Abstract
This paper provides new insights into the geochemical cycling of rare earth elements (REEs) in acid sulfate soils developed on salt marsh sediments of the Huelva estuary (Spain) as a result of sulfide mineral oxidation in abandoned ore stockpiles. The study was aimed at determining the REE abundance, fractionation pattern and mineralogical control of the dispersal and retention of REEs in the soil system. Forty-one samples were collected at 13 core sampling sites along two transects extending across the degraded marshland, and they were subjected to XRD, ESEM-EDS and ICP-MS analyses. Measurements revealed that the soil receiving acid discharges has relatively high concentrations of ΣREEs (174.77 ± 19.77 mg kg−1) compared to local baseline concentrations. Shale-normalized REE patterns are generally flat, but a slight middle REE (MREE) enrichment is consistently apparent in all soil samples, involving relatively low LaN/GdN ratios (0.83 ± 0.08) and GdN/LuN ratios up to 1.42. The convex-upward REE pattern supports the possibility that iron oxy-hydroxide minerals play an important role in MREE retention through adsorption and co-precipitation mechanisms. Efflorescent sulfate salts left on the topsoil by the evaporation of acid waters show a strong depletion of light REEs (LaN/GdN = 0.16 ± 0.10) and act as a temporary reservoir of labile MREEs and heavy REEs during dry periods.
1. Introduction
Rare earth elements (REEs) are a group of transition metals with similar atomic structure and chemical properties, and are comprised of the lanthanide series (La to Lu) plus scandium (Sc) as well as yttrium (Y) [1], although in most geochemical discussions the term is restricted to the lanthanides. REEs are arbitrarily segregated into light REEs (LREEs: La to Nd), middle REEs (MREEs: Sm to Dy), and heavy REEs (HREEs: Ho to Lu) based on their atomic mass and the gradual reduction in ionic radius across the series (lanthanide contraction). REE abundance in Earth systems decreases with an increase in atomic weights, and elements of even atomic number are more abundant than adjacent elements with an odd atomic number according to the Oddo–Harkins rule. REEs are being increasingly used for high-tech applications, and recent studies [2] suggest they are anthropogenic contaminants of emerging concern.
Geochemically, REEs are lithophile refractory elements that display similar behavior through many geological processes [3]. During Earth surface processes, such as weathering and pedogenesis, REEs undergo significant fractionation [4], which reflects the depletion or enrichment of individual REEs, or subgroups of REEs, relative to a standard reference material. In soil science the reference commonly used to normalize lanthanide concentrations is either internal (parent material or geological substratum) or external (upper continental crust or shale composites used as proxies) to the surface environment under investigation [4]. Ln3+ (Ln refers to any lanthanide element) is the most stable lanthanide ion found in nature except for Eu, which is found mostly as Eu2+, and Ce, which may be partially oxidized to Ce4+. Anomalies can occur due to redox lability for Ce and Eu within the Eh-pH regime of water stability [5].
The fractionation patterns of REEs are either inherited from their parent materials (source-related), controlled by natural geochemical processes (adsorption, precipitation, ion exchange, and complexation), or influenced by anthropogenic sources [6,7,8,9,10,11,12,13,14]. Therefore, they provide a powerful tool for unravelling the geochemical history of a site [15,16]. In particular, REE fractionation signatures are increasingly being used as tracers of water–rock interactions in streams and sediments receiving acid sulfate soil drainage [17,18,19,20,21] and watersheds impacted by acid mine drainage (AMD) [22,23,24,25,26]. In these acidic settings, REEs provide clues for understanding the biogeochemical processes involved in the generation, monitoring, and remediation of AMD, including the reactive transport and transfer of potentially toxic trace elements from soil to biota [27]. The mobility of lanthanides in acidic soils is much higher than in natural soils because the low-pH conditions of the soil solution tend to enhance REE removal [28], resulting in high total REE concentrations in soil water and MREE enrichments in the dissolved REE signature [23,25,29]. The origin of and the mechanisms that control the development of MREE enrichments are still a matter of debate, and it is not sufficiently clear whether the REE patterns relate to the aqueous processes or are source-related [30]. Acid sulfate soils and mine soils are, therefore, highly favorable environments for REE mobilization, and their occurrence is a major factor that influences the concentration of REEs in naturally and anthropogenically acidified stream waters.
Despite the extensive use of REE fractionation patterns to trace natural geochemical processes and anthropogenic influences, relatively few studies have explored lanthanide dynamics in estuarine soil systems [15,31]. Processes controlling REE abundance and behavior are poorly constrained in the solid phase of marsh soils and sediments receiving AMD [32], and this represents a significant gap in our understanding of REE cycling in coastal wetland systems [21]. In fact, our knowledge of the geochemical behavior of lanthanides in soils is still far from complete [33], especially in wetland environments where REE-rich acidic water interacts with saline estuarine water.
The estuary of Huelva, on the Atlantic coast of southwestern Spain, provides an ideal opportunity to extend the knowledge concerning the behavior of REEs in secondary acid sulfate soils developed by long-term acidic drainage from legacy sulfide mine wastes, as will be described later. The pH values of the leachates emanating from abandoned sulfide heaps are in the range of 1.66–2.16 [34], and the soil receiving AMD discharges appears to be strongly acidified and contaminated with elevated levels of sulfates and heavy metals, thus limiting their quality, functions, and capability to support vegetation [35]. While there are numerous studies that consider the REE dynamics in surface waters and Holocene estuarine sediments [36,37,38,39,40,41], the cycling of REEs in the marsh soils remains poorly understood and further investigations are needed to address the legacy impacts of improper mine waste disposal on the receiving environment. In order to address this issue, the current research intended (1) to determine the abundance, spatial variability, and geochemical signature of REEs in the wetland soil adversely affected by continuous discharges of AMD from the sulfidic mine wastes, and (2) to ascertain the mineralogical controls on the dispersal, enrichment, and fractionation patterns of REEs.
2. Site Description and History
The former industrial site of Corrales lies on the west side of the upper estuary of Huelva, right across from the salt marshes of the river Odiel (Figure 1). These marshes form part of a set of coastal wetlands with high ecological value, declared in 1983 as a Biosphere Reserve by UNESCO. They are considered a wetland of international importance (RAMSAR Convention 1989), and a bird special protection area within the European Natura 2000 network. The area is characterized by a Mediterranean climate moderated by the influence of the Atlantic Ocean. The average annual precipitation is 525 mm and the mean temperatures range from 11.0 °C (January) to 25.8 °C (July–August).
