1. Introduction
The emergence of the field of supramolecular chemistry was recognised with the award of the 1987 Nobel Prize in Chemistry to Cram, Lehn and Pedersen “for their development and use of molecules with structure-specific interactions of high selectivity” [
1]. In their work, each of these researchers developed molecular systems that exhibited selectivity in binding to alkali metal ions [
2,
3,
4,
5,
6,
7,
8]. In the following decades, the field of metallosupramolecular chemistry proved to be a rich and fertile area with researchers such as Fujita producing a stunning array of cage-type compounds in which metal-centres are linked by bridging ligands [
9,
10]. In addition to metal-ligand assemblies, expansion of the field of supramolecular chemistry included the area of crystal engineering. Gautum Desiraju’s pioneering work encouraged researchers to consider the crystal as a supramolecular entity in which a variety of complementary interactions control the arrangement of molecules [
11,
12]. In this context, Ward exploited complementary hydrogen bonding interactions between anionic sulphonates and guanidinium cations to generate an impressive series of network materials that represent an outstanding example of true crystal engineering [
13,
14]. All of the above examples rely upon different types of complementary interactions between components to yield supramolecular assemblies. In this current work, the serendipitous formation of a novel series of crystalline, supramolecular compounds is described, in which the tris (catechol) molecule, cyclotricatechylene (H
6ctc,
I,
Scheme 1), participates in all of the types of associations indicated in the examples listed above i.e., alkali metal binding, cage formation and hydrogen bonding within a highly symmetric extended network. The resulting compounds represent remarkable examples of high symmetry multi-component supramolecular assemblies in which a synergy exists between the different types of complementary interactions that govern their formation.
Cyclotricatechylene possesses 3-fold symmetry and commonly adopts a bowl-shaped conformation with hydroxylic groups located on the rim of the bowl. The compound is closely related to its hexamethyl counterpart, cyclotriveratrylene (
II,
Scheme 1), which has received considerable attention with respect to its ability to host guest molecules within the bowl-shaped cavity [
15,
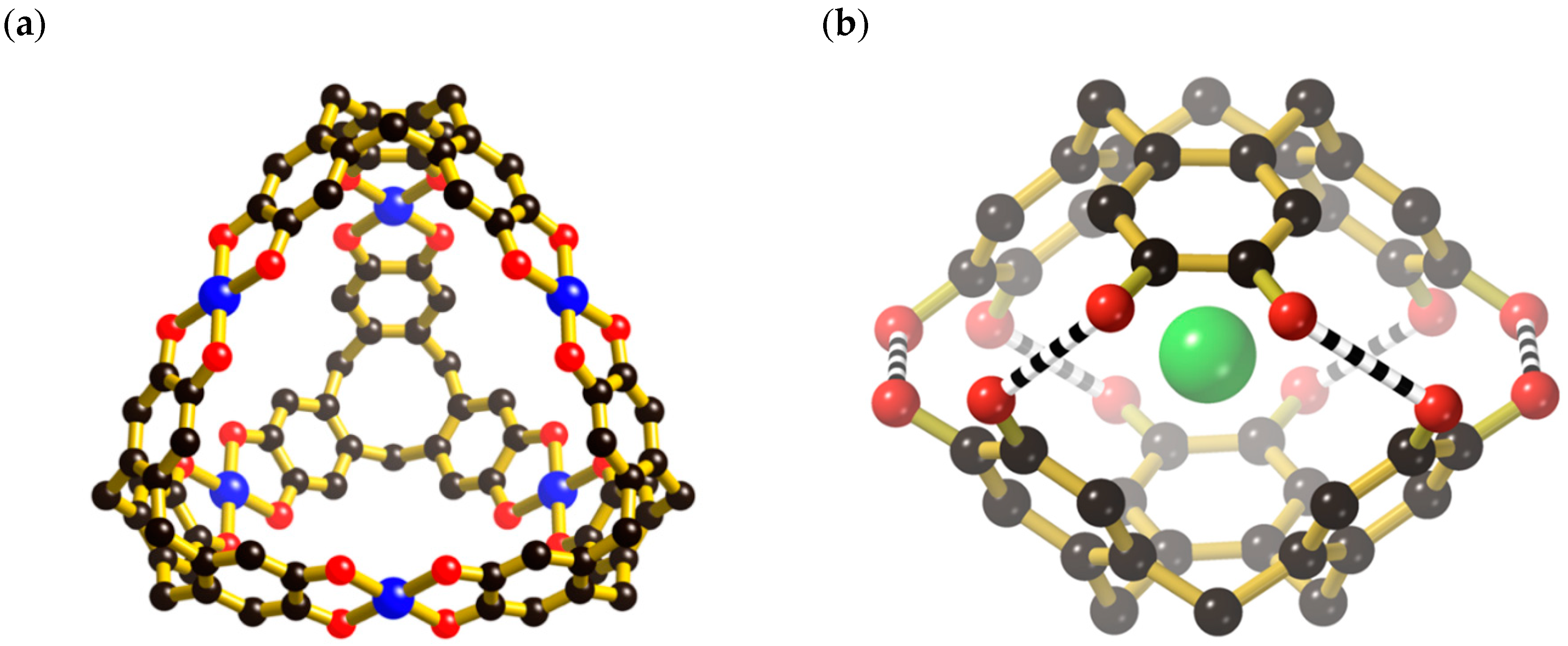
16]. In the last 12 years, interest in the supramolecular chemistry of cyclotricatechylene has grown and a variety of metal-based derivatives have been synthesised and structurally characterised. The structures of these assemblies depend upon the manner in which the metal ion interacts with the tris (catechol) ligand. For example, transition metal ions (M) such as V
IV (in the form of vanadyl) [
17] and Cu
II [
18] combine with ctc
6− to produce M
6ctc
4 anionic tetrahedral cages in which ctc hexaanions are located at the vertices of a tetrahedron. The catecholate arms extend along the tetrahedron edges and are linked by metal centres, each of which is linked to a pair of catecholate groups as indicated in
Figure 1a. Recent work has demonstrated that 5-coordinate Si centres can fulfil the role of the metal centre to generate robust covalent cages [
19,
20].
In addition to the cages of the type described above, H
6ctc, in various stages of deprotonation, is able to form fascinating clam-like structures when it is combined with large alkali metal ions, in particular, Rb
+ and Cs
+ [
21]. In these structures, pairs of partially deprotonated H
nctc
(6−n)− units assemble through complementary hydrogen bonding interactions between hydroxyl groups which results in a rim-to-rim arrangement as shown in
Figure 1b. A Rb
+ or Cs
+ ion occupies the internal cavity of the assembly and interacts only with the aromatic surfaces of the H
nctc
(6−n)− units. A computational investigation of the stability of these clam-like structures, under different levels of protonation, was recently reported [
22]. The affinity of ctc units for alkali metal ions is also apparent in the metal-like assemblies of the type depicted in
Figure 1a with the large alkali metal cations associating with the internal aromatic surfaces of the cage.
In this current work, we report further investigations into supramolecular assemblies formed from the combination of alkali metal cations with H6ctc, this time in the presence of the tetrahedral oxyanions, sulphate and phosphate. The original intention of the study was to investigate the interaction of large alkali metal cations (Rb+ or Cs+) with the internal aromatic surfaces of cyclotricatechylene, in various states of deprotonation, and determine how simple anions such as phosphate and sulphate impact upon the nature of the assembly. The high symmetry products obtained from this synthetic investigation were completely unexpected and display a remarkable level of complementarity involving the six components from which each of the compounds is assembled.
