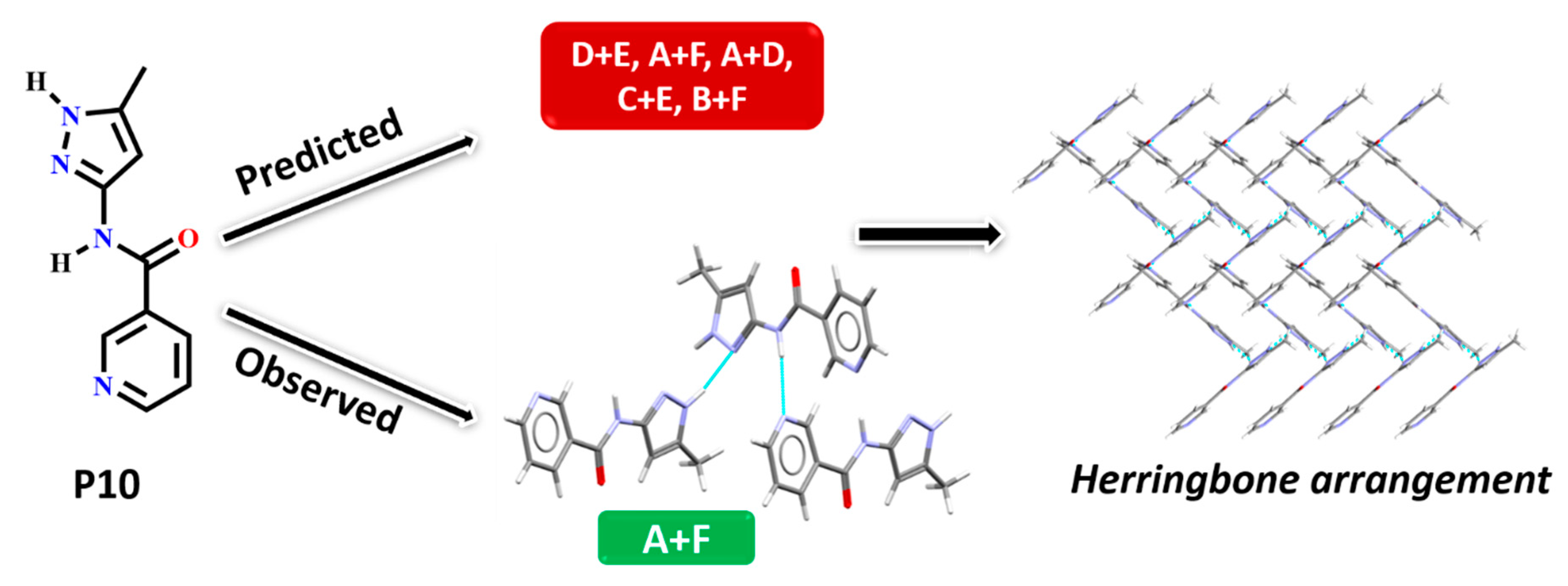
| P10 | Synthon (A + F) | No | Yes | Yes | No |
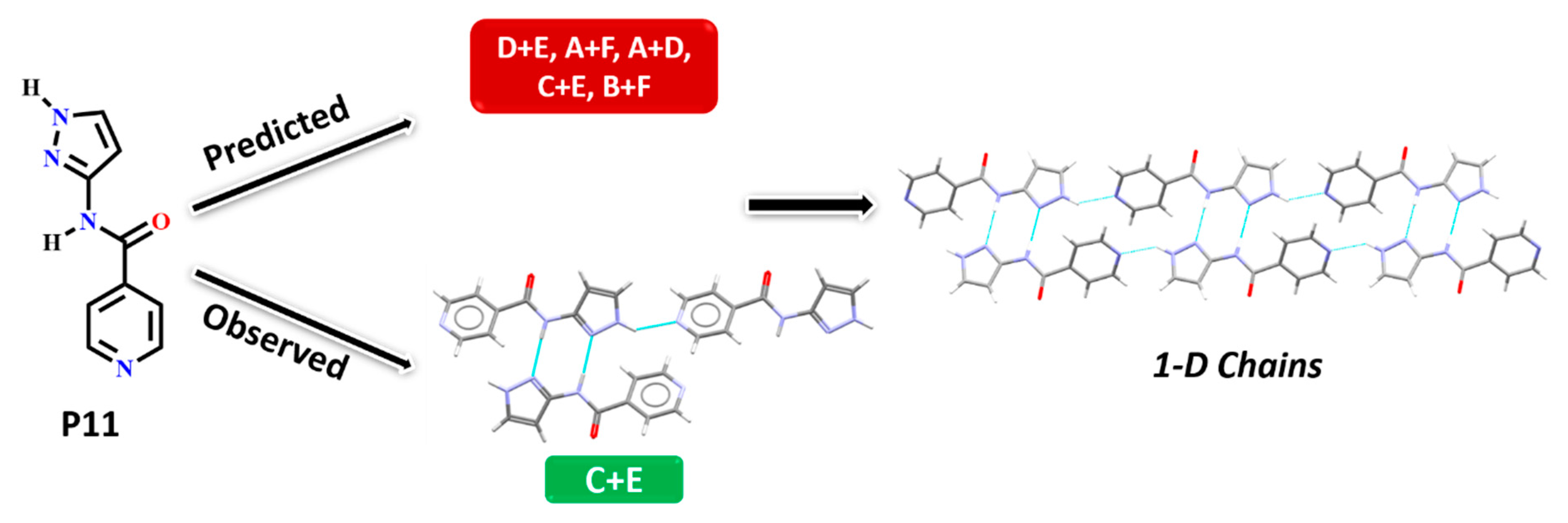
| P11 | Synthon (C + E) | No | Yes | No | No |
| Overall | 62.5% | 100% | 87.5% | 62.5% | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandhu, B.; McLean, A.; Sinha, A.S.; Desper, J.; Aakerӧy, C.B. Assessment of Computational Tools for Predicting Supramolecular Synthons. Chemistry 2021, 3, 612-629. https://doi.org/10.3390/chemistry3020043
Sandhu B, McLean A, Sinha AS, Desper J, Aakerӧy CB. Assessment of Computational Tools for Predicting Supramolecular Synthons. Chemistry. 2021; 3(2):612-629. https://doi.org/10.3390/chemistry3020043
Chicago/Turabian StyleSandhu, Bhupinder, Ann McLean, Abhijeet S. Sinha, John Desper, and Christer B. Aakerӧy. 2021. "Assessment of Computational Tools for Predicting Supramolecular Synthons" Chemistry 3, no. 2: 612-629. https://doi.org/10.3390/chemistry3020043
APA StyleSandhu, B., McLean, A., Sinha, A. S., Desper, J., & Aakerӧy, C. B. (2021). Assessment of Computational Tools for Predicting Supramolecular Synthons. Chemistry, 3(2), 612-629. https://doi.org/10.3390/chemistry3020043