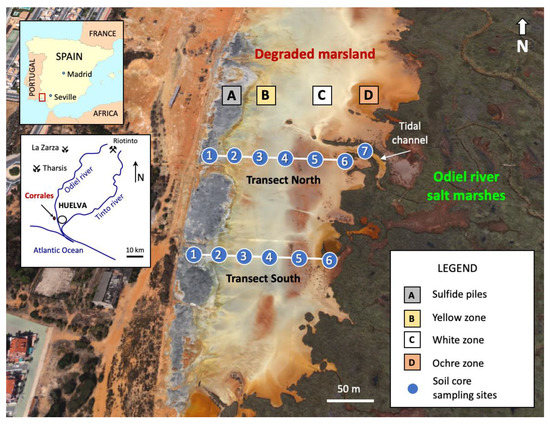
Figure 1.
Geographical setting of the Huelva estuary and satellite view (from Google Earth) of the degraded wetland, showing the location of the core sampling sites along two linear transects spanning from the abandoned sulfide heaps to the fresh salt marshes of the river Odiel.
The estuarine marsh soil (Salic Fluvisol, according to the WRB [42]) developed on fluvio-marine silty clayey sediments. The soil profile is of an AC or ABC type, with a salic horizon within 50 cm from the surface and hydromorphic features in the lower horizons. In dry periods the soil usually contains salt deposits at the surface, forming a coating layer. The wetland vegetation is dominated by salt-tolerant plant species, such as Spartina maritima, Salicornia ramosissima, Sarcocornia perennis, Halimione portulacoides and Spartina densiflora [43], except in the vicinity of the sulfide waste piles where the salt marsh is entirely devoid of vegetation.
Past mineral-processing operations by Tharsis Sulphur and Copper (TSC) have left a legacy of large amounts of pyrite concentrates stockpiled on the banks of the river Odiel. TSC was a major world trader in sulfur and one of the most active mining companies for over a century (1866–1978) in the Iberian Pyrite Belt [44], focusing its activity on the Tharsis and La Zarza mines, two world-class massive sulfide deposits located at distances of about 50 km and 65 km, respectively, from the port of Huelva where TSC had its own loading dock. All the ores extracted from the mines, comprised primarily of pyrite (FeS2) with minor amounts of chalcopyrite (CuFeS2), sphalerite (ZnS), and galena (PbS), were transported by railway to the estuary to be shipped overseas. Prior to shipment, the ore was crushed, ground, screened and classified to standard mesh sizes in processing plants located at the railway terminal of Corrales (Figure 2a), near the ore loading dock, facing the city of Huelva.

Figure 2.
Field pictures of the former industrial site of Corrales: (a) postcard view of the mineral crushing plant when it was operating (ca. 1910); (b) abandoned piles of crushed pyrite ore (2005); and (c) salt marsh soils affected by the disposal of sulfide wastes and reclaimed land area (2010).
The most extensive mining and mineral processing operations took place from the late 19th century to the middle of the 20th century, when pyrite became the main source of raw material used for sulfuric acid production by the European chemical industry. The last mining operation closed at the end of December 2000 for economic reasons linked to the growing competition of sulfuric acid being generated as a by-product of the smelting industry. Consequently, the pyrite concentrates were left on the site without any remediation plan to mitigate the environmental impacts on the adjacent soil (Figure 2b). During the operational phase and especially since the closure of the ore processing plants, the pollutants have been, and still are being, transferred from the sulfide heaps to nearby marsh soils by both chemical and physical processes (e.g., leaching, runoff, and atmospheric deposition of wind-blown dust), resulting in the deterioration of the environmental quality. It is particularly concerning that a considerable portion of the waste disposal land was converted for urban development.
From the entry into force of the contaminated land regime in Spain [45], the parcels that had been re-classified for residential use were declared as polluted in 2007 because the site posed unacceptable risks to human health by exposure to potentially toxic trace elements. In 2009, the sulfide wastes were removed from the private parcels and the underlying soil was lime-treated to neutralize acidity, and sealed with a low-permeability layer of compacted clay to prevent the seepage and release of pollutants (Figure 2c). Nowadays, the abandoned ore stockpiles, occupying an area of about 15,000 m2 within the maritime–terrestrial public domain, still remain unclaimed and continue to act as a source of acidity, posing a risk to human health and the environment [46].
3. Materials and Methods
The study area was subdivided into three floodplain zones based on the dominant color of the surface soil, namely a yellow zone, a white zone, and an ochre zone, which are aligned in a north–south direction, nearly parallel to the sulfide stockpiles (Figure 1). Six undisturbed soil cores (sites 1 to 6) were extracted with a gouge auger (100 cm length and 30 mm diameter) along two linear transects of approximately 150 m in length spaced 120 m apart. Line transects were positioned perpendicular to the riverbank and randomly setup across the degraded marshland. An additional soil core (site 7) was augered in the tidal channel near location 6 of the northern transect. The cores were sectioned according to variations in color and texture to obtain a total of forty-one samples (Figure 3), including sulfide waste and efflorescent salt samples, then placed in zip-lock plastic bags and immediately transported to the laboratory for analysis. The soil core samples were air-dried, gently disaggregated with a wooden roller, passed through a 10-mesh nylon sieve, and blended to achieve a high degree of homogeneity. After homogenization, aliquots of sieved material (<2 mm) were ground in an agate mortar and pestle until a nearly uniform fine powder (<63 μm) was produced for mineralogical and chemical analyses.
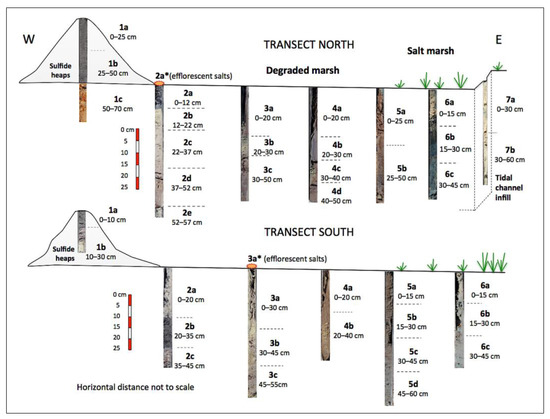
Figure 3.
Schematic depiction of the soil sections from the extracted core samples.
Mineralogical analysis was performed by X-ray diffraction (XRD) on a Bruker AXS D8 ADVANCE diffractometer (Central Research Services, University of Huelva, Spain) using CuKα radiation at 40 kV and 30 mA. Randomly oriented powders were scanned with a step size of 0.02° and a counting time of 0.6 s per step. The XRD patterns were processed using DIFFRAC plus software (Bruker AXS) linked with a reference database, and relative mineral abundance was estimated by empirical intensity factors, weighting the integrated peak areas of diagnostic reflections [47]. Selected samples were examined by environmental scanning electron microscopy (ESEM), using a Fei Quanta 200 (Central Research Services, University of Huelva) instrument operated at 20 kV and equipped with an energy-dispersive spectrometer (EDS) microanalytical system, by combining back-scattered electron imaging with EDS microanalysis to assist in mineral identification. Before the ESEM examination, the specimens were mounted onto stubs by electrically conductive double-sided tape and coated with a thin layer of sputtered carbon.