3. Results and Discussion
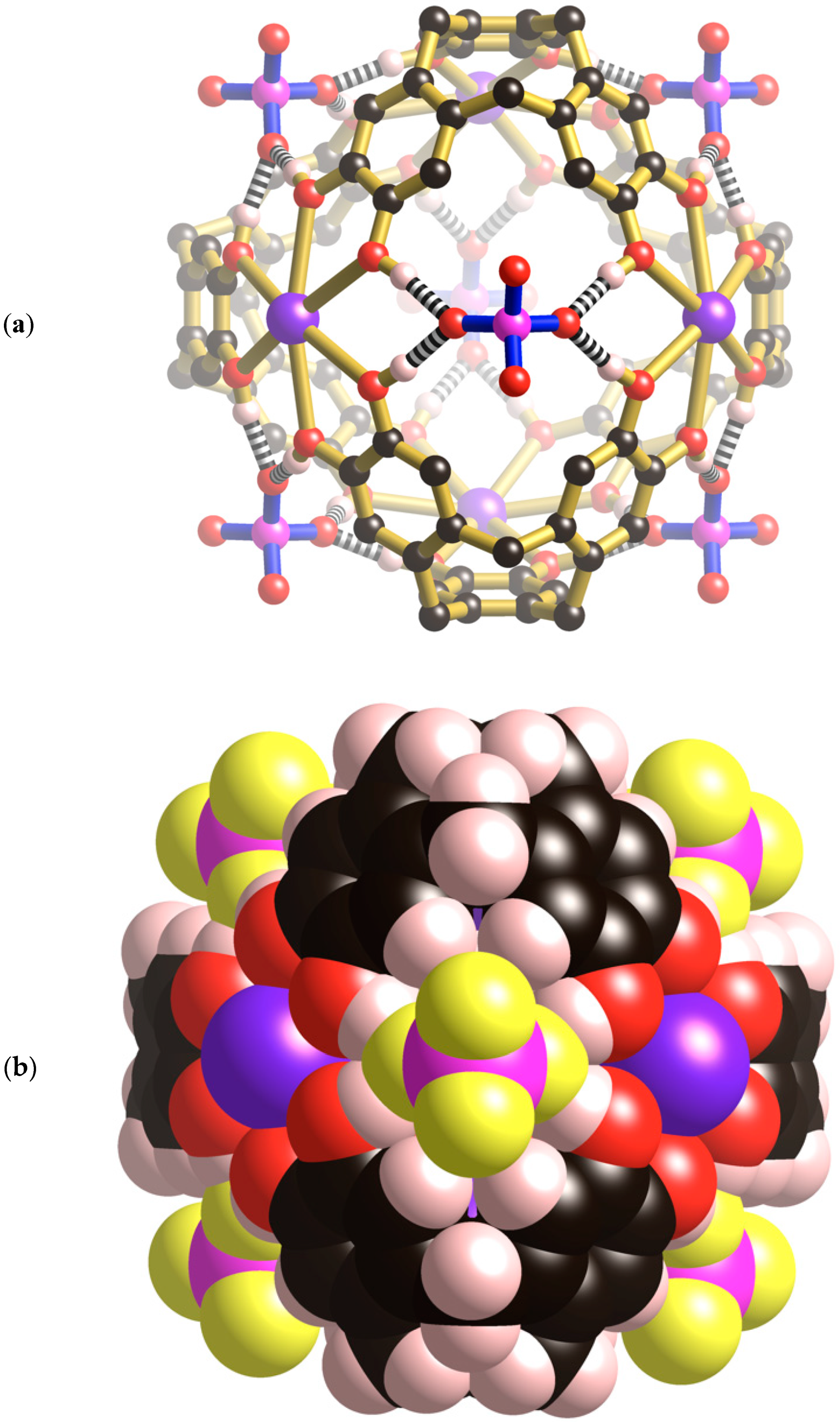
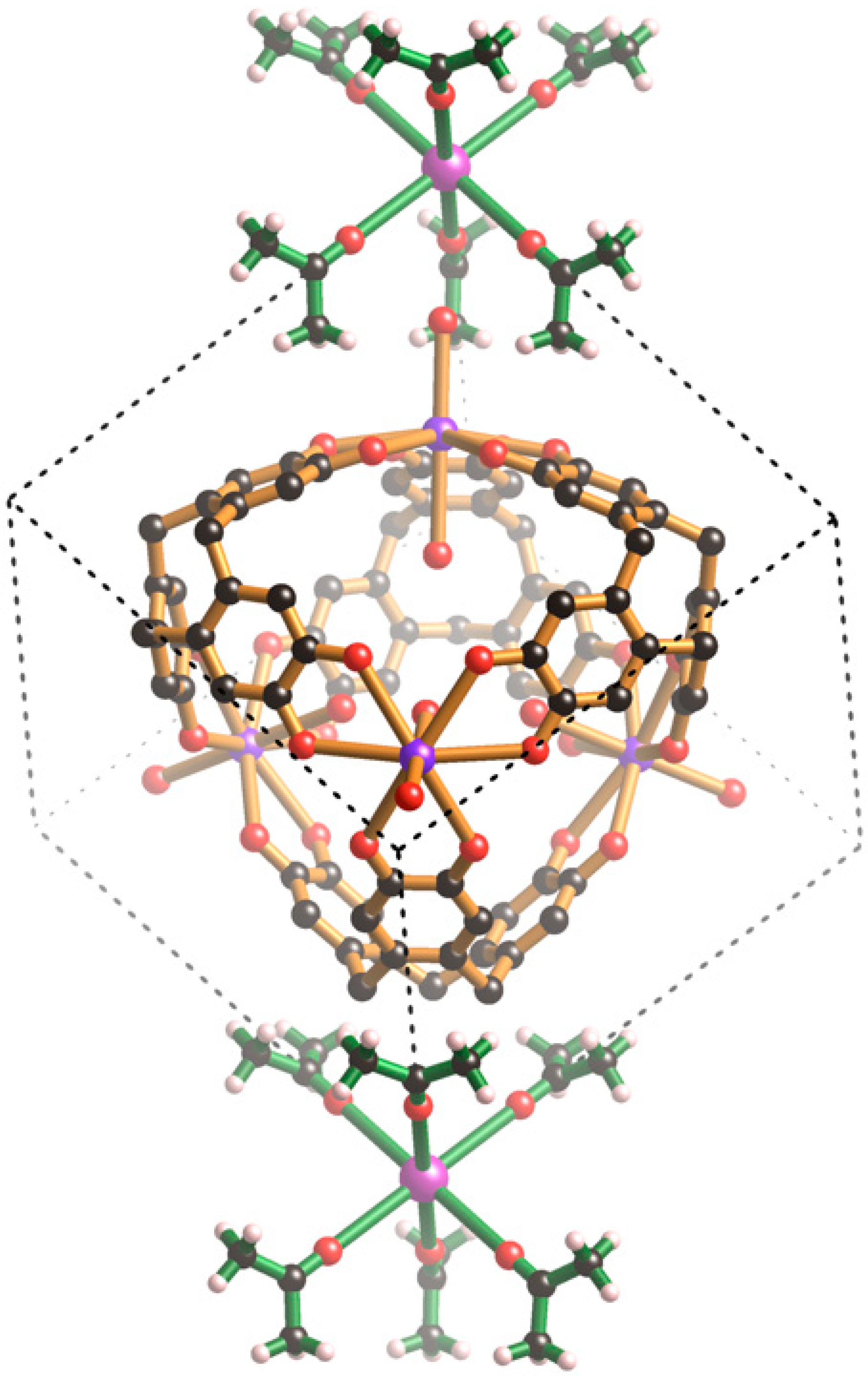
The combination of H6ctc with CsOH and K2HPO4 in aqueous acetone results in the overnight formation of large colourless crystals of composition, [Cs((CH3)2CO)6][K4(H6ctc)4(H2O)8]- [Cs4(H2O)6](PO4)3 (III). The crystals possess cubic symmetry and adopt the space group P-43m. A key feature of this structure is the complementary interactions that exist between the various cationic, anionic and neutral species.
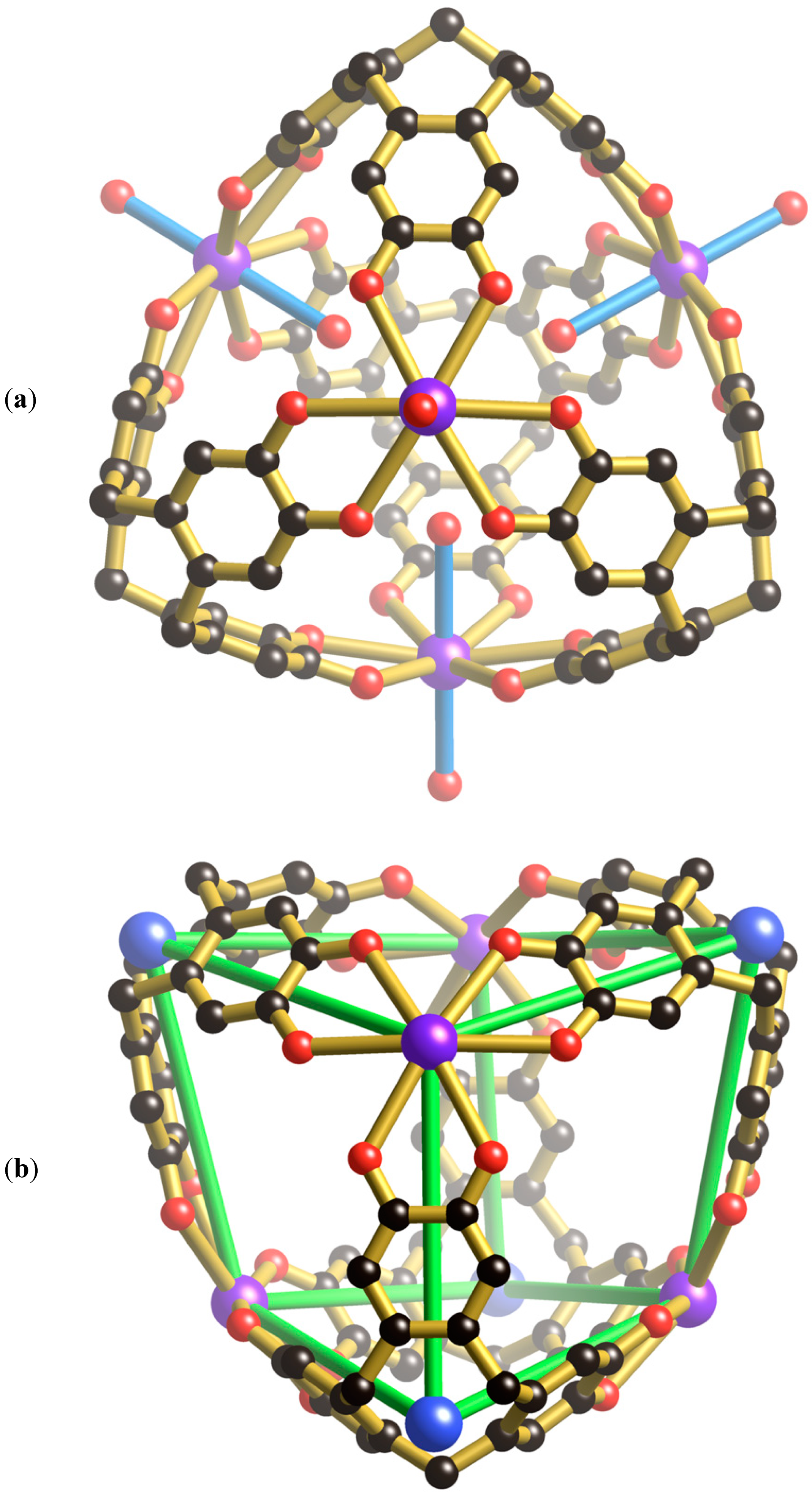
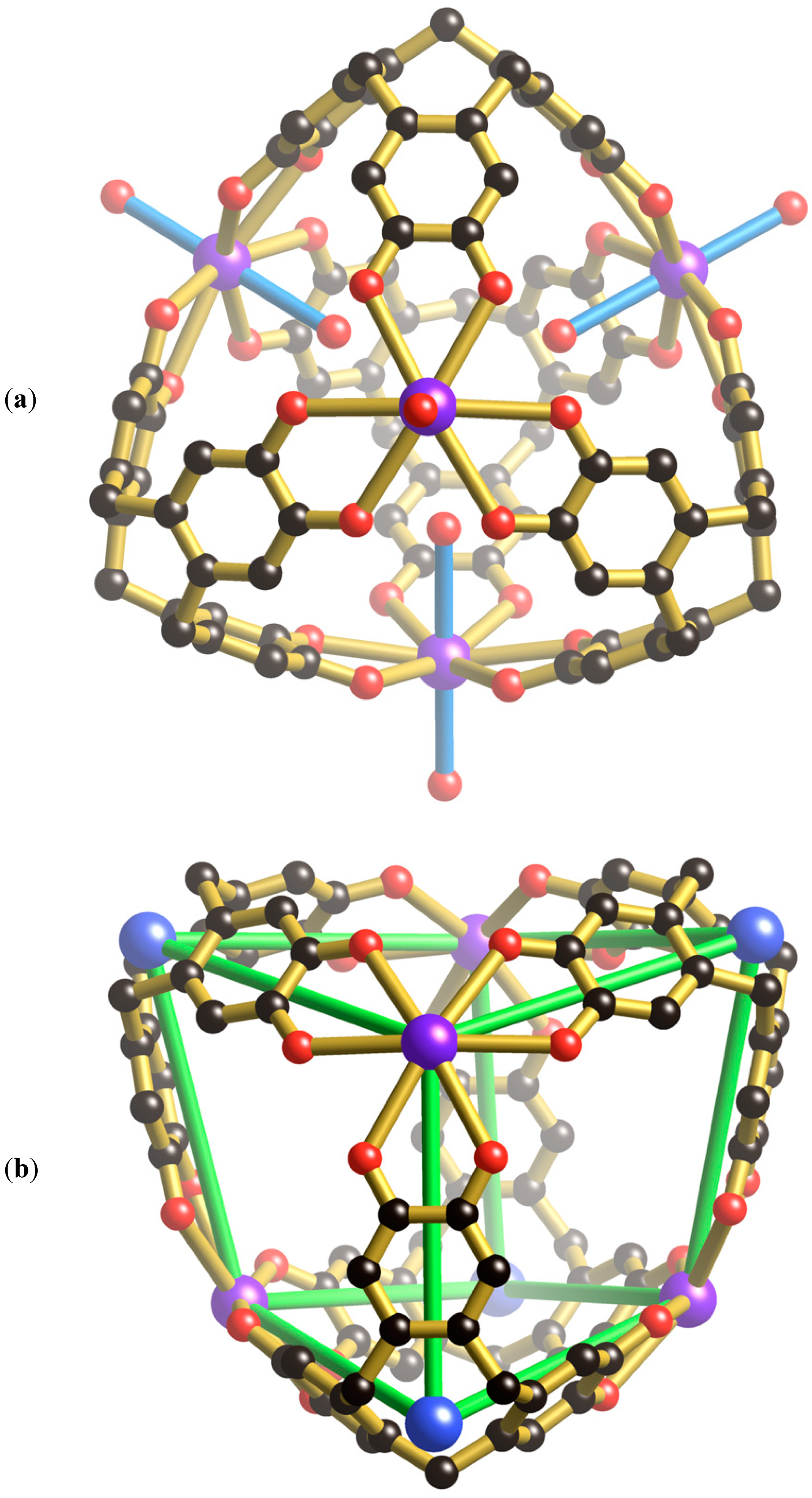
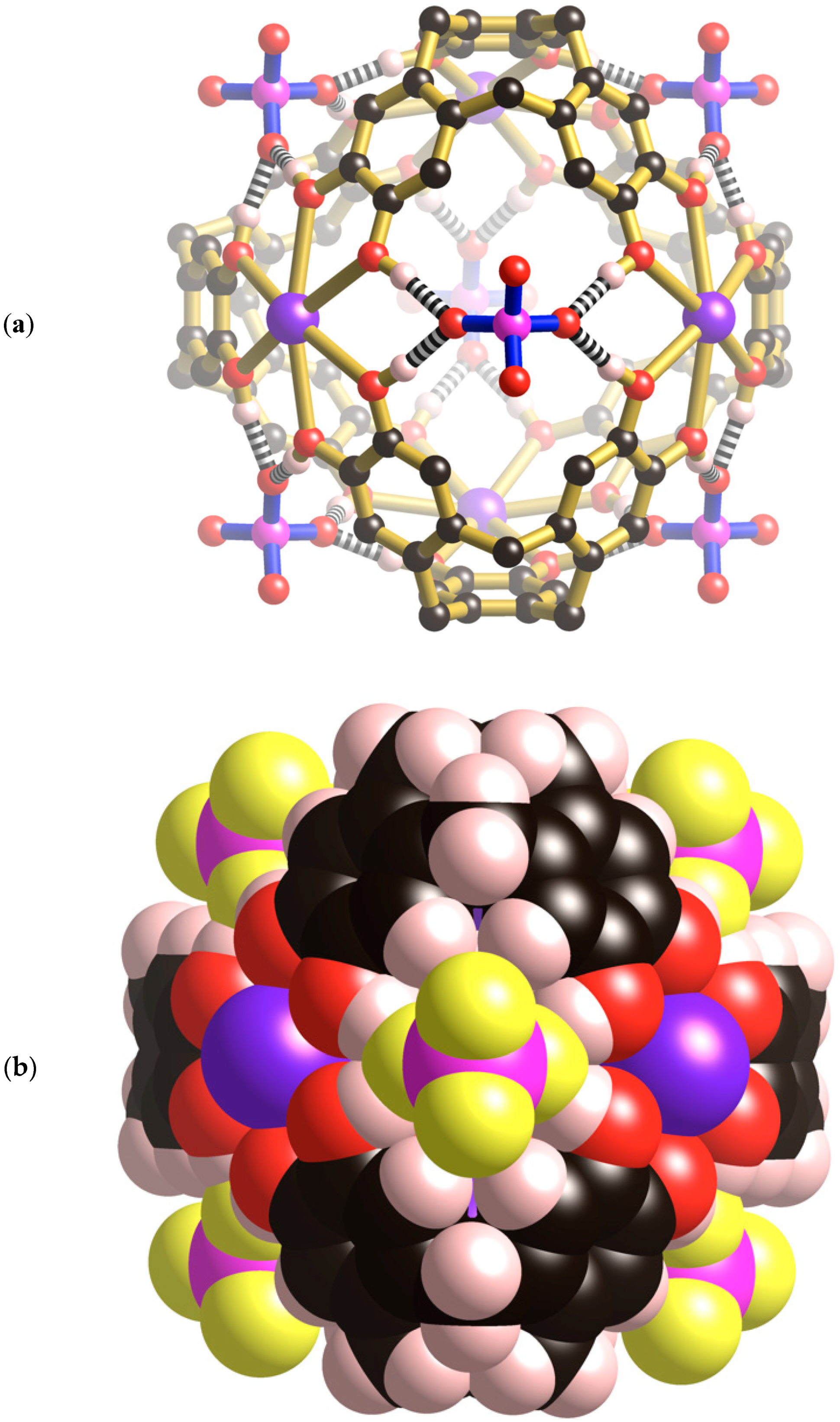
Within each unit cell there is a single [K
4(H
6ctc)
4(H
2O)
8]
4+ cage (
Figure 2a) which is topologically different from the M
6(ctc)
4 anionic cages described in the introduction. All K
+ ions are symmetry related and exist in an 8-coordinate, slightly distorted hexagonal bipyramidal environment formed from six symmetry related co-planar catechol oxygen atoms and two
trans water molecules. Each K
+ ion is located just above the plane of the six bound catechol oxygen atoms. The cage possesses the topology of a cube with four K
+ ions and four H
6ctc molecules units serving as the eight 3-connecting nodes and the catechol groups running along the cube edges. The topological relationship between the cube and the cage is apparent from inspection of
