Rainfall Alters Permafrost Soil Redox Conditions, but Meta-Omics Show Divergent Microbial Community Responses by Tundra Type in the Arctic



Abstract
1. Introduction
2. Materials and Methods
2.1. Mesocosm Design
2.2. Soil Properties
2.3. Soil Sample Collection
2.4. Modeling Soil O2 Accumulation and Consumption Rates
2.5. Nucleic Acid Extraction and Sequencing
2.6. 16S rRNA Gene Analysis
2.7. Metagenome Assembly
2.8. Metatranscriptome Assembly
2.9. Taxonomic Composition and Diversity
2.10. Genomic Potential and Functional Gene Expression
2.11. Differential Gene Expression
2.12. Oxygen-Regulated Gene Expression
2.13. Iron Redox Cycling Gene Expression
2.14. Microbial Respiration and Methane Production
3. Results
3.1. Soil Properties
3.2. Soil O2 Accumulation and Consumption Rates and Final Concentrations
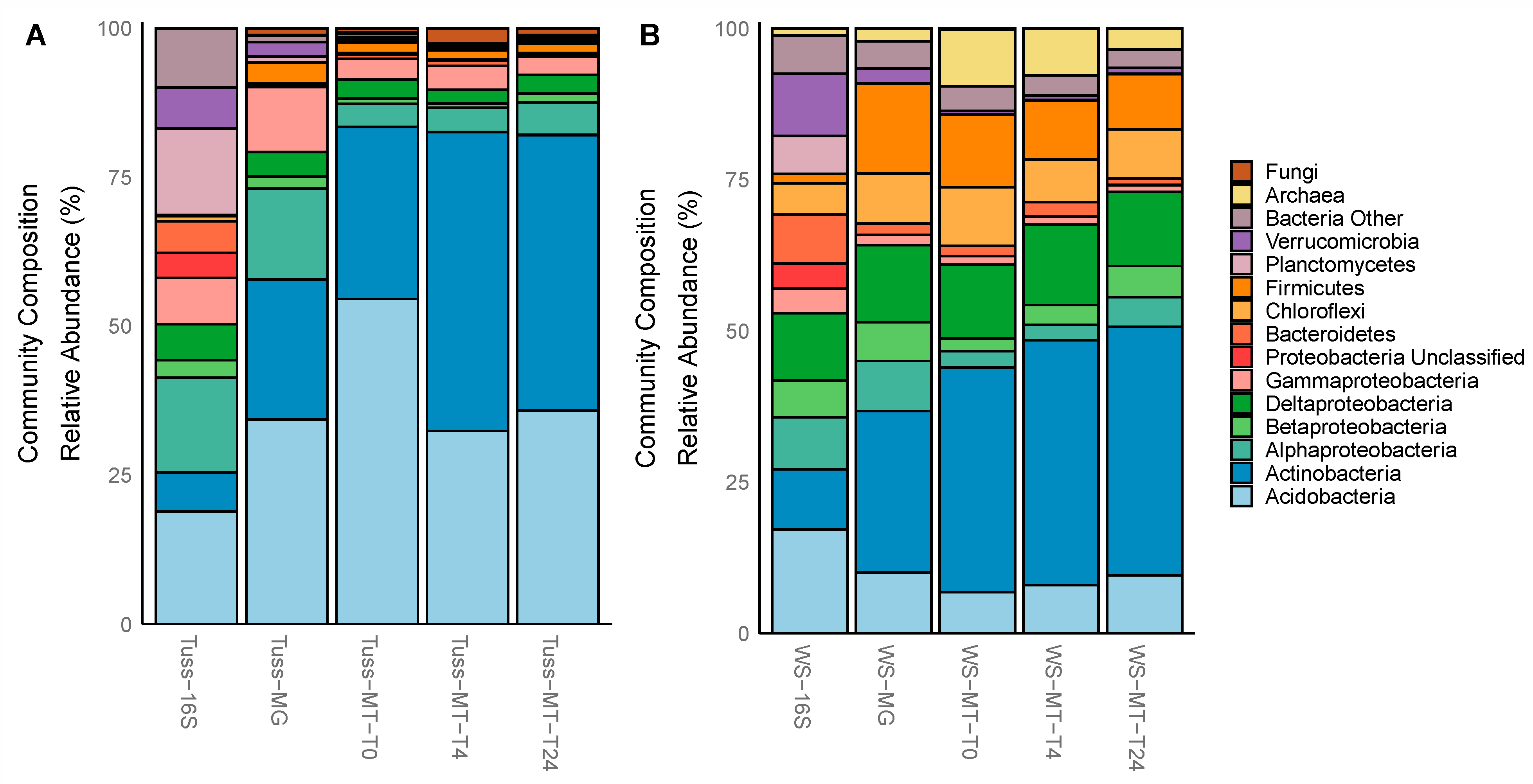
3.3. Taxonomic Composition and Diversity of the Total and Active Microbial Communities
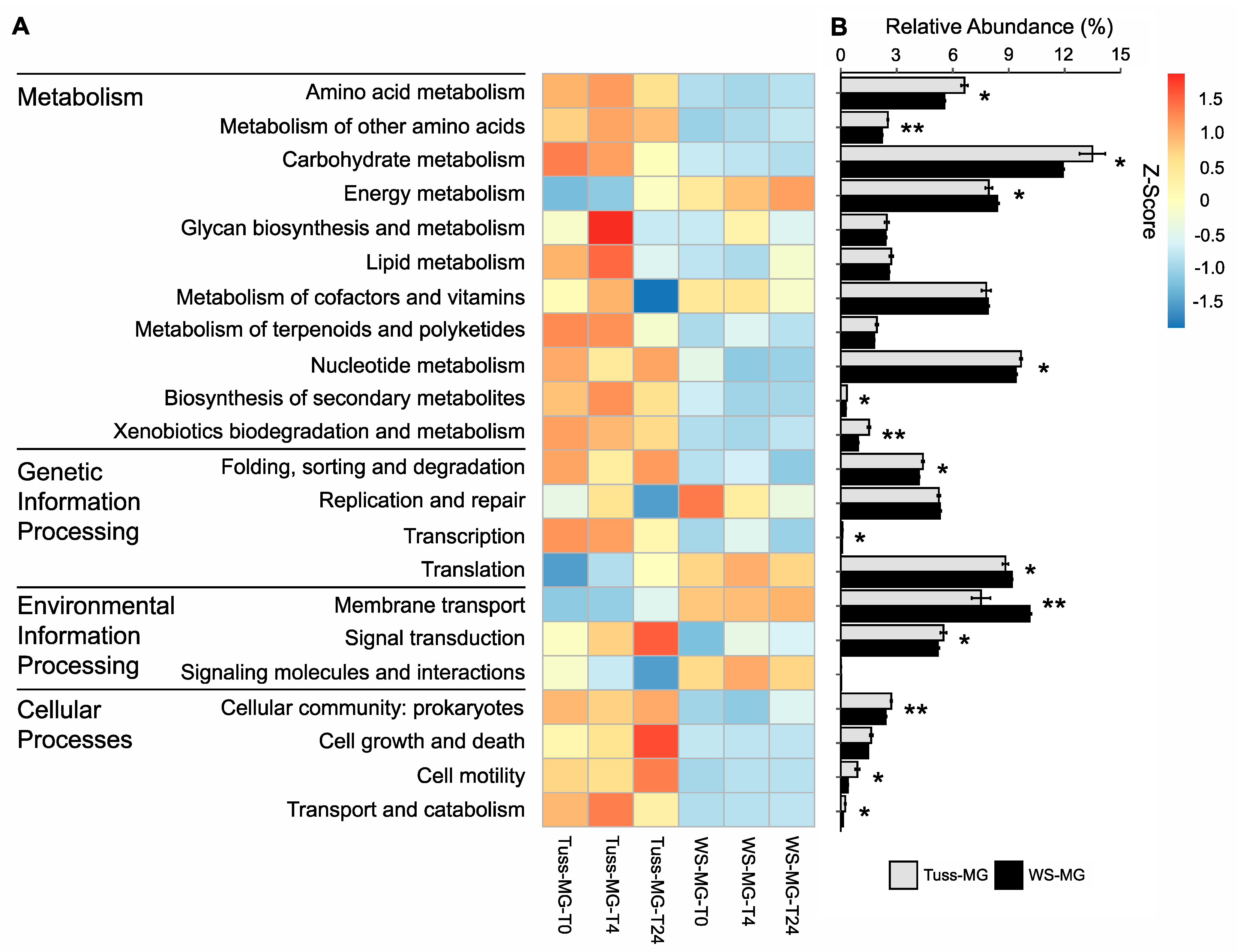
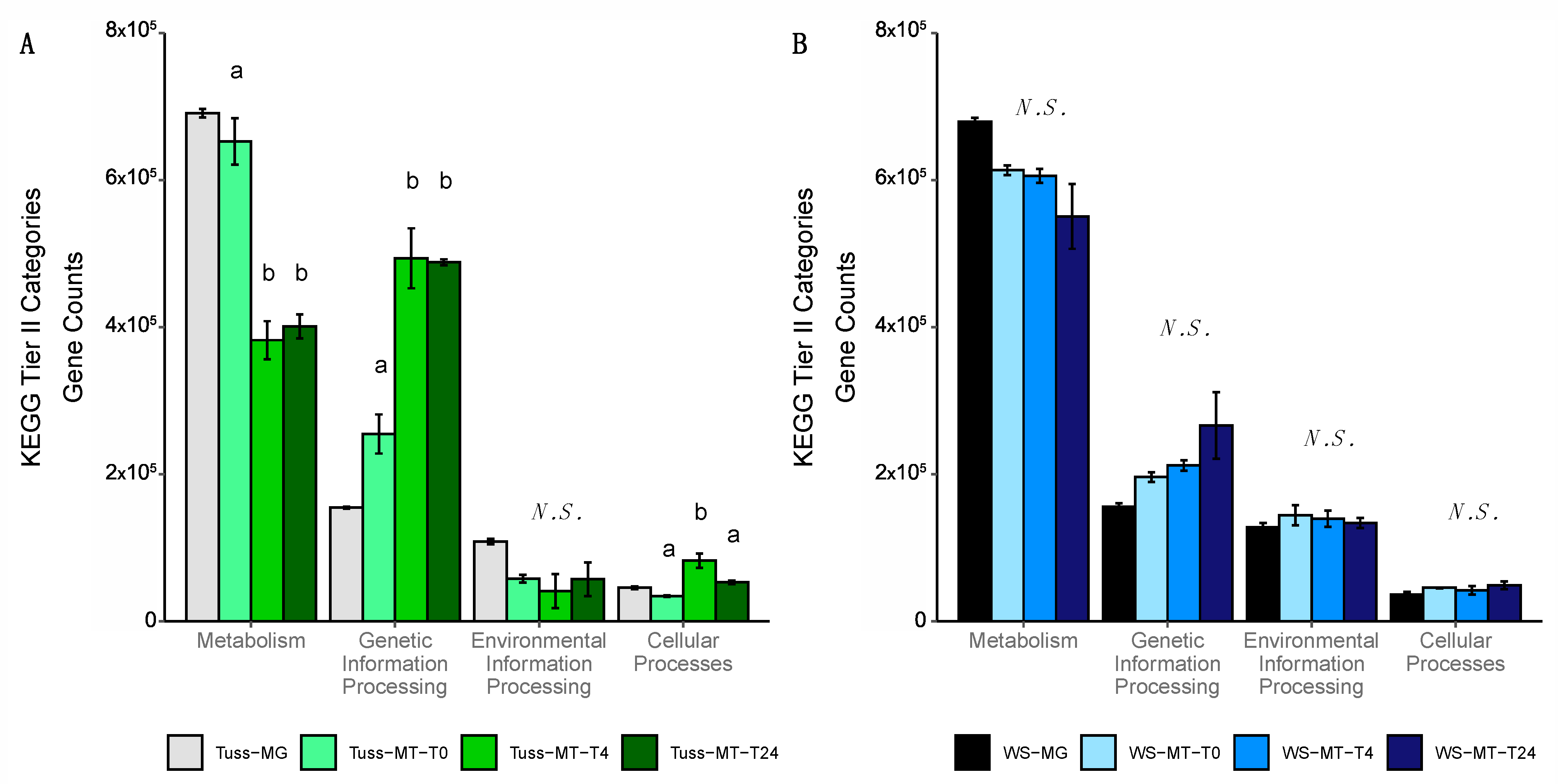
3.4. Genomic Potential and Functional Gene Diversity of the Microbial Communities
3.5. Microbial Community Expression in Response to Rainfall
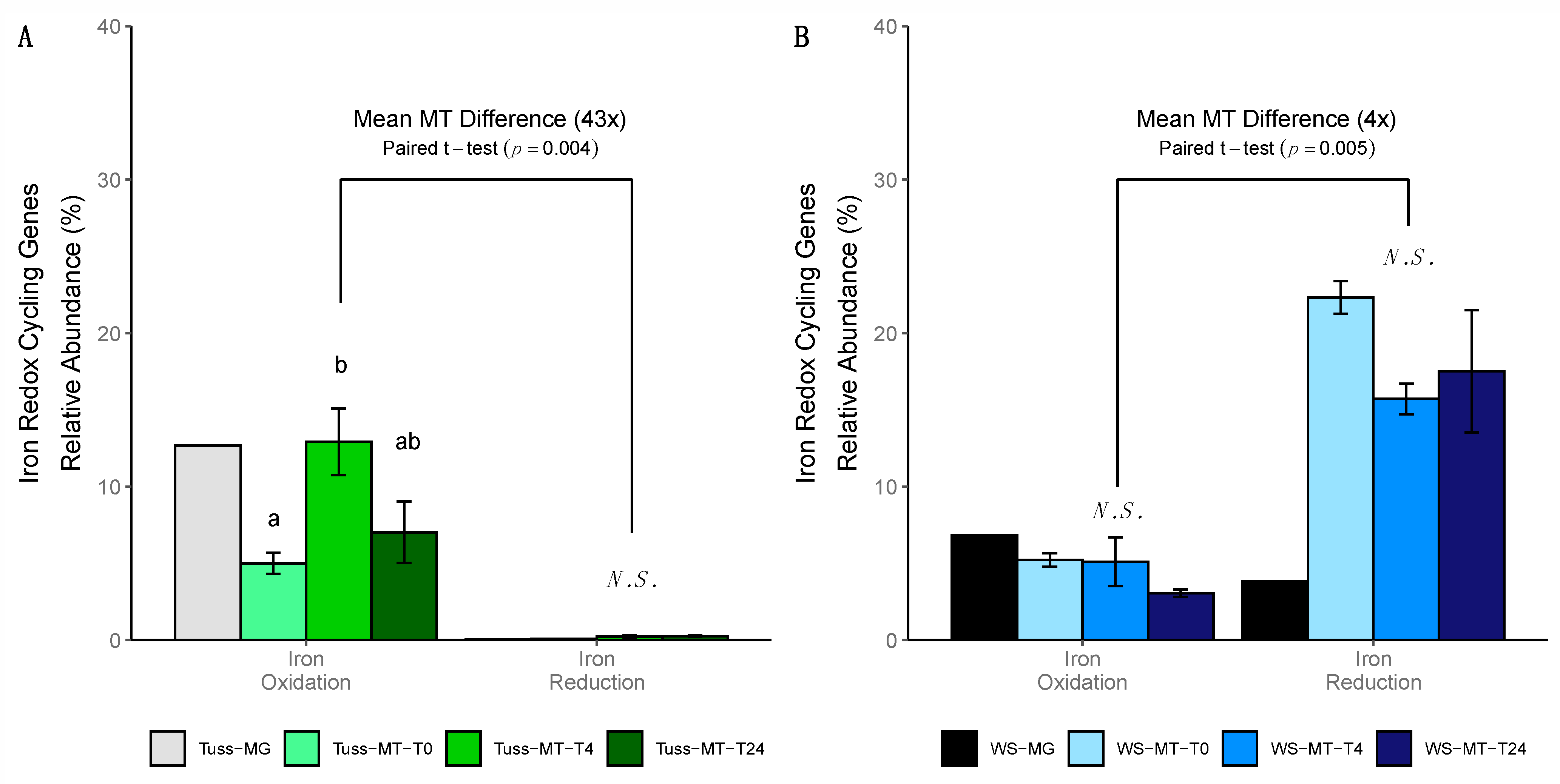
3.6. Iron Redox Cycling Gene Expression
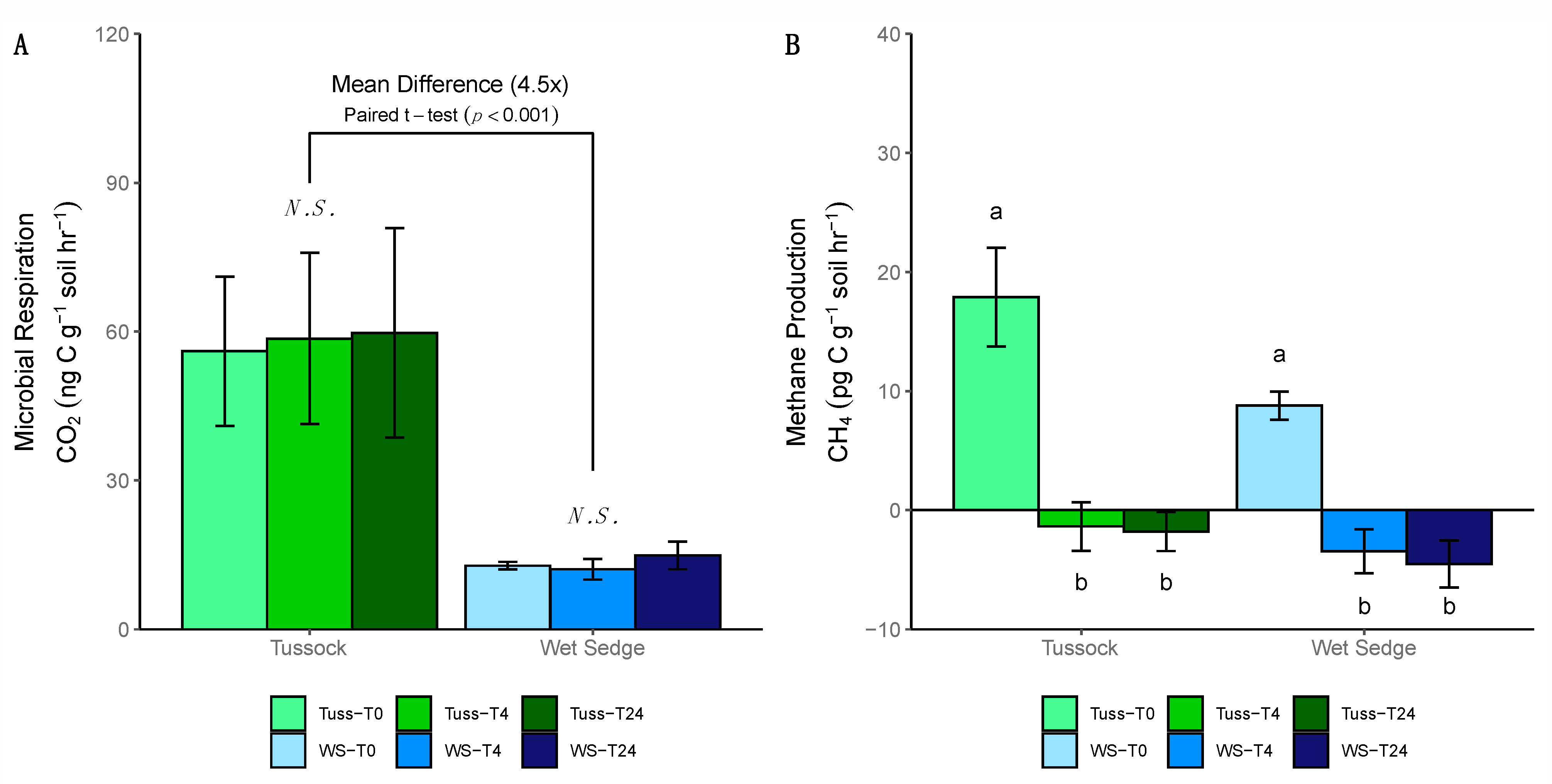
3.7. Soil Incubations
4. Discussion
4.1. Microbial Composition Differs among Tundra Communities
4.2. Genomic Potential Differs between Tundra Communities
4.3. Soil O2 Accumulation during Rainfall Differs among Tundra Soils
4.4. Tussock Tundra Community Responds to Rainfall-Induced Soil Oxidation
4.5. Wet Sedge Tundra Community Maintains Anaerobic Gene Expression Following Rainfall
4.6. Relating Microbial Expression to Rates of Respiration and CH4 Production
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hugelius, G.; Strauss, J.; Zubrzycki, S.; Harden, J.W.; Schuur, E.A.G.; Ping, C.L.; Schirrmeister, L.; Grosse, G.; Michaelson, G.J.; Koven, C.D.; et al. Estimated stocks of circumpolar permafrost carbon with quantified uncertainty ranges and identified data gaps. Biogeosciences 2014, 11, 6573–6593. [Google Scholar] [CrossRef]
- Gorham, E. Northern peatlands: Role in the carbon cycle and probable responses to climatic warming. Ecol. Appl. 1991, 1, 182–195. [Google Scholar] [CrossRef]
- Margesin, R. Permafrost soils. In Soil Biology; Margesin, R., Ed.; Springer: Berlin/Heidelberg, Germany, 2019; Volume 16. [Google Scholar]
- Zona, D. Long-term effects of permafrost thaw. Nature 2016, 537, 625–626. [Google Scholar] [CrossRef]
- Trusiak, A.; Treibergs, L.A.; Kling, G.W.; Cory, R.M. The controls of iron and oxygen on hydroxyl radical (OH) production in soils. Soil. Syst. 2019, 3, 1. [Google Scholar] [CrossRef]
- Herndon, E.M.; Kinsman-Costello, L.; Godsey, S. Biogeochemical cycling of redox-sensitive elements in permafrost-affected ecosystems. In Biogeochemical Cycles: Ecological Drivers and Environmental Impact, Geophysical Monograph; Dontsova, K., Balogh-Brunstad, Z., Le Roux, G., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2020. [Google Scholar]