Total REE concentrations were analyzed after a wet digestion of 0.1 g dry weight with 8 mL of HF and 3 mL of HNO3 in closed Teflon vessels, heating until nearly dry. After adding 3 mL of HNO3 the solution was evaporated close to dryness again. This step was repeated using 3 mL of HCl. The residue was dissolved with 2 mL of HNO3. The REE analysis was conducted by inductively coupled plasma mass spectrometry (ICP-MS) on an Agilent 7900 ICP-MS instrument (Center for Research in Sustainable Chemistry, University of Huelva) with detection limits close to 0.01 μg L−1 for all the determinations. The isotopes determined were: 139La, 140Ce, 141Pr, 146Nd, 147Sm, 153Eu, 157Gd, 159Tb, 163Dy, 165Ho, 166Er, 169Tm, 172Yb, and 175Lu. Doubly charged and oxide interferences were less than 1.5% and 0.5%, respectively, and minimized during the tuning operation of the instrument. Analytical quality control was monitored using international certified reference materials (AGW-1 and SARM-4), blanks, and duplicates. The average precision and accuracy of data fall in the range of 5–10% relative standard deviation.
To obliterate the Oddo–Harkins effect and to characterize the fractionation patterns, the measured concentrations of REEs were normalized to the reference values of the European Shale Composite (EUS) [48] and to an internal reference material (natural background). In this study, the REE concentrations of nearby salt marsh sediments [39] served as a suitable baseline reference material, under the assumption that they are not affected by acid discharges emanating from the sulfide ore stockpiles. REE data of the sulfide-rich samples were also normalized to chondrite values [49]. Fractionation between LREEs and HREEs was quantified by the ratio LaN/LuN, with the subscript N denoting normalized concentration. Anomalies of Ce, Eu, and Gd in REE patterns were quantified by comparing the measured EUS-normalized concentrations (Ln) with the theoretical background concentrations (Ln*). A positive anomaly (enrichment) is defined as a calculated value (Ln/Ln*) > 1, while a negative anomaly (depletion) is < 1. The background Ce* concentration was achieved by interpolating between the normalized values of La and Pr (Equation (1)). Similarly, the background Eu* concentration was interpolated from Sm and Tb (Equation (2)), and the normalized concentrations of Nd and Dy were used to interpolate Gd* (Equation (3)) as Eu is often anomalous [16].
4. Results and Discussion
4.1. Field Observations
The marshland directly affected by AMD discharges has a surface area of about 50 ha, and shows a well-defined chromatic zonation. The sulfide heaps have a distinct grayish color (GLEY 1 hue in the Munsell color chart) and are composed essentially of fine-grained pyrite, but also contain quartz pebbles and a wide variety of artifacts and materials of anthropic origin, such as fragments of bricks, glass, wood, pottery, etc. The yellow zone (2.5Y to 5Y) contains abundant secondary products of pyrite oxidation and yellow efflorescent sulfate salts. The soil of the white zone (10YR to 2.5Y) has more frequent interactions with the tidal dynamics and also exhibits efflorescent salt deposits. The surface soil of the most distal zone is reddish-ochre in color (5YR to 7.5YR), which denotes a high degree of oxidation of iron. The floodplain is cut by tidal channels filled with yellowish (2.5Y to 5Y) fine-grained sediment.
4.2. Soil pH and Mineralogy
The sulfide waste samples showed ultra-acidic pH values (Table 1), ranging from 1.10–1.25 (top samples) to 1.60–1.73 (bottom samples) with an average of 1.3–1.5. The efflorescent sulfate salts also provided an important source of acidity (1–2 pH units) upon dissolution. Soil acidity varied greatly along the transects and with depth, from pH values around 2.0 in the yellow zone adjacent to the sulfide heaps (core two) to about 5.5 in distal locations (cores five and six), where the soil becomes moderately acidic.

Table 1.
Soil pH values in water and semi-quantitative mineral composition of the soil core samples determined by combining XRD and ESEM-EDS analyses. Mineral abbreviations: Amp (amphiboles); Ang (anglesite); Ba (barite); Cp (copiapite); Cpy (chalcopyrite); Fd (feldspars); Fs (phyllosilicates); Gy (gypsum); Hlt (halotrichite); Ha (halite); Hm (hematite); Js (jarosite); Me (melanterite); Mz (monazite); Py (pyrite); Qz (quartz); Sph (sphalerite); Sz (szomolnokite); and Zr (zircon).
The results from the XRD analysis (Table 1) confirmed that pyrite is the dominant sulfide phase in the waste stockpiles. Other accessory minerals identified by ESEM-EDS are chalcopyrite, sphalerite, anglesite, and barite. Pyrite is also present in some topsoil samples collected in the yellow zone and within the subsurface soil layers of the white zone. Jarosite is the most abundant and widespread mineral in the soil around the waste disposal area, and responsible for giving the distinctive yellow color of the proximal zone. The tidal channels are filled with jarosite-rich (over 70%) mud. Jarosite also occurs as a subordinate mineral in the white zone, whereas it is lacking in the ochre one. In most samples, the XRD pattern fits well with the standard pattern of natrojarosite, which is another jarosite-group mineral with Na instead of K.
Quartz, illite, kaolinite, and feldspars were present in all samples in varying amounts. These silicates are dominant in soil on which the sulfide wastes were stockpiled and in the deeper core samples of the distal zones, as they are primary constituents of the original soil. Besides, a variety of resistant accessory minerals, such as amphiboles, barite, monazite, and zircon were detected by ESEM-EDS. Iron oxides, notably hematite, occur mainly in the most distal zone, giving rise to the reddish-ochre color of the soil. Halite was found in the core sampling sites closest to the salt marsh, that is, in the white and ochre zones, where the tidal influence is strongest.
During prolonged periods of dryness, the exposed surfaces of the sulfide piles and the marshland degraded by the effects of AMD are covered with a multicolored efflorescence of readily water-soluble sulfate salts. A variety of evaporitic sulfate minerals with different hydration degrees were identified by XRD and ESEM-EDS analyses (Table 1), with copiapite, szomolnokite, and halotrichite being the most common precipitates.
4.3. REE Abundance and Distribution
Table 2 and Table 3 contain the results of the REE analyses including some relevant ratios, and Table 4 summarizes the descriptive statistics of the dataset.
Table 2.
Chemical composition of REEs (mg kg−1) in the core samples collected along the northern transect. EUS-normalized values are denoted with the subscript N.