Figure 2b. The formation of the cube rather than the tetrahedron, of the type depicted in
Figure 1a, reflects the ability of K
+ ions to adopt an 8-coordinate hexagonal bipyramidal geometry.
In addition to the four water molecules that extend towards the centre of the cubic cage from the K
+ ions, there are four Cs
+ ions that associate with the internal aromatic surfaces of the [K
4(H
6ctc)
4(H
2O)
8]
4+ cage. Each of the Cs
+ ions is located on a 3-fold axis that passes through one of the K
+ ions, its two
trans water molecules and along the central axis of a H
6ctc molecule with which it is in contact (
Figure 2c). The location of the Cs
+ cation within the H
6ctc bowl is similar to the arrangement of Cs
+ ions found in the tetrahedral cages, [(VO)
6(ctc)
4]
12− and [(PhSi)
6(ctc)
4]
6−. As with the tetrahedral cages, the closest contact is made with the aromatic C atoms bound to the methylene groups of the H
6ctc. As well as interacting with the internal surface of the H
6ctc, each Cs
+ ion is bound to a trio of symmetry-related water molecules each of which bridges a pair of Cs
+ ions. The result is a [Cs
4(H
2O)
6]
4+ cationic assembly located inside each cage. From a structural perspective, this cationic unit resembles an adamantane-type skeleton which is apparent in molecules such as P
4O
6.