- Bubier, J.L.; Moore, T.R.; Bellisario, L.; Comer, N.T.; Crill, P.M. Ecological controls on methane emissions from a northern peatland complex in the zone of discontinuous permafrost, Manitoba, Canada. Glob. Biogeochem. Cycles. 1995, 9, 455–470. [Google Scholar] [CrossRef]
- Turetsky, M.R.; Treat, C.C.; Waldrop, M.P.; Waddington, J.M.; Harden, J.W.; McGuire, A.D. Short-term response of methane fluxes and methanogen activity to water table and soil warming manipulations in an Alaskan peatland. J. Geophys. Res.-Biogeo. 2008, 113, G00A10. [Google Scholar] [CrossRef]
- Olefeldt, D.; Turetsky, M.R.; Crill, P.M.; McGuire, A.D. Environmental and physical controls on northern terrestrial methane emissions across permafrost zones. Glob. Chang. Biol. 2013, 19, 589–603. [Google Scholar] [CrossRef]
- Miller, P.C.; Kendall, R.; Oechel, W.C. Simulating carbon accumulation in northern ecosystems. Simulation 1983, 40, 119–131. [Google Scholar] [CrossRef]
- McGuire, A.D.; Anderson, L.G.; Christensen, T.R.; Dallimore, S.; Guo, L. Sensitivity of the carbon cycle in the arctic to climate change. Ecol. Monogr. 2009, 79, 523–555. [Google Scholar] [CrossRef]
- Oechel, W.C.; Hastings, S.J.; Vourlitis, G.; Jenkins, M.; Riechers, G.; Grulke, N. Recent change of Arctic tundra ecosystems from a net carbon dioxide sink to a source. Nature 1993, 361, 520–523. [Google Scholar] [CrossRef]
- Euskirchen, E.S.; Bret-Harte, M.S.; Shaver, G.R.; Edgar, C.W.; Romanovsky, V.E. Long-term release of carbon dioxide from arctic tundra ecosystems in Alaska. Ecosystems 2017, 20, 960–974. [Google Scholar] [CrossRef]
- Osterkamp, T.E.; Romanovsky, V.E. Evidence for warming and thawing of discontinuous permafrost in Alaska. Permafr. Periglac. 1999, 10, 17–37. [Google Scholar] [CrossRef]
- Brown, J.G.; Hinkel, K.M.; Nelson, F.E. The circumpolar active layer monitoring (CALM) program: Research designs and initial results. Polar. Geogr. 2000, 24, 165–258. [Google Scholar] [CrossRef]
- Jorgenson, M.T.; Shur, Y.L.; Pullman, E.R. Abrupt increase in permafrost degradation in Arctic Alaska. Geophys. Res. Lett. 2006, 33, D22103. [Google Scholar] [CrossRef]
- Kumar, M.; Want, R.; Link, T.E. Effects of more extreme precipitation regimes on maximum seasonal snow water equivalent. Geophys. Res. Lett. 2012, 39, 1–6. [Google Scholar] [CrossRef]
- Spence, C.; Phillips, R.W. Refining understanding of hydrological connectivity in a boreal catchment. Hydrol. Process. 2015, 29, 3491–3505. [Google Scholar] [CrossRef]
- Bintanja, R.; Andry, O. Towards a rain-dominated Arctic. Nat. Clim. Chang. 2017, 7, 263–268. [Google Scholar] [CrossRef]
- Douglas, T.A.; Turetsky, M.R.; Koven, C.D. Increased rainfall stimulates permafrost thaw across a variety of interior Alaskan boreal ecosystems. Npj. Clim. Atmos. Sci. 2020, 28, 1–7. [Google Scholar] [CrossRef]