Table 3.
Chemical composition of REEs (mg kg−1) in the core samples collected along the southern transect. EUS-normalized values are denoted with the subscript N.
Table 4.
Descriptive statistics of the REE dataset summarizing range and measures of central tendency and variability in each group of samples. EUS-normalized values are denoted with the subscript N.
For all samples, the REE concentration distribution typically obeys the Oddo–Harkins rule, depicting zigzag patterns (Figure 4) with the LREE concentrations being higher than the HREE contents. In terms of REE abundance, the sum of total concentrations (ΣREEs) ranged from 12.67 to 88.72 mg kg−1 in the sulfide waste samples, with a mean of 48.15 mg kg−1, whereas the ΣREEs of the jarosite-rich samples from the adjacent acidified soil environment was 83.10 mg kg−1 on average. The ephemeral products of the evaporation of the sulfate-rich solutions derived from oxidizing pyrite (efflorescent sulfate salts) showed the lowest ΣREE concentrations (21.59–33.46 mg kg−1, average of 27.53 mg kg−1).
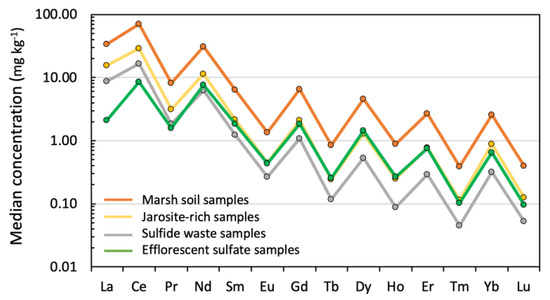
Figure 4.
Median concentration of individual REEs for each group of samples showing the saw-tooth effect explained by the Oddo–Harkins rule.
Overall, the ΣREE concentrations measured in the sulfide heaps are similar to those of the massive sulfide orebodies of the Iberian Pyrite Belt [50] as well as in other acid sulfate soil profiles [20], although a noticeable amount of REEs was lost during sulfide weathering. This is indicated by the decrease in the ΣREE content upwards in the profile of the pyrite heaps, from 28.04 to 12.67 mg kg−1 in the northern transect and from 88.72 to 36.89 mg kg−1 in the southern transect, thus suggesting the remobilization of REEs during sulfide oxidation. Indeed, the development of acidic and oxidizing conditions promoted a suitable environment for REE leaching.
Interestingly, the marsh soil affected by the leaching of sulfide heaps and acid sulfate soil showed relatively elevated concentrations of ΣREEs (174.77 ± 19.77 mg kg−1), with the highest values observed at sampling site five of the southern transect. The ΣREEs levels generally increased when increasing the distance from the abandoned sulfide piles. The ΣREE contents of the soil cores recovered in the distal zone were markedly higher than those registered in the acid sulfate (jarosite-rich) soil, reaching maximum values of about 200 mg kg−1 at pHs between 5.3 and 6.3. These values were also higher than those observed in sediments of the upper estuary of Huelva (Figure 5). Reported measurements of ΣREEs in nearby salt marsh sediments from non-impacted sites range between 52 and 60 mg kg−1 [39]. Therefore, the results suggest that the wetland soil receiving AMD inputs displays a relatively strong enrichment of total REE concentrations when compared to local baseline concentrations. This finding supports earlier studies elsewhere [15,16,17,18,19,20,23,25] which show that acid drainage produced during sulfide oxidation mobilizes REEs, in addition to iron, sulfate, and heavy metals, which then accumulate in soils and sediments closest to the acidic source.
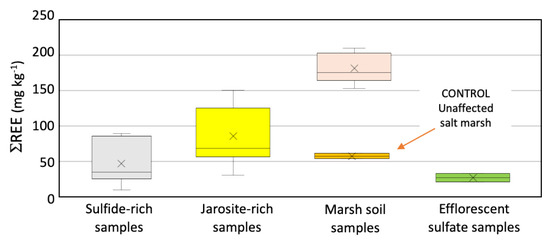
Figure 5.
Boxplots of the sum of total concentrations (ΣREEs) by groups of samples, including the reported values [39] of nearby salt marsh sediments not affected by acid leachates emanating from the sulfide heaps.
4.4. REE Fractionation Patterns
The chondrite-normalized REE profiles of the sulfide-rich samples (Figure 6) revealed an overall fractionation pattern (LaN/LuN = 7.2–17.4; average 13.8) with a relative enrichment of LREEs (LaN/GdN = 3.5–8.5; average 6.0) and slight negative Eu anomalies (Eu/Eu * = 0.83 on average), which are typical features of the REE distribution in regional massive sulfide orebodies [50].
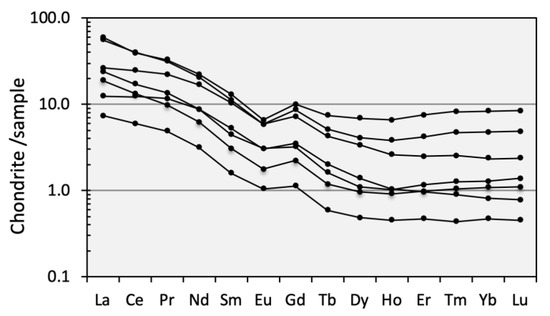
Figure 6.
Chondrite-normalized spider diagram for the sulfide-rich samples.
By contrast, the sulfide ore samples from the waste stockpiles (samples 1a and 1b in both transects) are characterized by relatively flat patterns when normalized against EUS (Figure 7a), with all REEs less than EUS values, as observed in other massive sulfide samples normalized to an average Earth surface reservoir [51,52]. This is consistent with little fractionation of the REEs from La to Lu (LaN/LuN = 1.22–1.83; average 1.65) due to the ultra-acidic conditions (pH values below 2) prevailing within the sulfide waste environment. In these acidic settings, REE speciation is dominated by free metal ions (Ln3+) and sulfate complexes, whose stability constants are relatively uniform across the entire REE series [9,53], and so limited fractionation of REEs would be expected from an aqueous complexation with sulfate.
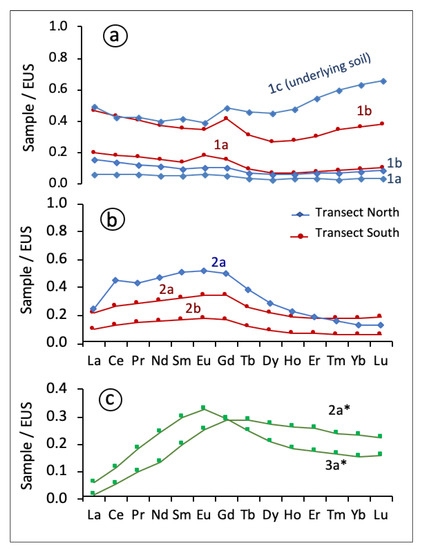
Figure 7.