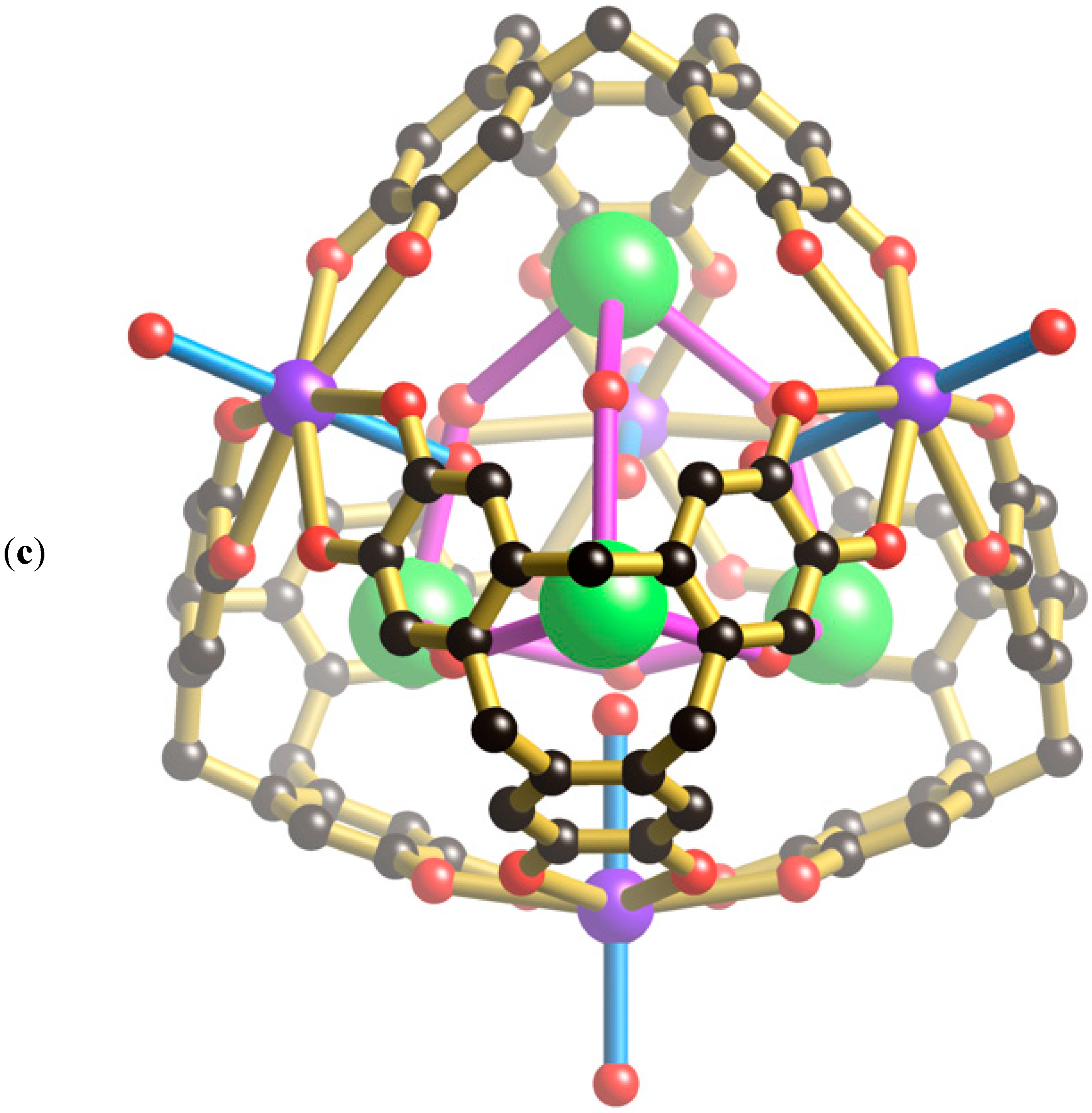
The [K
4(H
6ctc)
4(H
2O)
8]
4+ cage has six windows corresponding to the faces of a cube. As indicated in
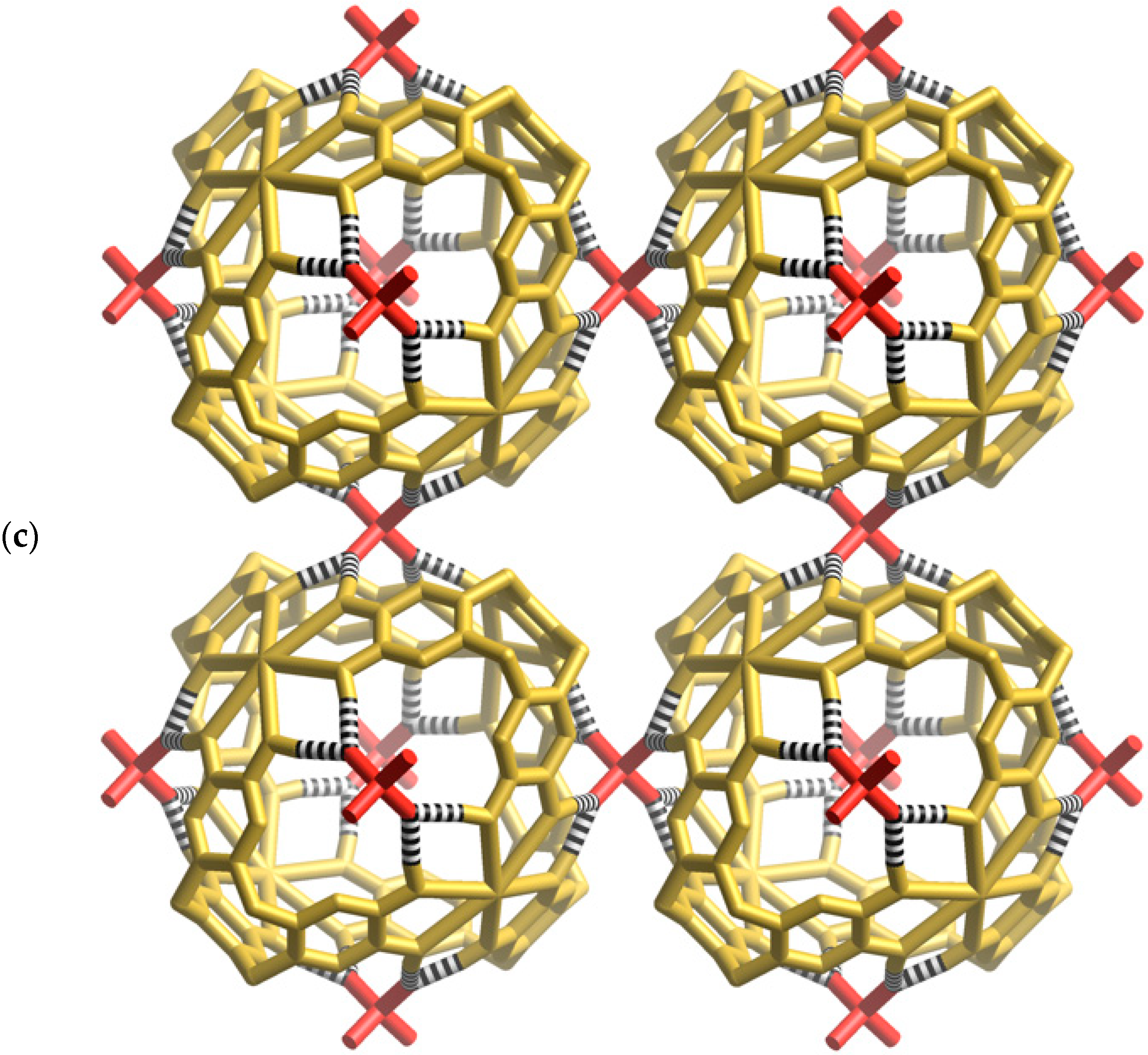
Figure 3a, two oxygen atoms of a phosphate ion are involved in forming four equivalent hydrogen bonds with phenolic groups of the cage (O···O, 2.59 Å); the phosphorus atom sits above the mid-point of each window. The highly complementary nature of this interaction is apparent in the space-filling representation (
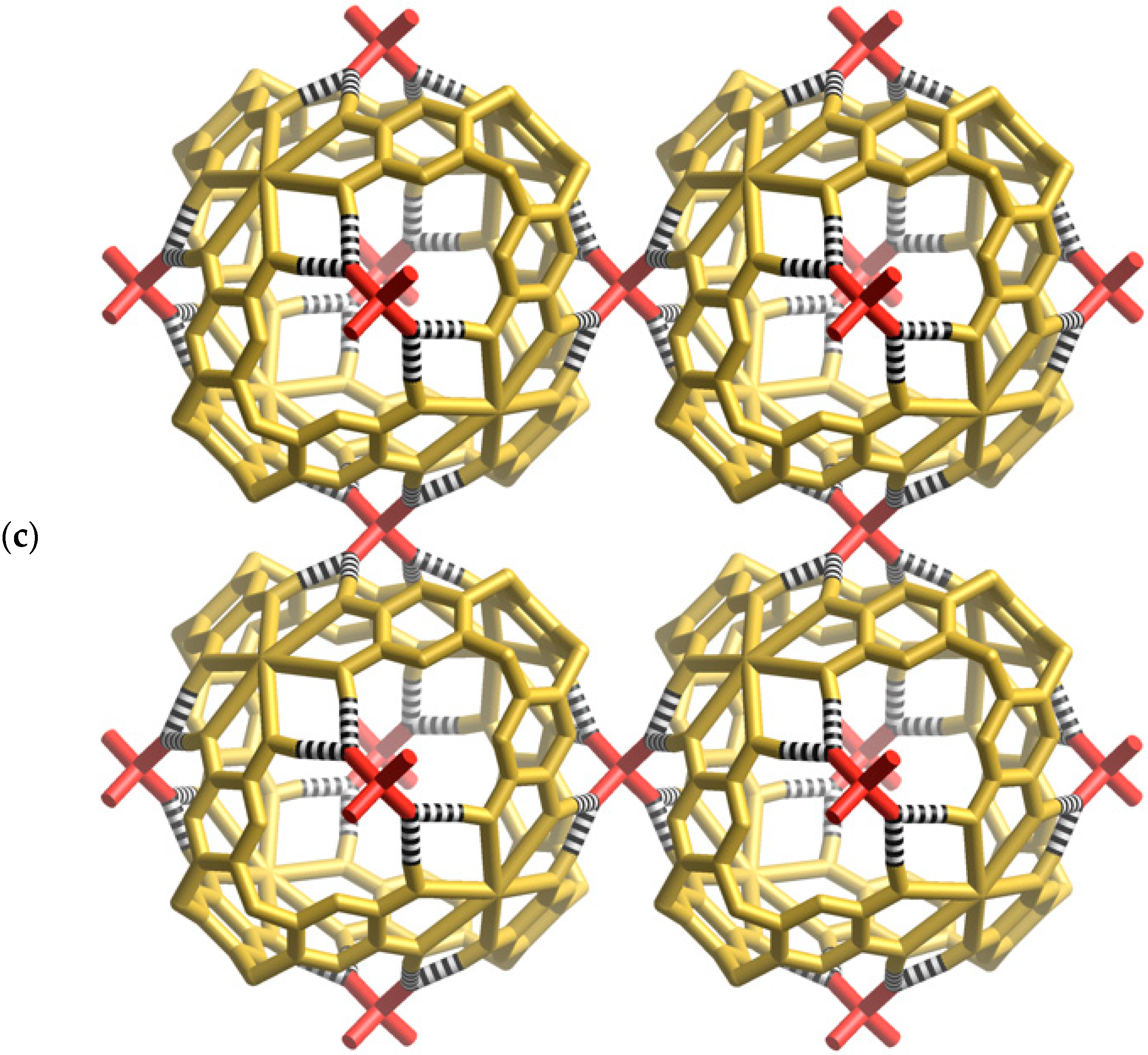
Figure 3b) in which the phosphate ion neatly plugs each cage window. In addition to the four hydrogen bonds in a single window, each phosphate forms a quartet of hydrogen bonds to the phenolic hydrogen atoms lying in a window of a neighbouring cage. Thus, each phosphate anion participates, as an acceptor, in eight equivalent hydrogen bonds as shown in
Figure 3c. Through the bridging phosphate ions, each [K
4(H
6ctc)
4(H
2O)
8]
4+ cage is linked to six equivalent cages each located in a neighbouring, face sharing, unit cell. The result is a three-dimensional primitive cubic network.