- Neumann, R.B.; Moorber, C.J.; Lundquist, J.D.; Turner, J.C.; Waldrop, M.P.; McFarland, J.W.; Euskirchen, E.S.; Edgar, C.W.; Turetsky, M.R. Warming effects of spring rainfall increase methane emissions from thawing permafrost. Geophys. Res. Lett. 2019, 46, 1393–1401. [Google Scholar] [CrossRef]
- Schuur, E.A.G.; Vogel, J.G.; Crummer, K.G.; Lee, H.; Sickman, J.O.; Osterkamp, T.E. The effect of permafrost thaw on old carbon release and net carbon exchange from tundra. Nature 2009, 459, 556–559. [Google Scholar] [CrossRef]
- Grosse, G.; Romanovsky, V.; Jorgenson, T.; Anthony, K.W.; Brown, J.; Overduin, P.P. Vulnerability and feedbacks of permafrost to climate change. Eos. Trans. Am. Geophys. Union. 2011, 92, 73–74. [Google Scholar] [CrossRef]
- Mackelprang, R.; Waldrop, M.P.; DeAngelis, K.M.; David, M.M.; Chavarria, K.L.; Blazewicz, S.J.; Rubin, E.M.; Jansson, J.K. Metagenomic analysis of a permafrost microbial community reveals a rapid response to thaw. Nature 2011, 480, 368–371. [Google Scholar] [CrossRef]
- Xue, K.; Yuan, M.M.; Shi, Z.J.; Qin, Y.; Deng, Y.; Cheng, L.; Wu, L.; He, Z.; Van Nostrand, J.D.; Bracho, R.; et al. Tundra soil carbon is vulnerable to rapid microbial decomposition under climate change. Nat. Clim. Chang. 2016, 6, 595–600. [Google Scholar] [CrossRef]
- Wickland, K.P.; Striegl, R.G.; Neff, J.C.; Sachs, T. Effects of permafrost melting on CO2 and CH4 exchange of a poorly drained black spruce lowland. J. Geophys. Res. Biogeo. 2006, 111, G2. [Google Scholar] [CrossRef]
- Merbold, L.; Kutsch, W.L.; Corradi, C.; Kolle, O.; Rebmann, C.; Stoy, P.C.; Schulze, E.D. Artificial drainage and associated carbon fluxes (CO2/CH4) in a tundra ecosystem. Glob. Chang. Biol. 2009, 15, 2599–2614. [Google Scholar] [CrossRef]
- Huemmrich, K.F.; Kinoshita, G.; Gamon, J.A.; Houston, S.; Kwon, H.; Oechel, W.C. Tundra carbon balance under varying temperature and moisture regimes. J. Geophys. Res. 2010, 115, 1–8. [Google Scholar] [CrossRef]
- Zona, D.; Lipson, D.A.; Paw, U.K.T.; Oberbauer, S.F.; Olivas, P.; Gioli, B.; Oechel, W.C. Increased CO2 loss from vegetated drained lake tundra ecosystems due to flooding. Glob. Biogeochem. Cycles 2012, 26, 1–16. [Google Scholar] [CrossRef]
- Elberling, B.; Michelsen, A.; Schadel, C.; Schuur, E.A.G.; Christiansen, H.H.; Berg, L.; Tamstorf, M.P.; Sigsgaard, C. Long-term CO2 production following permafrost thaw. Nat. Clim. Chang. 2013, 3, 890–894. [Google Scholar] [CrossRef]
- Lawrence, D.M.; Koven, C.D.; Swenson, S.C.; Riley, W.J.; Slater, A.G. Permafrost thaw and resulting soil moisture changes regulate projected high-latitude CO2 and CH4 emissions. Environ. Res. Lett. 2015, 10, 094011. [Google Scholar] [CrossRef]