Rare earth element (REE) fractionation patterns normalized against the European Shale Composite (EUS) for (a) sulfide waste piles and underlying soil; (b) sulfide-rich samples from soil adjacent to the sulfide piles; and (c) efflorescent sulfate salts (sample 2a * and 3a *).
However, it is noteworthy that most of the sulfide-rich soil samples showed some preferential enrichment in MREEs over both LREEs and HREEs, especially those collected in sites directly adjacent to the oxidizing pyrite heaps (Figure 7b), where the MREE enrichment is more apparent (LaN/GdN = 0.50–0.63 and GdN/LuN = 1.87–3.85). Moreover, the EUS-normalized REE pattern for the substrate layer (sample 1c, northern transect, Figure 7a) indicated a relative enrichment of HREEs compared to LREEs (GdN/LuN = 0.73), suggesting that the HREEs released into solution during sulfide weathering were immobilized to a greater extent than LREEs as they have a greater affinity for the solid phase in AMD systems [10,11]. Cation sorption and exchange on clay particles of the soil underlying the sulfide piles is probably of scant significance at low ambient pH, as they have positively charged surfaces on both basal (001) and edge faces. Alternatively, the HREEs could be preferentially taken up by bacteria [54] or scavenged by iron oxyhydroxysulfates [55]. REE anomalies are lacking or inconspicuous in the REEEUS diagrams of the sulfide-rich material, except for a few samples (e.g., sample 2a, northern transect) that showed minor positive anomalies of cerium (Ce/Ce * = 1.33;) and gadolinium (Gd/Gd * = 1.34), respectively.
The EUS-normalized REE distribution pattern of the two efflorescent sulfate samples is characterized by a strong depletion of LREEs (Figure 7c). The sample 3a * showed a distinct enrichment of MREEs relative to LREEs (LaN/GdN = 0.23) and HREEs (GdN/LuN = 1.81), manifested by a bell-shaped pattern with upward convexity around Sm-Eu-Gd elements. The sample 2a * displayed a negative slope from Gd to Lu that tended to be slightly enriched in MREEs compared to HREEs (GdN/LuN = 1.25). Accordingly, the REEs are potentially removed from the acid sulfate solution through salt-induced precipitation, thus leading to some fractionation. The REE signatures seem to be defined by the nature of the sulfate phases. Thus, the highest retention of MREEs was observed in the Fe-dominant sulfate (copiapite) sample while the highest retention of HREEs was found in sample 2a *, which contains a significant proportion (>25%) of Al-bearing sulfate (halotrichite). This selective affinity of MREEs for Fe-rich sulfates and HREEs for Al-rich sulfates has been previously reported in AMD systems [52].
The EUS-normalized REE patterns of the soil samples are essentially flat, with slightly more depletion in LREEs and HREEs than in MREEs. However, in the jarosite-rich samples from the yellow zone the REE abundance is substantially lower than in the marsh soil samples, with all REEs below EUS values (Figure 8a). The results showed that LREE/HREE fractionation is not of great significance, as indicated by the low mean values of the LaN/LuN ratio in both marsh soil samples (0.98 ± 0.07) and jarosite-rich samples (1.30 ± 0.15). Therefore, the REE retention was not accompanied by large fractionation trends across the lanthanide series. The spider diagrams display convex-upward morphologies defined by a steady increase in the EUS-normalized values toward the MREE series, involving relatively low LaN/GdN ratios (0.83 ± 0.08 in the marsh soil samples and 1.10 ± 0.15 in the jarosite-rich samples) and GdN/LuN ratios up to 1.42. This upward convexity centered on Gd reveals an enrichment of MREEs in comparison to the other REEs, thus tracing the geochemical signature of the soil–water interaction in the AMD-impacted marshland. The soil samples generally did not show Eu nor Ce anomalies, and when present they were negligible (Eu/Eu * = 1.06 ± 0.01; Ce/Ce * = 1.04 ± 0.02). Overall, the enrichment and fractionation of REEs in the studied estuarine marsh soil are similar to those that received acid sulfate soil drainage [15].
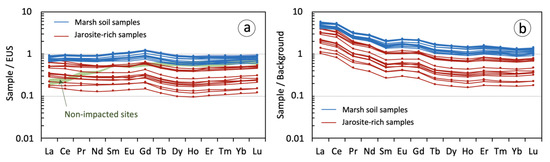
Figure 8.
Rare earth element (REE) fractionation patterns for acid sulfate (jarosite-rich) samples and marsh soil samples normalized against (a) European Shale Composite (EUS) values and (b) background values (mean REE concentration measured by López-González et al. [39] in nearby salt marsh sediments unaffected by acid discharges). The EUS-normalized REE pattern of the baseline reference material is also shown in Figure 8a for comparative purposes.
It should be also noted that the soil affected by AMD inputs appears to be enriched in LREEs compared to salt marsh sediments from non-impacted sites (Figure 8a). After normalization to background values (Figure 8b), the REE patterns of the soil receiving acidic drainage displayed a remarkable enrichment in LREEs (LaN/GdN = 3.18 ± 0.58) and a nearly flat trend in MREEs and HREEs (GdN/LuN = 1.51 ± 0.09). This finding suggests that the LREEs released during sulfide weathering were removed from the solution and retained onto soil particles to a greater extent than the HREEs, thus enhancing the total content of REEs in the degraded marshland.
4.5. Mineralogical Controls on REE Retention
The oxidative dissolution of pyrite exposed in the stockpile disposal area and the subsequent soil acidification is the principal driving force for REE mobilization and, therefore, the main source of aqueous REEs to the estuarine wetland. The lack of substantial REE fractionation is consistent with relatively large water–rock ratios during sulfide oxidation [51]. Although the lanthanides tend to remain in solution in sulfate waters with pH values lower than 5.5, with sulfate complexes being the dominant aqueous species [10,12], our data suggest that REEs are retained, at least in part, in newly formed minerals resulting from sulfide oxidation, such as evaporitic sulfate minerals, jarosite-group minerals, and iron oxide as well as oxy-hydroxide minerals.
Although sulfate complexation does not fractionate REEs significantly [9], in periods of dryness sulfate ions lead to the removal of MREEs and HREEs from the AMD solution by the formation of efflorescent sulfate minerals. Therefore, salt precipitation provides a mechanism for the temporary storage of REEs. Because of the high solubility of these salts [56], the efflorescences left on the surface soil layer by the evaporation of acid drainage and pore soil waters are transient reservoirs of REEs and potential sources of labile MREEs and HREEs upon dissolution by rain or high-tide flooding. Less soluble secondary sulfates such as jarosite-group minerals can accommodate REEs in their crystal structures, preferentially LREEs rather than MREEs and HREEs, although limited ionic substitution would be expected as the ionic radii of the Ln3+ ions are somewhat larger than that of the Fe3+ ion [57]. Dissolution experiments have shown that the REEs that are incorporated into the jarosite structure can be released upon reaction with acidic solutions generating MREE-enriched patterns [58].