The hydrogen bonded network carries a net negative charge with the phosphate ions located in each face of the unit cell contributing 9- per unit cell whilst the [K
4(H
6ctc)
4(H
2O)
8]
4+ cage, along with the four internal Cs
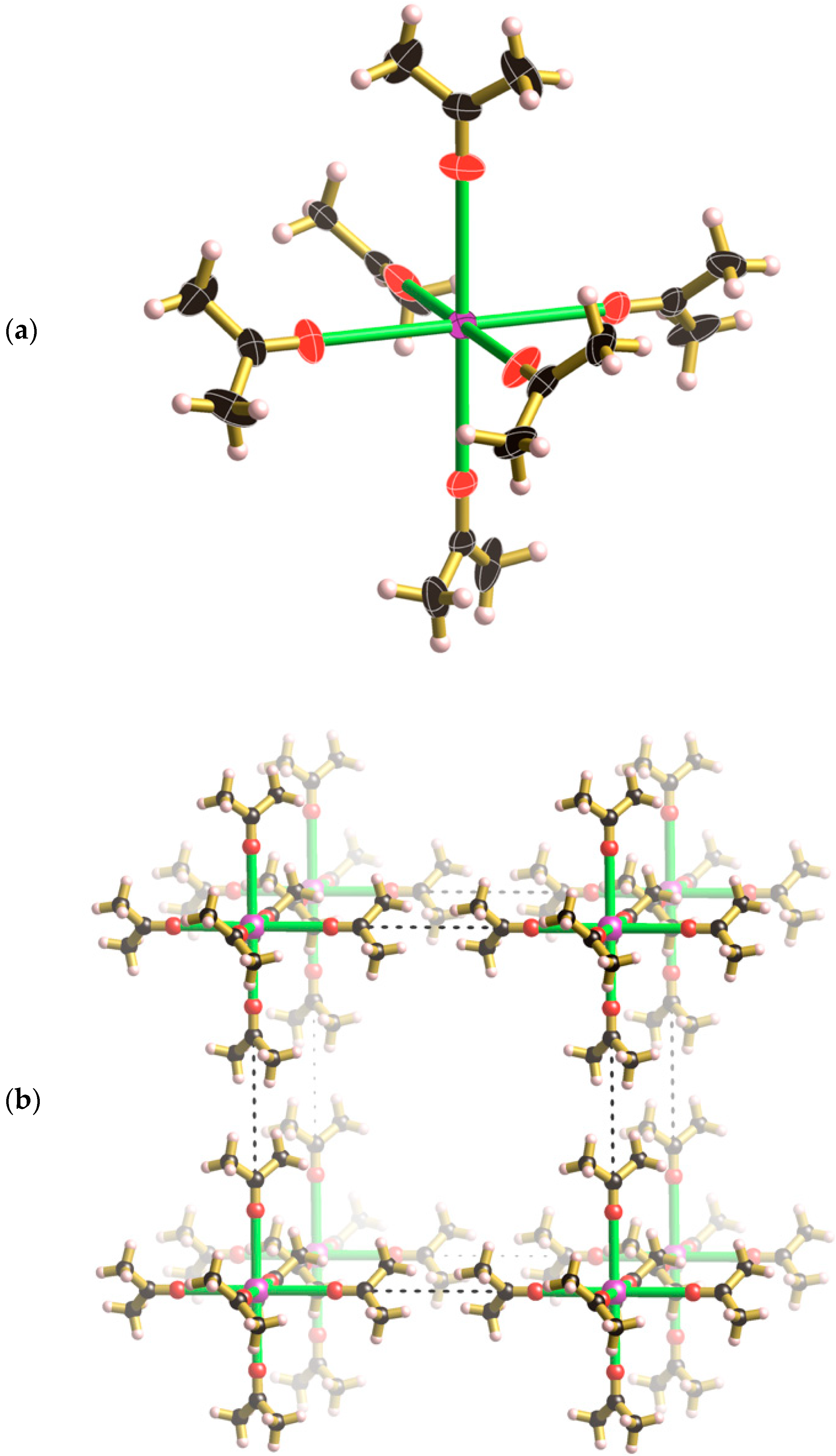
+ ions, has a net charge of 8+. Charge balance is achieved by the incorporation of a metal complex in which a Cs
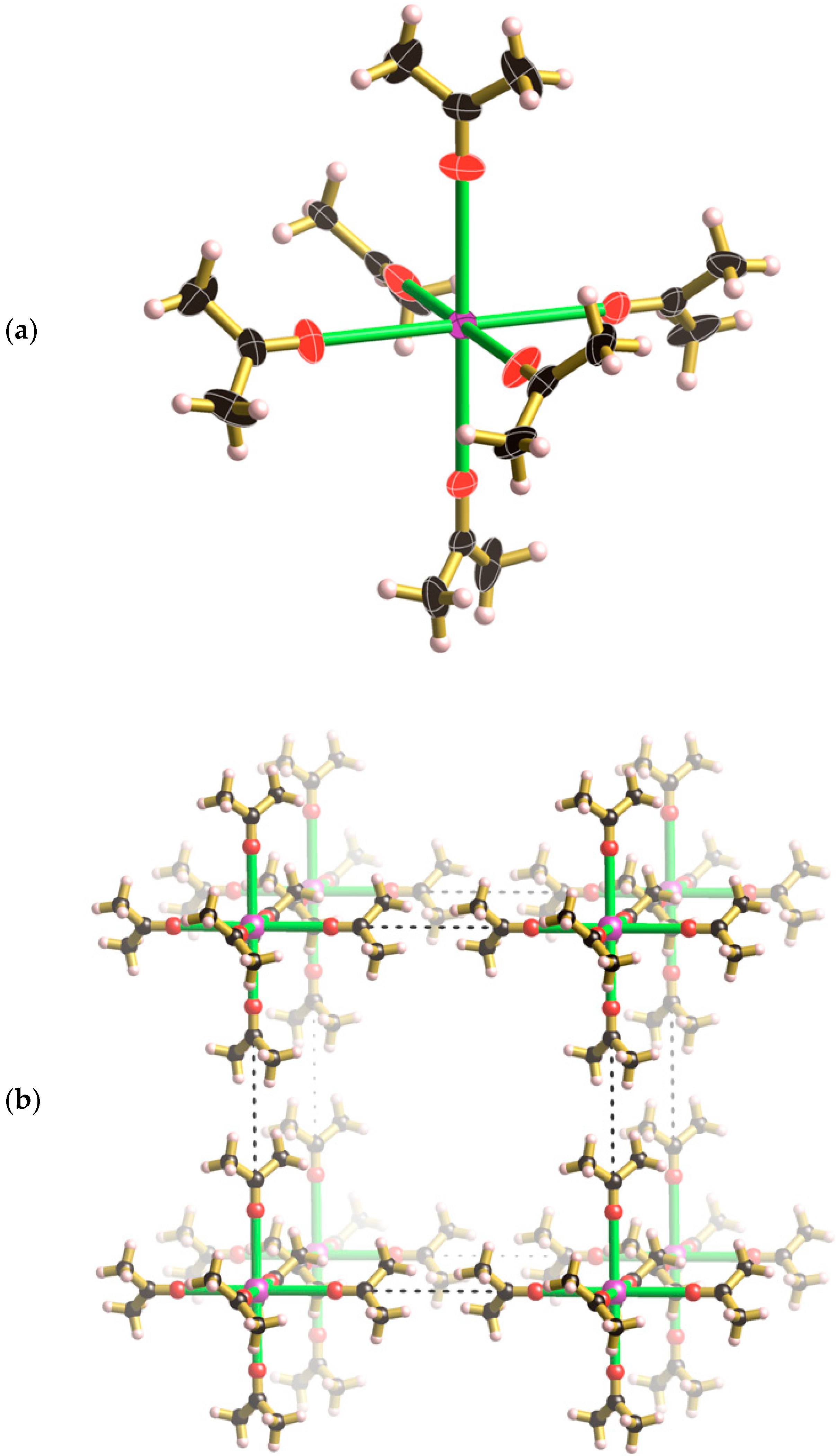
+ ion is coordinated by six symmetry related acetone molecules that lie along the unit cell axes (see
Figure 4a). Although examples of alkali metal ions coordinated by six acetone molecules are known, they are part of aggregates involving more than one alkali metal ion e.g., cationic clusters such as [Na
3((CH
3)
2CO)
9]
3+ [
27]. A search of the Cambridge Crystallographic Database reveals only three discrete M[(CH
3)
2CO]
6n+ complexes, [Mg((CH
3)
2CO)
6]
2+ which serves as a counter cation for bis((μ2-iodo)-iodo-copper(I)) [
28], [Ni[(CH
3)
2CO]
6]
2+ which accompanies a Ni-Cu carbide carbonyl cluster [
29] and [Fe((CH
3)
2CO)
6]