- Lipson, D.A.; Haggerty, J.M.; Srinivas, A.; Raab, T.K.; Sathe, S.; Dinsdale, E.A. Metagenomic insights into anaerobic metabolism along an arctic peat soil profile. PLoS ONE 2013, 8, e64659. [Google Scholar] [CrossRef]
- Ivanova, A.A.; Wegner, C.E.; Kim, Y.; Liesack, W.; Dedysh, S.N. Identification of microbial populations driving biopolymer degradation in acidic peatlands by metatranscriptomic analysis. Mol. Ecol. 2016, 25, 4818–4835. [Google Scholar] [CrossRef]
- Kolb, S. Aerobic methanol-oxidizing bacteria in soil. Fems. Microbiol. Lett. 2009, 300, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.D.; Walker, D.A.; Auerback, N.A. Plant communities of a tussock tundra landscape in the Brooks Range foothills, Alaska. J. Veg. Sci. 1994, 5, 843–866. [Google Scholar] [CrossRef]
- Judd, K.E.; Kling, G.W. Production and export of dissolved C in arctic tundra mesocosms: The roles of vegetation and water flow. Biogeochemistry 2002, 60, 213–234. [Google Scholar] [CrossRef]
- Judd, K.E.; Crump, B.C.; Kling, G.W. Environmental drivers control eco- system function in bacteria through changes in community composition. Ecology 2006, 87, 2068–2079. [Google Scholar] [CrossRef]
- Zak, D.R.; Kling, G.W. Microbial community composition and function across an arctic tundra landscape. Ecology 2006, 87, 1659–1670. [Google Scholar] [CrossRef]
- Hobbie, J.E. A Changing Arctic: Ecological Consequences for Tundra, Streams, and Lakes; Kling, G.W., Ed.; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Ward, C.P.; Cory, R.M. Chemical composition of dissolved organic matter draining permafrost soils. Geochim. Cosmochim. Acta 2015, 167, 63–79. [Google Scholar] [CrossRef]
- Tas, N.; Prestat, E.; Wang, S.; Wu, Y.; Ulrich, C.; Kneafsey, T.; Tringe, S.G.; Torn, M.S.; Hubbard, S.S.; Jansson, J.K. Landscape topography structures the soil microbiome in arctic polygonal tundra. Nat. Commun. 2018, 9, 777. [Google Scholar] [CrossRef]
- Deng, J.; Gu, Y.; Zhang, J.; Xue, K.; Qin, Y.; Yuan, M.; Yin, H.; He, Z.; Wu, L.; Schuur, E.A.G.; et al. Shifts of tundra bacterial and archaeal communities along a permafrost thaw gradient in Alaska. Mol. Ecol. 2015, 24, 222–234. [Google Scholar] [CrossRef]
- Hultman, J.; Waldrop, M.P.; Mackelprang, R.; David, M.M.; McFarland, J.; Blazewicz, S.J.; Harden, J.; Turetsky, M.R.; McGuire, A.D.; Shah, M.B.; et al. Multi-omics of permafrost, active layer and thermokarst bog soil microbiomes. Nature 2015, 521, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, B.M.; Kim, M.; Kim, Y.; Byun, E.; Yang, J.W.; Ahn, J.; Lee, Y.K. Variations in bacterial and archaeal communities along depth profiles of Alaskan soil cores. Sci. Rep. 2018, 8, 504. [Google Scholar] [CrossRef]
- McMahon, S.K.; Wallenstein, M.D.; Schimel, J.P. A cross-seasonal comparison of active and total bacterial community composition in arctic tundra soil using bromodeoxyuridine labeling. Soil. Biol. Biochem. 2011, 43, 287–295. [Google Scholar] [CrossRef]