The fact that ΣREEs concentrations are significantly higher in the marsh soil of the distal zone, where attenuation processes related to neutralization reactions are occurring, suggests that REEs are scavenged from the water under near-neutral conditions, and then immobilized or fixed by soil components. Recent studies have highlighted the role of iron in the REE scavenging by Fe-Al-precipitates in AMD systems [55,59]. Leaching experiments performed on sediments of the Huelva estuary showed that, at a pH of 6, most of the REEs are found concentrated in ochre precipitates [40], hence adsorption onto or co-precipitation with iron oxy-hydroxides could be responsible for REE retention, just as with heavy metals, as pointed out by Grantcharova and Fernández-Caliani [35]. The convex-upward REE pattern further supports the possibility that iron oxide and oxy-hydroxide minerals played an important role in the MREE enrichment of the ochre soil samples from the distal zone. The MREE-enriched pattern shown by the soil receiving AMD from the sulfide heaps, evidenced by LaN/GdN ratios that are less than 1, is consistent with those of the sediments generated as a result of acid mixing processes [32,36,37]. Another plausible mechanism for the enhancement of REEs in the marsh soil could be adsorption onto clay minerals such as illite and kaolinite [4,15,60]. However, REE fractionation by clay minerals does not generate MREE-enriched signatures [61], so they only have a minor influence on REE retention compared to oxide and oxy-hydroxide minerals.
5. Conclusions
The conclusions drawn from this study can be summarized as follows:
- The pyrite concentrates stockpiled on the banks of the estuarine salt marshes are a legacy source of leachable REEs in the AMD-impacted marshland.
- REEs are subject to geochemical cycling processes in the acid sulfate soil developed on the wetland by sulfide mineral oxidation and acidic drainage over decades.
- Abundance and fractionation patterns of REEs in the marsh soil that receives inputs from the sulfide heaps are distinct from those reported in nearby non-impacted sites. The receiving soil was found enriched in ΣREEs relative to local baseline concentrations, and reflects to some extent the source-related REE signal of the soil–water interaction.
- The EUS-normalized REE patterns of the marsh soil samples did not display a significant LREE/HREE fractionation, but rather a slight MREE enrichment, suggesting that AMD precipitates (notably iron oxyhydroxides) are able to scavenge and act as sinks for MREEs.
- The efflorescent sulfate salts represent a transient reservoir of readily leachable MREEs and HREEs for the wetland.
Author Contributions
J.C.F.-C.: conceptualization, methodology, validation, formal analysis, investigation, visualization, and writing—original draft; M.M.G.: investigation, formal analysis, and writing—review. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been partially supported by the Andalusian Regional Government (Spain) through the Research Group on Geology and Environmental Geochemistry (RNM-347).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the manuscript and its supplementary material.
Acknowledgments
We thank Jesús de la Rosa (University of Huelva, Spain) for his collaboration during fieldwork and for assistance with chemical analysis. The manuscript benefited from valuable reviews by three anonymous reviewers.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hartshorn, R.M.; Hellwich, K.-H.; Yerin, A.; Damhus, T.; Hutton, A.T. Brief guide to the nomenclature of inorganic chemistry. Pure Appl. Chem. 2015, 87, 1039–1049. [Google Scholar] [CrossRef]
- Gwenzi, W.; Mangori, L.; Danha, C.; Chaukura, N.; Dunjana, N.; Sanganyado, E. Sources, behaviour, and environmental and human health risks of high-technology rare earth elements as emerging contaminants. Sci. Total Environ. 2018, 636, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Chakhmouradian, A.R.; Wall, F. Rare Earth Elements: Minerals, Mines, Magnets (and More). Elements 2012, 8, 333–340. [Google Scholar] [CrossRef]
- Laveuf, C.; Cornu, S. A review on the potentiality of Rare Earth Elements to trace pedogenetic processes. Geoderma 2009, 154, 1–12. [Google Scholar] [CrossRef]
- Brookins, D.G. Chapter 8. Aqueous Geochemistry of Rare Earth Elements. Geochem. Mineral. Rare Earth Elem. 1989, 21, 201–226. [Google Scholar] [CrossRef]
- Elderfield, H.; Upstill-Goddard, R.; Sholkovitz, E. The rare earth elements in rivers, estuaries, and coastal seas and their significance to the composition of ocean waters. Geochimica Cosmochimica Acta 1990, 54, 971–991. [Google Scholar] [CrossRef]
- Johannesson, K.H.; Lyons, W.; Yelken, M.A.; Gaudette, H.E.; Stetzenbach, K.J. Geochemistry of the rare-earth elements in hypersaline and dilute acidic natural terrestrial waters: Complexation behavior and middle rare-earth element enrichments. Chem. Geol. 1996, 133, 125–144. [Google Scholar] [CrossRef]
- Bau, M. Scavenging of dissolved yttrium and rare earths by precipitating iron oxyhydroxide: Experimental evidence for Ce oxidation, Y-Ho fractionation, and lanthanide tetrad effect. Geochimica Cosmochimica Acta 1999, 63, 67–77. [Google Scholar] [CrossRef]
- Gimeno, M.J.; Auqué, L.F.; Nordstrom, D. REE speciation in low-temperature acidic waters and the competitive effects of aluminum. Chem. Geol. 2000, 165, 167–180. [Google Scholar] [CrossRef]
- Verplanck, P.L.; Nordstrom, D.; Taylor, H.E.; Kimball, B.A. Rare earth element partitioning between hydrous ferric oxides and acid mine water during iron oxidation. Appl. Geochem. 2004, 19, 1339–1354. [Google Scholar] [CrossRef]