2+ which is the countercation for [FeCl
4]
− [
30]. In each of these hexaacetone complexes there is a bend at the carbonyl oxygen atom whereas in
III the Cs
+-O=C angle is 180° with the carbonyl oxygen group aligned along the unit cell axes. As indicated in
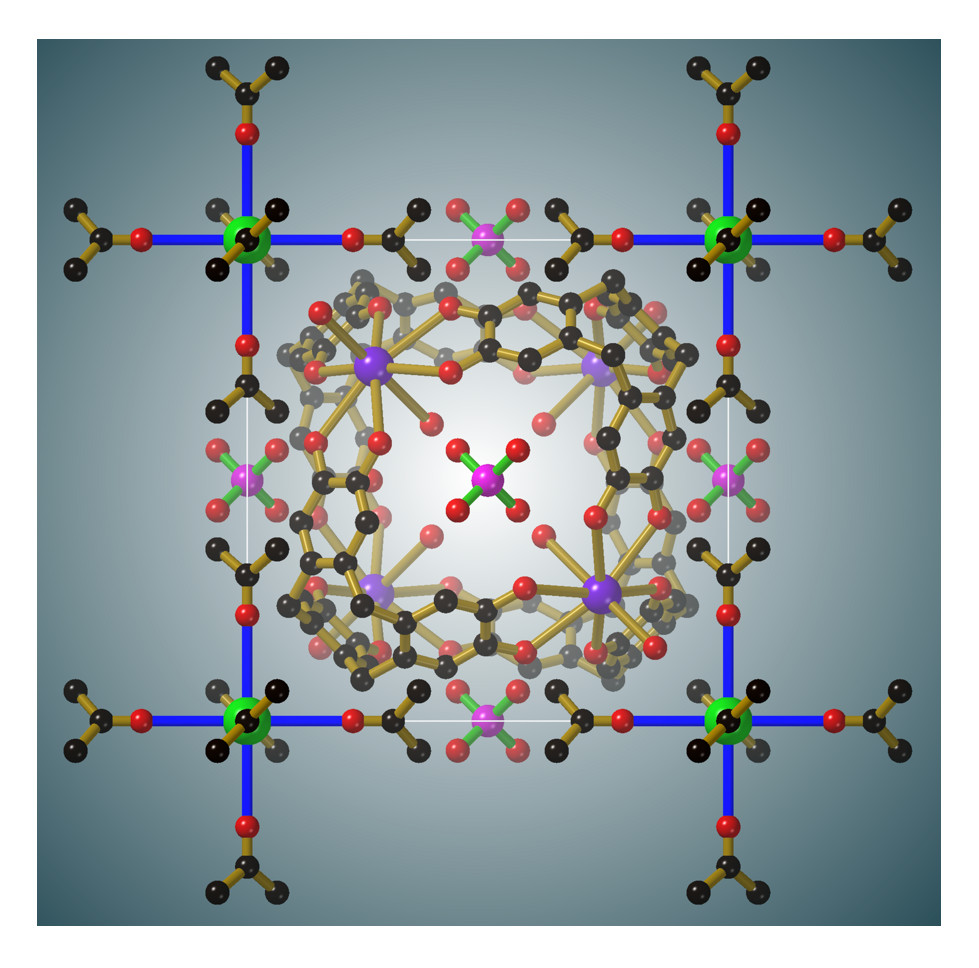
Figure 4a the thermal ellipsoid of the carbonyl oxygen is somewhat elongated in a direction perpendicular to the Cs-O bond, however, attempts to model the oxygen atom over two symmetry related sites were unsuccessful.
A Cs[(CH
3)
2CO]
6+ complex is located at each corner of the cubic unit cell as depicted in
Figure 4b. The methyl hydrogen atoms make relatively close contact along the edges of the unit cell to create a cubic framework. The mean planes of the two acetone molecules that approach each other along each cell edge are rotated by 90° relative to each other.
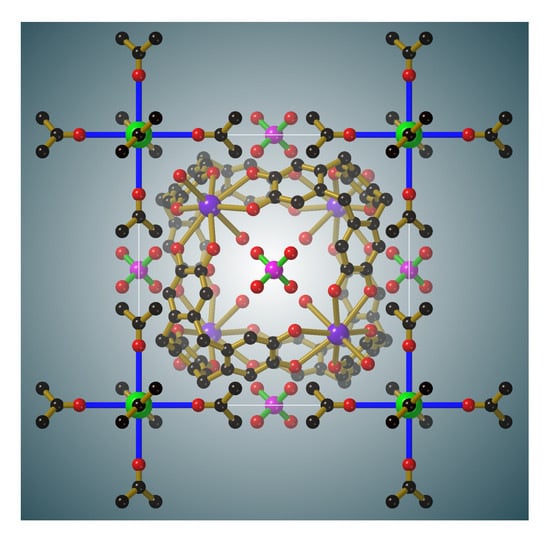
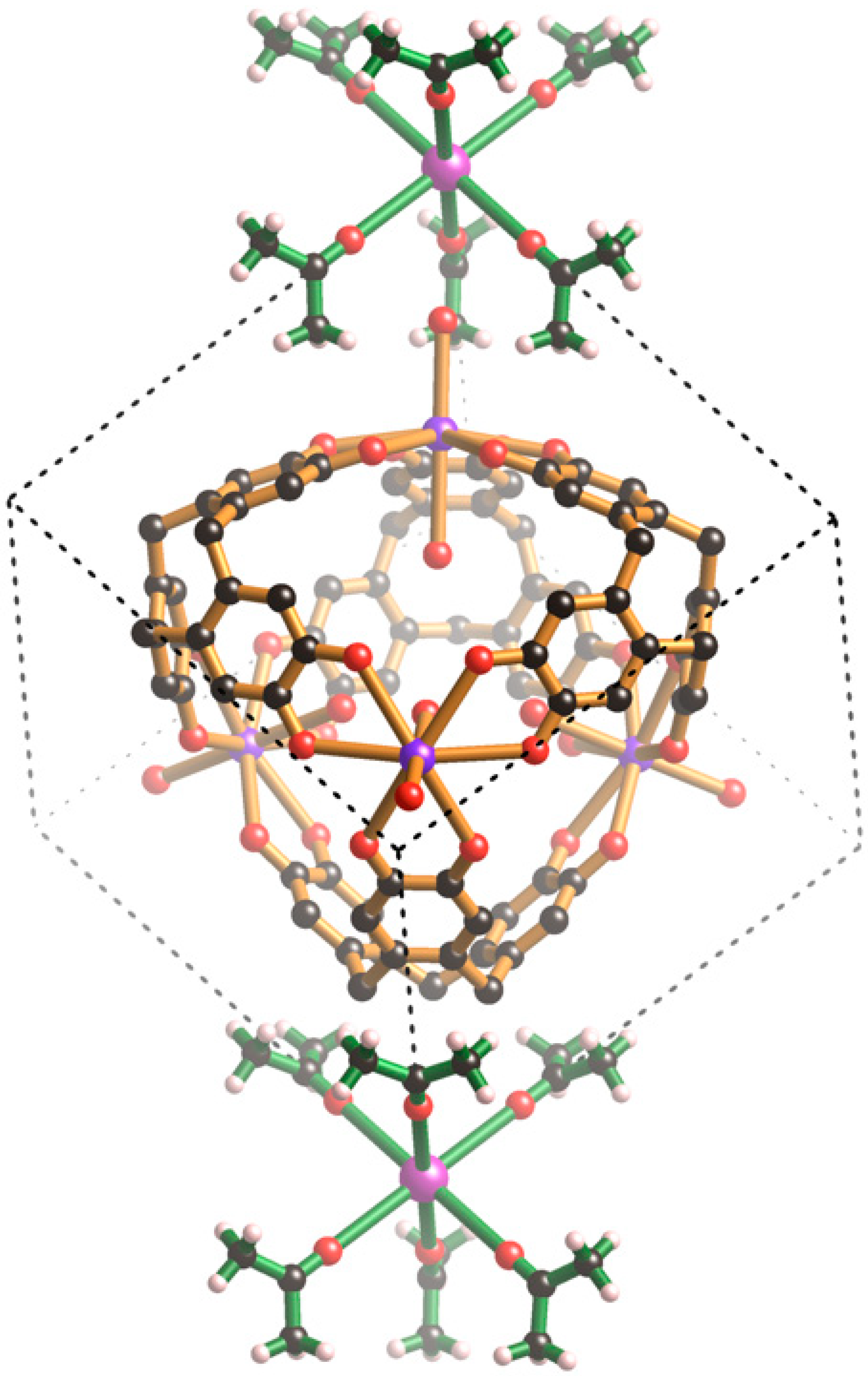
From a crystallographic perspective one of the most appealing aspects of the structure is the high level of symmetry that arises from the assembly of the components.