- Jansson, J.K.; Hofmockel, K.S. The soil microbiome—From metagenomics to metaphenomics. Curr. Opin. Microbiol. 2018, 43, 162–168. [Google Scholar] [CrossRef]
- Page, S.E.; Logan, J.R.; Cory, R.M.; McNeill, K. Evidence for dissolved organic matter as the primary source and sink of photochemically produced hydroxyl radical in arctic surface waters. Environ. Sci. Proc. Impacts 2014, 16, 807–822. [Google Scholar] [CrossRef] [PubMed]
- Trusiak, A.; Treibergs, L.A.; Kling, G.W.; Cory, R.M. The role of iron and reactive oxygen species in the production of CO2 in arctic soil water. Geochim. Cosmochim. Acta 2018, 224, 80–95. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic. Acids. Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahe, F. VSEARCH: A versatile open source tool for metagenomics. Peer. J. 2016, 4, e2584. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microb. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap Short-Read. Aligner and Other Bioinformatics Tools. 2015. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 10 March 2021).
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie. Nat. Methods 2012, 9, 357–359. [Google Scholar]
- Eren, A.M.; Esen, O.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ‘omics data. Peer. J. 2015, 3, e1319. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. Bmc. Bioinform. 2010, 11, 1–11. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Campbell, J.H.; O’Donoghue, P.; Alisha, G.C.; Schwientek, P.; Sczyrba, A.; Woyke, T.; Söll, D.; Podar, M. UGA is an additional glycine codon in unculutured SR1 bacteria from the human microbiota. Proc. Natl. Acad. Sci. USA 2013, 110, 5540–5545. [Google Scholar] [CrossRef]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A. Insights into phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Wagner, G.P.; Kin, K.; Lynch, V.J. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory. Biosci. 2012, 131, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Alneberg, J.; Bjarnason, B.S.; De Bruijn, I.; Schirmer, M.; Quick, J.; Ijaz, U.Z.; Lahti, L.; Loman, N.J.; Andersson, A.F.; Quince, C. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. Peer. J. 2015, 3, e1165. [Google Scholar] [CrossRef]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Chaumeil, P.A.; Rinke, C.; Mussig, A.J.; Hugenholtz, P. A complete domain-to-species taxonomy for Bacteria and Archaea. Nat. Biotechnol. 2020, 38, 1079–1086. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R Package version 2.5–5. 2019. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 10 March 2021).
- R Core Development Team. R: A Language and Environment for Statistical Computing; R Core Development Team: Vienna, Austria, 2017. [Google Scholar]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Kolde, R.; Kolde, M.R. Pheatmap: Pretty Heatmaps. 2015. Available online: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed on 10 March 2021).