- Gammons, C.H.; Wood, S.A.; Nimick, D. Diel behavior of rare earth elements in a mountain stream with acidic to neutral pH. Geochimica et Cosmochimica Acta 2005, 69, 3747–3758. [Google Scholar] [CrossRef]
- Wood, S.A.; Gammons, C.H.; Parker, S.R. The behavior of rare earth elements in naturally and anthropogenically acidified waters. J. Alloys Compd. 2006, 418, 161–165. [Google Scholar] [CrossRef]
- Noack, C.W.; Dzombak, D.A.; Karamalidis, A.K. Rare Earth Element Distributions and Trends in Natural Waters with a Focus on Groundwater. Environ. Sci. Technol. 2014, 48, 4317–4326. [Google Scholar] [CrossRef] [PubMed]
- Soyol-Erdene, T.; Valente, T.; Grande, J.; DE LA Torre, M.L. Mineralogical controls on mobility of rare earth elements in acid mine drainage environments. Chemosphere 2018, 205, 317–327. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.; Rate, A.W.; Burton, E.; Smirk, M.N. Enrichment and fractionation of rare earth elements in FeS- and organic-rich estuarine sediments receiving acid sulfate soil drainage. Chem. Geol. 2012, 308-309, 60–73. [Google Scholar] [CrossRef]
- Xu, N.; Morgan, B.; Rate, A. From source to sink: Rare-earth elements trace the legacy of sulfuric dredge spoils on estuarine sediments. Sci. Total Environ. 2018, 637-638, 1537–1549. [Google Scholar] [CrossRef]
- Åström, M. Abundance and fractionation patterns of rare earth elements in streams affected by acid sulphate soils. Chem. Geol. 2001, 175, 249–258. [Google Scholar] [CrossRef]
- Åström, M.E.; Nystrand, M.; Gustafsson, J.P.; Österholm, P.; Nordmyr, L.; Reynolds, J.K.; Peltola, P. Lanthanoid behaviour in an acidic landscape. Geochimica Cosmochimica Acta 2010, 74, 829–845. [Google Scholar] [CrossRef]
- Welch, S.; Christy, A.; Isaacson, L.; Kirste, D. Mineralogical control of rare earth elements in acid sulfate soils. Geochimica et Cosmochimica Acta 2009, 73, 44–64. [Google Scholar] [CrossRef]
- Gröger, J.; Proske, U.; Hanebuth, T.J.; Hamer, K. Cycling of trace metals and rare earth elements (REE) in acid sulfate soils in the Plain of Reeds, Vietnam. Chem. Geol. 2011, 288, 162–177. [Google Scholar] [CrossRef]
- Morgan, B.; Johnston, S.; Burton, E.; Hagan, R.E. Acidic drainage drives anomalous rare earth element signatures in intertidal mangrove sediments. Sci. Total Environ. 2016, 573, 831–840. [Google Scholar] [CrossRef]
- Worrall, F.; Pearson, D. Water-rock interaction in an acidic mine discharge as indicated by rare earth element patterns. Geochimica Cosmochimica Acta 2001, 65, 3027–3040. [Google Scholar] [CrossRef]
- Fernández-Caliani, J.; Barba-Brioso, C.; De la Rosa, J. Mobility and speciation of rare earth elements in acid minesoils and geochemical implications for river waters in the southwestern Iberian margin. Geoderma 2009, 149, 393–401. [Google Scholar] [CrossRef]
- Pérez-López, R.; Delgado, J.; Nieto, J.M.; García, B.M. Rare earth element geochemistry of sulphide weathering in the São Domingos mine area (Iberian Pyrite Belt): A proxy for fluid–rock interaction and ancient mining pollution. Chem. Geol. 2010, 276, 29–40. [Google Scholar] [CrossRef]
- Grawunder, A.; Merten, D.; Büchel, G. Origin of middle rare earth element enrichment in acid mine drainage-impacted areas. Environ. Sci. Pollut. Res. 2014, 21, 6812–6823. [Google Scholar] [CrossRef] [PubMed]
- Edahbi, M.; Plante, B.; Benzaazoua, M.; Ward, M.; Pelletier, M. Mobility of rare earth elements in mine drainage: Influence of iron oxides, carbonates, and phosphates. Chemosphere 2018, 199, 647–654. [Google Scholar] [CrossRef]
- Merten, D.; Geletneky, J.; Bergmann, H.; Haferburg, G.; Kothe, E.; Büchel, G. Rare earth element patterns: A tool for understanding processes in remediation of acid mine drainage. Geochemistry 2005, 65, 97–114. [Google Scholar] [CrossRef]
- Goldstein, S.J.; Jacobsen, S.B. Rare earth elements in river waters. Earth Planet. Sci. Lett. 1988, 89, 35–47. [Google Scholar] [CrossRef]
- Johannesson, K.H.; Zhou, X. Origin of middle rare earth element enrichments in acid waters of a Canadian High Arctic lake. Geochimica et Cosmochimica Acta 1999, 63, 153–165. [Google Scholar] [CrossRef]
- Verplanck, P.L.; Nordstrom, D.K.; Taylor, H.E. Overview of rare earth element investigations in acid waters of U.S. Geological Survey abandoned mine lands watersheds. U.S. Geol. Surv. Wat. Resourc. Invest. 1999, 99-4018A, 83–92. [Google Scholar]
- Chevis, D.; Johannesson, K.H.; Burdige, D.; Cable, J.; Martin, J.B.; Roy, M. Rare earth element cycling in a sandy subterranean estuary in Florida, USA. Mar. Chem. 2015, 176, 34–50. [Google Scholar] [CrossRef]
- Delgado, J.; Pérez-López, R.; Galván, L.; Nieto, J.M.; Boski, T. Enrichment of rare earth elements as environmental tracers of contamination by acid mine drainage in salt marshes: A new perspective. Mar. Pollut. Bull. 2012, 64, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Mihajlovic, J.; Bauriegel, A.; Stärk, H.-J.; Roßkopf, N.; Zeitz, J.; Milbert, G.; Rinklebe, J. Rare earth elements in soil profiles of various ecosystems across Germany. Appl. Geochem. 2019, 102, 197–217. [Google Scholar] [CrossRef]
- Dávila, J.M.; Sarmiento, A.M.; Santisteban, M.; Luís, A.; Fortes, J.C.; Diaz-Curiel, J.; Valbuena, C.; Grande, J.A. The UNESCO national biosphere reserve (Marismas del Odiel, SW Spain): An area of 18,875 ha affected by mining waste. Environ. Sci. Pollut. Res. 2019, 26, 33594–33606. [Google Scholar] [CrossRef] [PubMed]
- Grantcharova, M.M.; Fernández-Caliani, J.C. Geochemical and mineralogical reactions driven by acid generation and metal release from pyritic wastes improperly disposed on estuarine marsh soils. In Proceedings of the 31st Annual V.M. Goldschmidt Conference, Lyon, France, 4–9 July 2021; Available online: https://2021.goldschmidt.info/goldschmidt/2021/meetingapp.cgi/Paper/4050 (accessed on 20 September 2021).