Figure 5 shows the relative positions of the Cs[(CH
3)
2CO]
6+ complex, the [K
4(H
6ctc)
4(H
2O)
8]
4+ cage and the phosphate anions within a unit cell of
III. The centre of the unit cell corresponds to the midpoint of the cage and is on a site of −43 m symmetry as is the Cs
+ ion of the Cs[(CH
3)
2CO]
6+ complex. The phosphate ion, located in the middle of each face, is situated on a site of −42 m symmetry. The body diagonals of the unit cell are co-linear with 3-fold axes and pass through the K
+ ions and coordinated water molecules of the cage, the internal Cs
+ ion, the 9-membered ring of a H
6ctc in addition to the Cs
+ ion of the Cs[(CH
3)
2CO]
6+ complex.
In investigations of the clam type structure of the type depicted in
Figure 1b it was found that the clam would form both with Cs
+ and Rb
+ and it would thus appear that both ions are of an appropriate size to interact with the internal aromatic surfaces of the cyclotricatechylene. On this basis we attempted to synthesise compound
III but with Rb
+ in place of Cs
+. Unfortunately, it was not possible to obtain a homogenous bulk product but we were successful in transferring a crystal from the mother liquor and into a protective oil for structure determination. The combination of H
6ctc with K
2HPO
4 and RbOH in aqueous acetone yields crystals of composition [Rb((CH
3)
2CO)
6][K
2Rb
2(H
6ctc)
4(H
2O)
6][Rb
4(H
2O)
6](PO
4)
3 (
IV). Apart from the inclusion of Rb
+ in place of Cs
+, the metal ions that form the [M
4(H
6ctc)
4]
4+ cages are an equal mixture of disordered K
+ and Rb
+ ions. Another difference relates to the occupation of the cage cavity by water molecules. There are only two coordinated water molecules inside the cage which are disordered over four symmetry related sites. In addition, the non-coordinated water molecules are now disordered over two sites. Presumably the smaller radius of the Rb
+ ion results in the water molecules being unable to symmetrically bridge the internal Rb
+ ions. The crystalline product in this case initially appeared to be homogeneous but crystallinity of the material was lost when the crystals were removed from the mother liquor.
The successful generation of the cubic compounds described above, prompted an investigation to determine whether other tetrahedral oxyanions employed in place of phosphate would yield similar structures. The combination of H6ctc, KHSO4 and CsOH in aqueous acetone yields the compound [Cs((CH3)2CO)6][K4(H6ctc)4(H2O)8][Cs(H2O)9](SO4)3 (V). The structure closely resembles the phosphate analogue (III), except in regards to the contents of the [K4(H6ctc)4(H2O)8]4+ cage. Instead of four Cs+ ions inside the cage, only one Cs+ anion is present which is disordered over the four H6ctc bowl sites. The decrease in the number of internal cations is required to maintain charge balance following the inclusion of sulphate in place of phosphate. In the crystallographic refinement of the structure, the best modelling of the disorder corresponded to refinement of the bowl sites with 25% occupancy by Cs+ and 75% occupancy by the oxygen atom of a water molecule. The remaining six water molecules in the cage were each found to be disordered over a pair of sites.
The final compound considered in this series is [Rb((CH3)2CO)6][Rb2K2(H6ctc)4(H2O)6] [Rb(H2O)9](SO4)3 (VI) which is formed from the reaction of H6ctc with KHSO4 and RbOH, again in aqueous acetone. The internal contents of the cage are similar to that found in (V) but with Rb+ in place of Cs+ and only two coordinated water molecules instead of four. The cage itself is formed from a combination of H6ctc with Rb+ ions and K+ ions as found for (IV).
A summary of the components from which compounds
III–
VI are formed is presented in
Table 2.