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome. Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kieft, B.; Li, Z.; Bryson, S.; Crump, B.C.; Hettich, R.; Pan, C.; Mayali, X.; Mueller, R.S. Microbial community structure-function relationships in Yaquina Bay estuary reveal spatially distinct carbon and nitrogen cycling capacities. Front. Microbiol. 2018, 9, 1282. [Google Scholar] [CrossRef]
- Wu, H.; Moore, E. Association analysis of the general environmental conditions and prokaryotes’ gene distributions in various functional groups. Genomics 2010, 96, 27–38. [Google Scholar] [CrossRef]
- Garber, A.I.; Nealson, K.H.; Okamoto, A.; McAllister, S.M.; Chan, C.S.; Barco, R.A.; Merino, N. FeGenie: A comprehensive Tool for the Identification of Iron Genes and Iron Gene Neighborhoods in Genome and Metagenome Assemblies. Front. Microbiol. 2020, 11, 37. [Google Scholar] [CrossRef]
- Giblin, A.E.; Nadelhoffer, K.J.; Shaver, G.R.; Laundre, J.A.; McKerrow, A.J. Biogeochemical diversity along a river toposequence in arctic Alaska. Ecol. Monogr. 1991, 61, 415–435. [Google Scholar] [CrossRef]
- Nadelhoffer, K.J.; Giblin, A.E.; Shaver, G.R.; Laundre, J. Effects of temperature and organic matter quality on element mineralization in six arctic soils. Ecology 1991, 72, 242–253. [Google Scholar] [CrossRef]
- Gebauer, R.L.E.; Tenhunen, J.D.; Reynolds, J.F. Soil aeration in relation to soil physical properties, nitrogen availability and root characteristics within an arctic watershed. Plant Soil 1996, 178, 37–48. [Google Scholar] [CrossRef]
- Tveit, A.T.; Schwacke, R.; Svenning, M.M.; Urich, T. Organic carbon transformations in high-Arctic peat soils: Key functions and microorganisms. ISME J. 2013, 7, 299–311. [Google Scholar] [CrossRef]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community structure at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Rousk, J.; Baath, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef]
- Chapin, F.S., III; Fetcher, N.; Kielland, K.; Everett, K.R.; Linkins, A.E. Productivity and nutrient cycling of Alaskan tundra: Enhancement with flowing soil water. Ecology 1988, 69, 693–702. [Google Scholar] [CrossRef]
- Sistla, S.A.; Asao, S.; Schimel, J.P. Detecting microbial N-limitation in tussock tundra soil: Implications for Arctic soil organic carbon cycling. Soil. Biol. Biochem. 2012, 55, 78–84. [Google Scholar] [CrossRef]
- Lauro, F.M.; McDougald, D.; Thomas, T.; Williams, T.J.; Egan, S.; Rice, S.; DeMaere, M.Z.; Ting, L.; Ertan, H.; Johnson, J. The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 15527–15533. [Google Scholar] [CrossRef]
- Emerson, D.; Scott, J.J.; Benes, J.; Bowden, W.B. Microbial iron oxidation in the Arctic tundra and its implications for biogeochemical cycling. Appl. Environ. Microbiol. 2015, 81, 8066–8075. [Google Scholar] [CrossRef]
- Kim, H.M.; Lee, M.J.; Jung, J.Y.; Hwang, C.Y.; Kim, M.; Ro, H.M.; Chun, J.; Lee, Y.K. Vertical distribution of bacterial community is associated with the degree of soil organic matter decomposition in the active layer of moist acidic tundra. J. Microbiol. 2016, 54, 713–723. [Google Scholar] [CrossRef]
- Conrad, R.; Schutz, H.; Babbel, M. Temperature limitation on hydrogen turnover and methanogenesis in anoxic paddy soil. Fems. Microbiol. Ecol. 1987, 3, 281–289. [Google Scholar] [CrossRef]
- Metje, M.; Frenzel, P. Methanogenesis and methanogenic pathways in a peat from subarctic permafrost. Environ. Microbiol. 2007, 9, 954–964. [Google Scholar] [CrossRef]
- Tveit, A.T.; Urich, T.; Frenzei, P.; Svenning, M.M. Metabolic and trophic interactions modulate methane production by arctic peat microbiota in response to warming. Proc. Natl. Acad. Sci. USA 2015, 112, E2507–E2516. [Google Scholar] [CrossRef] [PubMed]
- Malard, L.A.; Pearce, D.A. Microbial diversity and biogeography in Arctic soils. Environ. Microbiol. Rep. 2018, 10, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Alexander, M. Introduction to Soil Microbiology, 2nd ed.; John Wiley and Sons: New York, NY, USA, 1976. [Google Scholar]
- Hobbie, J.E.; Hobbie, E.A.; Drossman, H.; Conte, M.; Weber, J.C.; Shamhart, J.; Weinrobe, M. Mycorrhizal fungi supply nitrogen to host plants in Arctic tundra and boreal forests: 15N is the key signal. Can. J. Microbiol. 2009, 55, 84–94. [Google Scholar] [CrossRef]
- Boberg, J.B.; Ihrmark, K.; Lindahl, B.D. Decomposing capacity of fungi commonly detected in Pinus sylvestris needle litter. Fungal. Ecol. 2011, 4, 110–114. [Google Scholar] [CrossRef]
- Crump, B.C.; Amaral-Zettler, L.A.; Kling, G.W. Microbial diversity in arctic freshwaters is structured by inoculation of microbes from soils. ISME J. 2012, 6, 1629–1639. [Google Scholar] [CrossRef]
- Moldrup, P.; Olesen, T.; Gamst, J.; Schjonning, P.; Yamaguchi, T.; Rolston, D. Predicting the gas diffusion coefficient in repacked soil: Water-induced linear reduction model. Soil. Sci. Soc. Am. J. 2000, 64, 1588–1594. [Google Scholar] [CrossRef]
- Thorbjorn, A.; Moldrup, P.; Blendstrup, H.; Komatsu, T.; Rolston, D.E. A gas diffusivity model based on air-, solid-, and water-phase resistance in variably saturated soil. Vadose. Zone. J. 2008, 7, 1276–1286. [Google Scholar] [CrossRef]
- Armstrong, W. Oxygen diffusion from the roots of some British bog plants. Nature 1964, 204, 801–802. [Google Scholar] [CrossRef]