- Elbaz-Poulichet, F.; Dupuy, C. Behaviour of rare earth elements at the freshwater–seawater interface of two acid mine rivers: The Tinto and Odiel (Andalucía, Spain). Appl. Geochem. 1999, 14, 1063–1072. [Google Scholar] [CrossRef]
- Borrego, J.; López-González, N.; Carro, B.; Lozano-Soria, O. Geochemistry of rare-earth elements in Holocene sediments of an acidic estuary: Environmental markers (Tinto River Estuary, South-Western Spain). J. Geochem. Explor. 2005, 86, 119–129. [Google Scholar] [CrossRef]
- Borrego, J.; Carro, B.; López-González, N.; De La Rosa, J.; Grande, J.A.; Gómez, T.; De La Torre, M.L. Effect of acid mine drainage on dissolved rare earth elements geochemistry along a fluvial–estuarine system: The Tinto-Odiel Estuary (S.W. Spain). Hydrol. Res. 2012, 43, 262–274. [Google Scholar] [CrossRef]
- López-González, N.; Borrego, J.; Carro, B.; Grande, J.A.; De la Torre, M.L.; Valente, T. Rare earth element fractionation patterns in estuarine sediments as a consequence of acid mine drainage: A case study in SW Spain. Bol. Geol. Min. 2012, 123, 55–64. [Google Scholar]
- LeComte, K.L.; Sarmiento, A.; Borrego, J.; Nieto, J. Rare earth elements mobility processes in an AMD-affected estuary: Huelva Estuary (SW Spain). Mar. Pollut. Bull. 2017, 121, 282–291. [Google Scholar] [CrossRef]
- Cánovas, C.R.; Basallote, M.D.; Macías, F. Distribution and availability of rare earth elements and trace elements in the estuarine waters of the Ría of Huelva (SW Spain). Environ. Pollut. 2020, 267, 115506. [Google Scholar] [CrossRef]
- WRB. World Reference Base for Soil Resources; World Soil Resources Reports 106; FAO: Rome, Italy, 2014; Available online: http://www.fao.org/3/i3794en/I3794en.pdf (accessed on 20 September 2021).
- Finkl, C.W.; Makowski, C. Coastal Wetlands: Alteration and Remediation; Springer International Publishing: Cham, Switzerland, 2017; pp. 215–241. [Google Scholar] [CrossRef] [Green Version]
- Checkland, S.G. The Mines of Tharsis. Roman, French and British Enterprise in Spain; Allen and Unwin: London, UK, 1967; p. 288. [Google Scholar]
- Tarazona, J.V.; Fernández, M.D.; Vega, M.M. Regulation of Contaminated Soils in Spain—A New Legal Instrument (4 pp). J. Soils Sediments 2005, 5, 121–124. [Google Scholar] [CrossRef]
- Fernández-Caliani, J.C. Risk-based assessment of multimetallic soil pollution in the industrialized peri-urban area of Huelva, Spain. Environ. Geochem. Health 2011, 34, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Kahle, M.; Kleber, M.; Jahn, R. Review of XRD-based quantitative analyses of clay minerals in soils: The suitability of mineral intensity factors. Geoderma 2002, 109, 191–205. [Google Scholar] [CrossRef]
- Bau, M.; Schmidt, K.; Pack, A.; Bendel, V.; Kraemer, D. The European Shale: An improved data set for normalisation of rare earth element and yttrium concentrations in environmental and biological samples from Europe. Appl. Geochem. 2018, 90, 142–149. [Google Scholar] [CrossRef]
- Taylor, S.R.; McLennan, S. Composition and Evolution of the Continental Crust; Cambridge University Press: Cambridge, UK, 2009; pp. 301–324. [Google Scholar]
- Ruiz, C.; Arribas, A. Mineralogy and geochemistry of the Masa Valverde blind massive sulphide deposit, Iberian Pyrite Belt (Spain). Ore Geol. Rev. 2002, 19, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Leybourne, M.I.; Peter, J.M.; Layton-Matthews, D.; Volesky, J.; Boyle, D.R. Mobility and fractionation of rare earth elements during supergene weathering and gossan formation and chemical modification of massive sulfide gossan. Geochimica et Cosmochimica Acta 2006, 70, 1097–1112. [Google Scholar] [CrossRef]
- Da Silva, E.F.; Bobos-Radu, I.; Matos, J.X.; Patinha, C.; Reis, A.; Fonseca, E. Mineralogy and geochemistry of trace metals and REE in volcanic massive sulfide host rocks, stream sediments, stream waters and acid mine drainage from the Lousal mine area (Iberian Pyrite Belt, Portugal). Appl. Geochem. 2009, 24, 383–401. [Google Scholar] [CrossRef]
- Wood, S.A. The aqueous geochemistry of the rare-earth elements and yttrium: 1. Review of available low-temperature data for inorganic complexes and the inorganic REE speciation of natural waters. Chem. Geol. 1990, 82, 159–186. [Google Scholar] [CrossRef]
- Breuker, A.; Ritter, S.F.; Schippers, A. Biosorption of Rare Earth Elements by Different Microorganisms in Acidic Solutions. Metals 2020, 10, 954. [Google Scholar] [CrossRef]
- Lozano, A.; Ayora, C.; Fernández-Martínez, A. Sorption of rare earth elements on schwertmannite and their mobility in acid mine drainage treatments. Appl. Geochem. 2020, 113, 104499. [Google Scholar] [CrossRef]
- Jambor, J.L.; Nordstrom, D.K.; Alpers, C. Metal-sulfate Salts from Sulfide Mineral Oxidation. Rev. Miner. Geochem. 2000, 40, 303–350. [Google Scholar] [CrossRef]
- Dutrizac, J. The behaviour of the rare earths during the precipitation of sodium, potassium and lead jarosites. Hydrometallurgy 2004, 73, 11–30. [Google Scholar] [CrossRef]
- Welch, S.A.; Christy, A.; Kirste, D.; Beavis, S.G.; Beavis, F. Jarosite dissolution I—Trace cation flux in acid sulfate soils. Chem. Geol. 2007, 245, 183–197. [Google Scholar] [CrossRef]
- De Moraes, M.L.B.; Ladeira, A.C.Q. The role of iron in the rare earth elements and uranium scavenging by Fe–Al-precipitates in acid mine drainage. Chemosphere 2021, 277, 130131. [Google Scholar] [CrossRef] [PubMed]
- Aström, M.E.; Yu, C.X.; Virtasalo, J.J.; Österholm, P.; Peltola, P.; Burton, E.D.; Hogmalm, K.J.; Ojala, A.E.K. Extensive accumulation of rare earth elements in estuarine sediments affected by leaching of acid sulfate soils. Boreal Environ. Res. 2020, 25, 105–120. [Google Scholar]
- Coppin, F.; Berger, G.; Bauer, A.; Castet, S.; Loubet, M. Sorption of lanthanides on smectite and kaolinite. Chem. Geol. 2002, 182, 57–68. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).