The concept of complementarity, in which certain specific favourable interactions lead to the self-assembly of components into larger structures, is a key theme in both supramolecular chemistry and crystal engineering. In the examples considered in this current work, the complementary interactions involve three neutral molecules (cyclotricatechylene, acetone and water), two cations (K+ and either Rb+ or Cs+) and an anion (either phosphate or sulphate) that participate in a variety of primary and secondary bonding interactions. Quite remarkably the six species combine to generate a series of crystalline materials exhibiting particularly high symmetry. It should be noted that whilst the initial formation of cubic crystals was serendipitous, the subsequent generation of related compounds was achieved through a recognition that some of the key interactions between components may persist upon the substitution of some components for structurally similar units.
Whilst prediction of the initial structure would seem most unlikely, the types of interactions between components are similar to those commonly encountered in the assembly of supramolecular species e.g., hydrogen bonding and metal coordination. Each of the compounds III–VI represents an additional example of the clear affinity of the larger alkali metal ions for the aromatic surfaces of cyclotricatechylene, although in contrast to previous examples the tris (catechol) is fully protonated.
Perhaps the most surprising aspect of the structures is the presence of the octahedral M[(CH
3)
2CO]
6+ (M = Rb, Cs) complexes in each structure. At first inspection it would seem quite remarkable that such a species, with linear M-C=O links, would be generated particularly given that the solvent from which the crystals were obtained contains water, which would normally be expected to have a much greater affinity for the alkali metal ions than acetone. One can only speculate about the reason for the presence of the unusual M[(CH
3)
2CO]
6+ octahedral complexes but it would seem likely that its size and shape neatly matches inter-cage spaces that are generated by the anionic hydrogen bonded network formed from the combination of the cages with the tetrahedral anions (PO
43− or SO
42−). As is apparent from inspection of
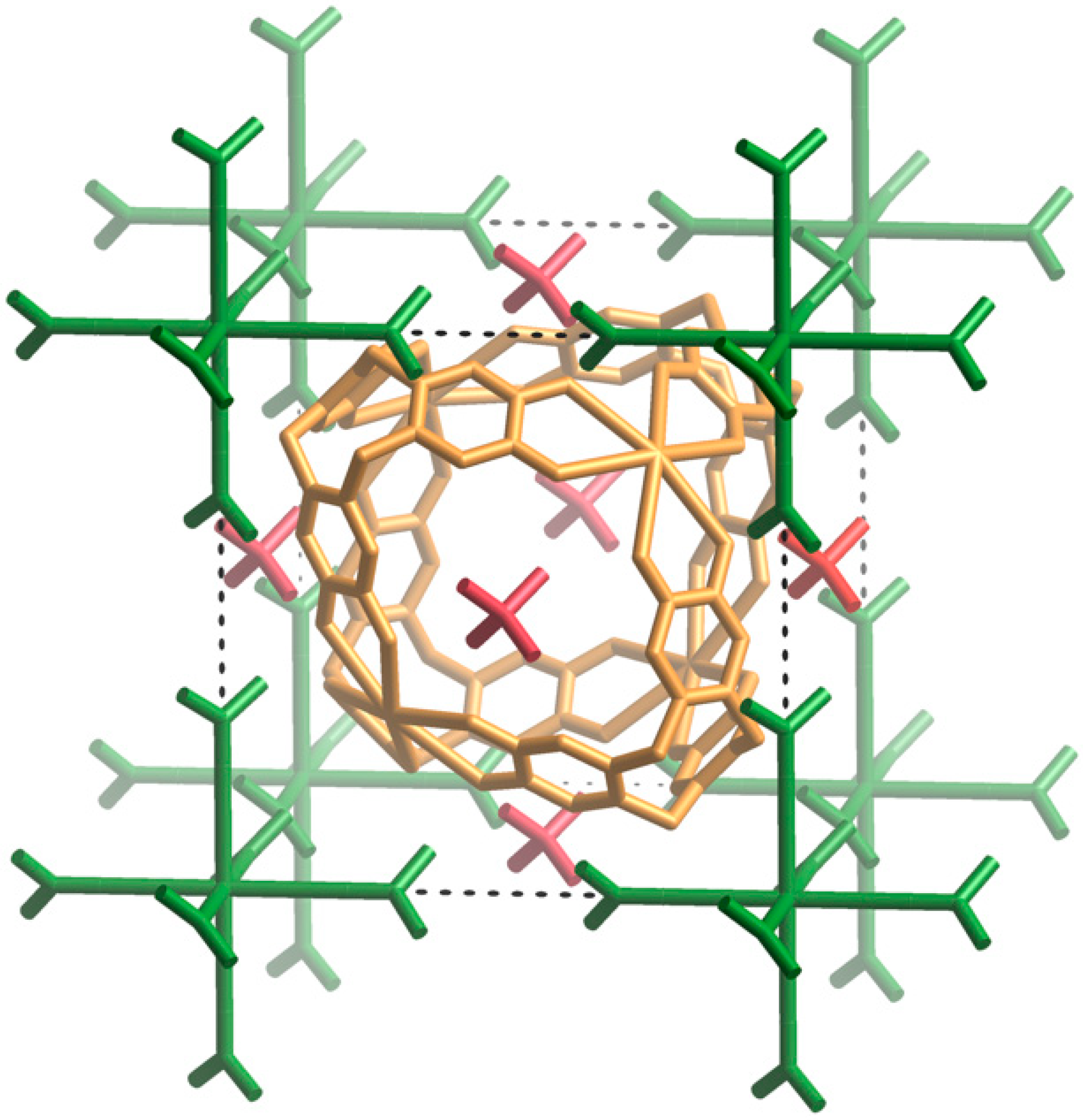
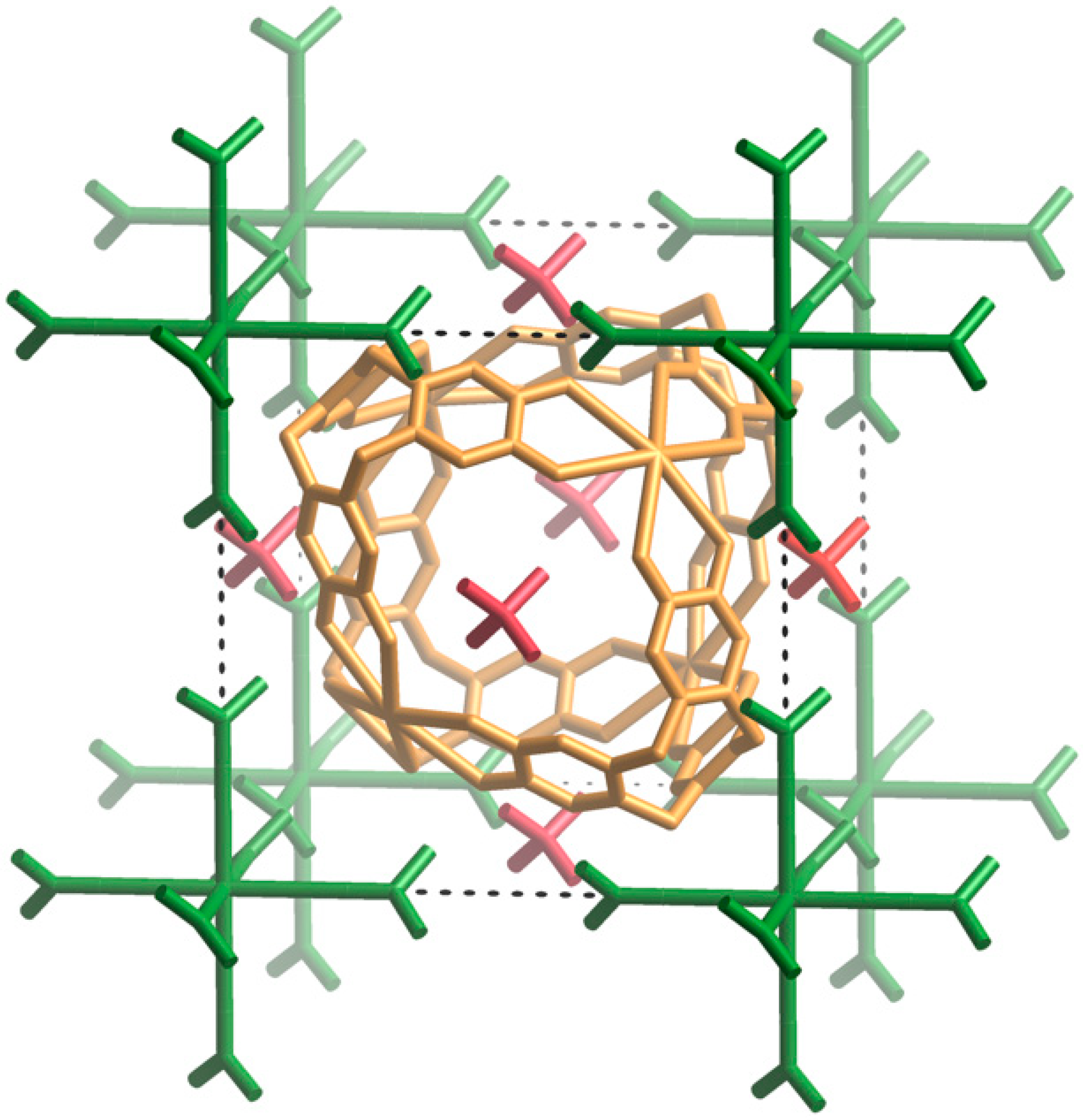
Figure 6, the van der Waals contact between coordinated acetone molecules half-way along each edge of the unit cell leads to a M···M separation equal to the unit cell edge length. This distance matches the separation between the centres of oxyanions on opposite faces of the cubic unit cell which form hydrogen bonds with the cage at the centre of the unit cell. Close inspection of each of the structures reveals complementary interactions between the M
4(H
2O)
8(H
6ctc)
44+ cages and the surrounding M[(CH
3)
2CO]
6+ octahedral complexes. Trios of acetone molecules orthogonal (
fac) to one another in the octahedral Cs[(CH
3)
2CO]
6+ complex are arranged in one of two ways. As is apparent from
Figure 6, in one arrangement, the methyl hydrogen atoms are pointing towards catechol oxygen atoms which are bound to a single K
+ centre. The oxygen atom of the externally coordinated water molecule sits in the middle of a trio of methyl groups. The second case provides a contrast with three acetone molecules forming a concave cavity which acts as a bowl for a H
6ctc unit. In this arrangement, the mean plane of the acetone molecule is close to parallel with the aromatic surface to which it makes close contact. Through these two types of interaction each cage associates with eight Cs[(CH
3)
2CO]
6+ complexes and each complex is surrounded by eight cages.
Inspection of
Table 1 reveals that the sulphate containing compounds,
V and
VI have longer unit cell lengths than
III and
IV. This would appear to be a consequence of the difference in the hydrogen bond distances. In the case of the SO
42− structures the O···O separation between the sulphate oxygen atom and the oxygen atom of the H
6ctc is 2.73 Å for
V and 2.72 Å for
VI. This is significantly longer than the separation of 2.59 Å found in
III and
IV and perhaps reflects a stronger interaction between the H
6ctc and the anion when the charge on the anion is 3-.

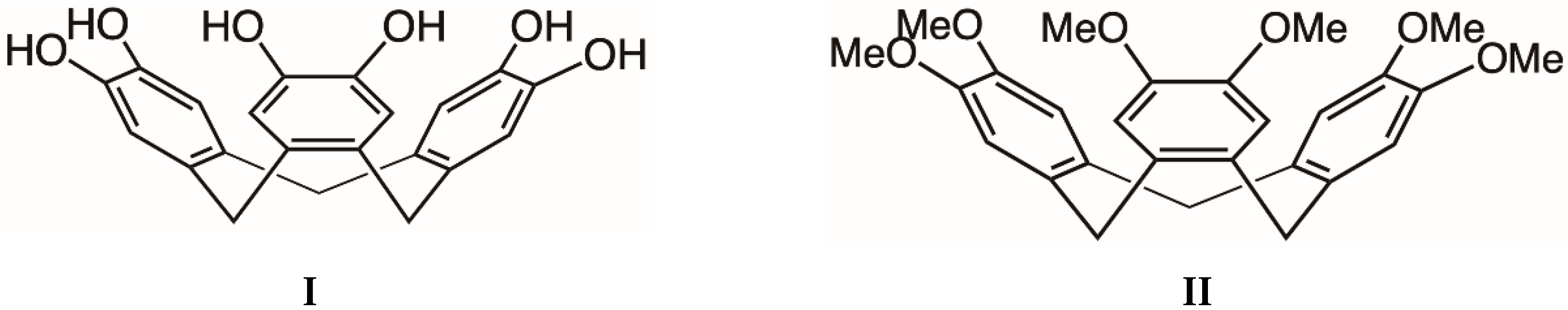


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
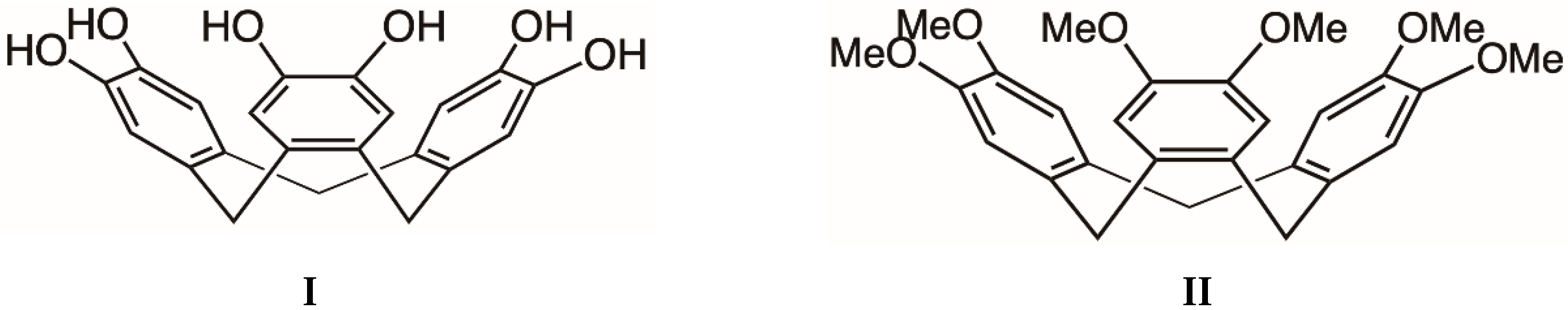{kind=link}