- King, J.Y.; Reeburgh, W.S.; Regli, S.K. Methane emission and transport by arctic sedges in Alaska: Results of a vegetation removal experiment. J. Geophys. Res. Atmos. 1998, 103, 29083–29092. [Google Scholar] [CrossRef]
- Stieglitz, M.; Shaman, J.; McNamara, J.; Engel, V.; Shanley, J.; Kling, G.W. An approach to understanding hydrologic connectivity on the hillslope and the implications for nutrient transport. Glob. Biogeochem. Cycles. 2003, 17, 1105. [Google Scholar] [CrossRef]
- O’Connor, M.T.; Cardenas, M.B.; Ferencz, S.B.; Wu, Y.; Neilson, B.T.; Chen, J.; Kling, G.W. Empirical models for predicting water and heat flow properties of permafrost soils. Geophys. Res. Lett. 2020, 47, 1–10. [Google Scholar] [CrossRef]
- Neilson, B.T.; Cardenas, M.B.; O’Connor, M.T.; Rasmussen, M.T.; King, T.V.; Kling, G.W. Groundwater flow and exchange across the land surface explain carbon export patterns in continuous permafrost watersheds. Geophys. Res. Lett. 2018, 45, 7596–7605. [Google Scholar] [CrossRef]
- Nalven, S.G.; Ward, C.P.; Payet, J.P.; Cory, R.M.; Kling, G.W.; Sharpton, T.J.; Sullivan, C.M.; Crump, B.C. Experimental metatranscriptomics reveals the costs and benefits of dissolved organic matter photo-alteration for freshwater microbes. Environ. Microbiol. 2020, 22, 3505–3521. [Google Scholar] [CrossRef] [PubMed]
- McKnight, D.M.; Boyer, E.W.; Westerhoff, P.K.; Doran, P.T.; Kulbe, T.; Andersen, D.T. Spectrofluorometric characterization of dissolved organic matter for indication of precursor organic material and aromaticity. Limnol. Oceanogr. 2001, 46, 38–48. [Google Scholar] [CrossRef]
- Shen, L.; Preiss, J. Biosynthesis of bacterial glycogen: I. Purification and properties of the adenosine diphosphoglucose pyrophosphorylase of Arthrobacter species NRRLB1973. J. Biol. Chem. 1965, 240, 2334–2340. [Google Scholar] [CrossRef]
- Sekar, K.; Linker, S.M.; Nguyen, J.; Grunhagen, A.; Stocker, R.; Sauer, U. Bacterial glycogen provides short-term benefits in changing environments. Appl. Environ. Microb. 2020, 86, 9. [Google Scholar] [CrossRef]
- Lennon, J.T. Microbial life deep underfoot. Mbio 2020, 11, e03201–e03219. [Google Scholar] [CrossRef]
- Jia, X.; Xi, B.; Li, M.; Liu, D.; Hou, J.; Hao, Y.; Meng, F. Metaproteomic analysis of the relationship between microbial community phylogeny, function and metabolic activity during biohydrogen-methane coproduction under short-term hydrothermal pretreatment from food waste. Bioresour. Technol. 2017, 245, 1030–1039. [Google Scholar] [CrossRef] [PubMed]
- Veith, N.; Feldman-Salit, A.; Cojocaru, V.; Henrich, S.; Kummer, U.; Wade, R.C. Organism-adapted specificity of the allosteric regulation of pyruvate kinase in lactic acid bacteria. Plos. Comput. Biol. 2013, 9, e1003159. [Google Scholar] [CrossRef]
- Marchitti, S.A.; Brocker, C.; Stagos, D.; Vasiliou, V. Non-P450 aldehyde oxidizing enzymes: The aldehyde dehydrogenase superfamily. Expert. Opin. Drug. Met. 2008, 4, 697–720. [Google Scholar] [CrossRef]
- Nosova, T.; Jokelainen, K.; Kaihovaara, P.; Jousimies-Somer, H.; Siitonen, A.; Heine, R.; Salaspuro, M. Aldehyde dehydrogenase activity and acetate production by aerobic bacteria representing the normal flora of human large intestine. Alcohol Alcohol. 1996, 31, 555–564. [Google Scholar] [CrossRef]
- Weber, K.A.; Achenbach, L.A.; Coates, J.D. Microorganisms pumping iron: Anaerobic microbial iron oxidation and reduction. Nat. Rev. Microbiol. 2006, 4, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Shi, W.; Li, X.; Hu, Y.; Yu, F.; Ma, H. Ferroptosis accompanied by OH generation and cytoplasmic viscosity increase revealed via dual-functioning fluorescence probe. J. Am. Chem. Soc. 2019, 141, 18301–18307. [Google Scholar] [CrossRef] [PubMed]
- Conrad, M.; Kagan, V.E.; Bayir, H.; Pagnussat, G.C.; Head, B.; Traber, M.G.; Stockwell, B.R. Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev. 2018, 32, 602–619. [Google Scholar] [CrossRef] [PubMed]
- Ratledge, C. (Ed.) Biochemistry of Microbial Degradation; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1994. [Google Scholar]
- Brune, A.; Frenzel, P.; Cypionka, H. Life at the oxic-anoxic interface: Microbial activities and adaptations. FEMS. Microbiol. Rev. 2000, 24, 691–710. [Google Scholar] [CrossRef]
- Emerson, D.; Revsbech, N. Investigation of an iron-oxidizing microbial mat community located near Aarhus, Denmark: Laboratory studies. Appl. Environ. Microbiol. 1994, 60, 4032–4038. [Google Scholar] [CrossRef]
- Doherty, S.J.; Barbato, R.A.; Grandy, A.S.; Thomas, W.K.; Monteux, S.; Dorrepaal, E.; Johansson, M.; Ernakovich, J.G. The transition from stochastic to deterministic bacterial community assembly during permafrost thaw succession. Front. Microbiol. 2020, 11, 596589. [Google Scholar] [CrossRef] [PubMed]
- Schadel, C.; Bader, M.K.F.; Schuur, E.A.; Biasi, C.; Bracho, R.; Capek, P.; De Baets, S.; Diakova, K.; Ernakovich, J.; Estop-Aragones, C.; et al. Potential carbon emissions dominated by carbon dioxide from thawed permafrost soils. Nat. Clim. Chang. 2016, 6, 950–953. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil Properties | Tundra Soil | Mean Difference between Tundra | |
|---|---|---|---|
| Tussock | Wet Sedge | ||
| Chemical Properties | |||
| pH | 4.58 ± 0.39 | 6.12 ± 0.06 | 1.54 * |
| Conductivity (µS cm−1) | 65.9 ± 18.6 | 123.6 ± 14.4 | 57.7 * |
| Physical Properties | |||
| Temperature (°C) | 7.8 ± 0.3 | 8.5 ± 0.3 | 0.7 NS |
| Moisture (% water) | 63.1 ± 7.6 | 70.1 ± 4.2 | 7.0 NS |
| Dissolved [O2] (mg O2 L−1) | 1.21 ± 0.23 | 1.04 ± 0.05 | 0.17 NS |
| Bulk Soil Properties | |||
| Organic Matter Content (%) | 67.6 ± 8.6 | 80.7 ± 4.5 | 13.1 * |
| Organic Carbon Content (%) | 33.8 ± 4.3 | 40.3 ± 2.3 | 6.5 * |
| Bulk density (g soil dry cm−3) | 0.53 ± 0.10 | 0.27 ± 0.04 | 0.26 * |
| Porosity (% soil volume) | 63.3 ± 6.7 | 81.2 ± 2.6 | 17.9 * |
| Soil Modeling Variables | Tundra Soil | Mean Difference between Tundra | |
|---|---|---|---|
| Tussock | Wet Sedge | ||
| Flush Variables | |||
| DI Water Flushed (L) | 20 ± 0 | 20 ± 0 | 0 NS |
| Duration of Flush (hr) | 3.77 ± 0.42 | 1.13 ± 0.19 | 2.64 * |
| Soil O2 Accumulation and Consumption Rates | |||
| Soil O2 Accumulation (Frain; mg O2 L−1 h−1) | 2.31 ± 0.95 | 2.61 ± 0.99 | 0.30 NS |
| Soil O2 Consumption (Sall; mg O2 L−1 h−1) | 0.04 ± 0.01 | 0.02 ± 0.01 | 0.02 * |
| Soil O2 Concentrations after Simulated Rainfall | |||
| Soil O2 Concentration after Flush (Mf; mg O2 L−1) | 10.26 ± 1.11 | 4.10 ± 0.53 | 6.16 * |
| Soil O2 Concentration at T4 (Mf+4; mg O2 L−1) | 10.09 ± 1.11 | 4.01 ± 0.53 | 6.08 * |
| Soil O2 Concentration at T24 (Mf+24; mg O2 L−1) | 9.20 ± 1.11 | 3.57 ± 0.53 | 5.63 * |
| Taxonomy | MG Rel. Abund. (%) | MG Mean Difference | MT Rel. Abund. (%) | MT Mean Difference | ||
|---|---|---|---|---|---|---|
| Tussock | Wet Sedge | Tussock | Wet Sedge | |||
| Bacteria | ||||||
| Acidobacteria | 34 ± 4.5 | 10 ± 0.3 | 24% * | 41 ± 12 | 8.2 ± 0.8 | 33% ** |
| Actinobacteria | 24 ± 5.7 | 27 ± 1.9 | 3.0% NS | 42 ± 12 | 40 ± 3.0 | 2.0% NS |
| Alphaproteobacteria | 15 ± 3.6 | 8.3 ± 0.6 | 6.7% NS | 4.5 ± 1.6 | 3.4 ± 1.5 | 1.1% ** |
| Betaproteobacteria | 2.0 ± 0.8 | 6.4 ± 0.7 | 4.4% ** | 1.0 ± 0.5 | 3.5 ± 1.3 | 2.5% ** |
| Deltaproteobacteria | 4.1 ± 0.4 | 13 ± 0.4 | 8.9% ** | 2.9 ± 0.8 | 13 ± 1.3 | 10% *** |
| Gammaproteobacteria | 11 ± 0.3 | 1.7 ± 0.2 | 9.3% *** | 3.5 ± 1.0 | 1.3 ± 0.5 | 2.2% ** |
| Bacteroidetes | 0.4 ± 0.0 | 1.9 ± 0.7 | 1.5% NS | 0.7 ± 0.1 | 1.7 ± 0.6 | 1.0% * |
| Chloroflexi | 0.2 ± 0.1 | 8.3 ± 0.3 | 8.1% *** | 0.2 ± 0.0 | 8.3 ± 1.8 | 8.1% *** |
| Firmicutes | 3.5 ± 0.5 | 15 ± 1.6 | 12% ** | 1.7 ± 0.2 | 10 ± 1.5 | 8.3% *** |
| Planctomycetes | 1.0 ± 0.2 | 0 ± 0 | 1.0% NS | 0 ± 0 | 0 ± 0 | 0.0% NS |
| Verrucomicrobia | 2.1 ± 0.6 | 2.1 ± 0.7 | 0.0% NS | 0 ± 0 | 1.2 ± 0.1 | 1.2% NS |
| Archaea | ||||||
| Euryarchaeota | 0 ± 0 | 1.7 ± 0.3 | 1.7% * | 0 ± 0 | 6.5 ± 6.3 | 6.5% * |
| Fungi | ||||||
| Ascomycetes | 1.1 ± 0.2 | 0 ± 0 | 1.1% * | 1.5 ± 0.5 | 0 ± 0 | 1.5% * |
| KEGG Tier Category | logFC |
|---|---|
| Carbohydrate Metabolism | |
| Glycolysis/Gluconeogenesis | |
| K00128 aldehyde dehydrogenase (NAD+) | 3.7 ± 0.0 |
| Pyruvate Metabolism | |
| K00656 formate C-acetyltransferase | −4.4 ± 1.8 |
| Starch and Sucrose Metabolism | |
| K00975 glucose-1-phosphate adenylyltransferase | −2.5 ± 0.0 |
| Cofactors and Vitamins Metabolism | |
| Nicotinate and Nicotinamide Metabolism | |
| K00763 nicotinate phosphoribosyltransferase | −3.5 ± 0.0 |
| Overview Metabolism | |
| Biosynthesis of Amino Acids | |
| K01915 glutamine synthetase | −3.2 ± 0.6 |
| Carbon Metabolism | |
| K00873 pyruvate kinase | −3.6 ± 0.0 |
| K01623 fructose-bisphosphate aldolase | −4.0 ± 0.0 |
| K00626 acetyl-CoA C-acetyltransferase | −2.6 ± 0.2 |
| Energy Metabolism | |
| Anaerobic Carbon Fixation | |
| K00174 2-oxoglutarate/2-oxoacid ferredoxin oxidoreductase | −3.9 ± 0.4 |
| K00855 phosphoribulokinase | −3.8 ± 0.0 |
| Methane Metabolism | |
| K00625 phosphate acetyltransferase | −5.3 ± 0.6 |
| K00925 acetate kinase | −3.0 ± 0.2 |
| K01895 acetyl-CoA synthetase | −3.7 ± 1.3 |
| Fermentation | |
| K00169 pyruvate ferredoxin oxidoreductase | −3.1 ± 0.0 |
| DGE Analysis | Unique KEGG Orthologs (KOs) | Significant Differences | ||
|---|---|---|---|---|
| Higher | Lower | Not Significant | ||
| Tussock vs. Wet Sedge | ||||
| T0 | 27,239 | 12,732 | 126,783 | 24% |
| T4 | 32,366 | 13,512 | 120,876 | 28% |
| T24 | 12,123 | 4774 | 149,857 | 10% |
| Tussock Tundra | ||||
| T4 vs. T0 | 667 | 588 | 100,055 | 1.3% |
| T24 vs. T0 | 75 | 67 | 101,168 | <0.1% |
| T24 vs. T4 | 2 | 5 | 101,303 | <0.01% |
| Wet Sedge Tundra | ||||
| T4 vs. T0 | 78 | 13 | 139,939 | <0.01% |
| T24 vs. T0 | 9 | 2 | 140,019 | <0.01% |
| T24 vs. T4 | 0 | 0 | 140,030 | 0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanowicz, K.J.; Crump, B.C.; Kling, G.W. Rainfall Alters Permafrost Soil Redox Conditions, but Meta-Omics Show Divergent Microbial Community Responses by Tundra Type in the Arctic. Soil Syst. 2021, 5, 17. https://doi.org/10.3390/soilsystems5010017
Romanowicz KJ, Crump BC, Kling GW. Rainfall Alters Permafrost Soil Redox Conditions, but Meta-Omics Show Divergent Microbial Community Responses by Tundra Type in the Arctic. Soil Systems. 2021; 5(1):17. https://doi.org/10.3390/soilsystems5010017
Chicago/Turabian StyleRomanowicz, Karl J., Byron C. Crump, and George W. Kling. 2021. "Rainfall Alters Permafrost Soil Redox Conditions, but Meta-Omics Show Divergent Microbial Community Responses by Tundra Type in the Arctic" Soil Systems 5, no. 1: 17. https://doi.org/10.3390/soilsystems5010017
APA StyleRomanowicz, K. J., Crump, B. C., & Kling, G. W. (2021). Rainfall Alters Permafrost Soil Redox Conditions, but Meta-Omics Show Divergent Microbial Community Responses by Tundra Type in the Arctic. Soil Systems, 5(1), 17. https://doi.org/10.3390/soilsystems5010017