Investigation of RNA Editing Sites within Bound Regions of RNA-Binding Proteins

 ,
, 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Data Sources
2.2. Culturing of Cells, qRT-PCR, and siRNAs
2.3. Western Blotting
2.4. RNA Immunoprecipitation (RIP)
2.5. RNA-seq
2.6. Statistics
3. Results
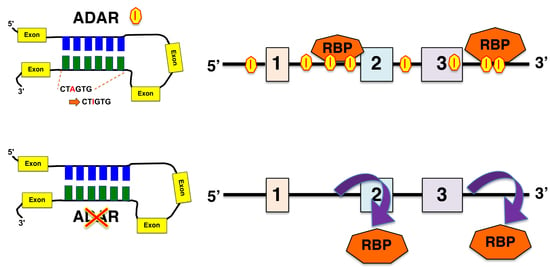
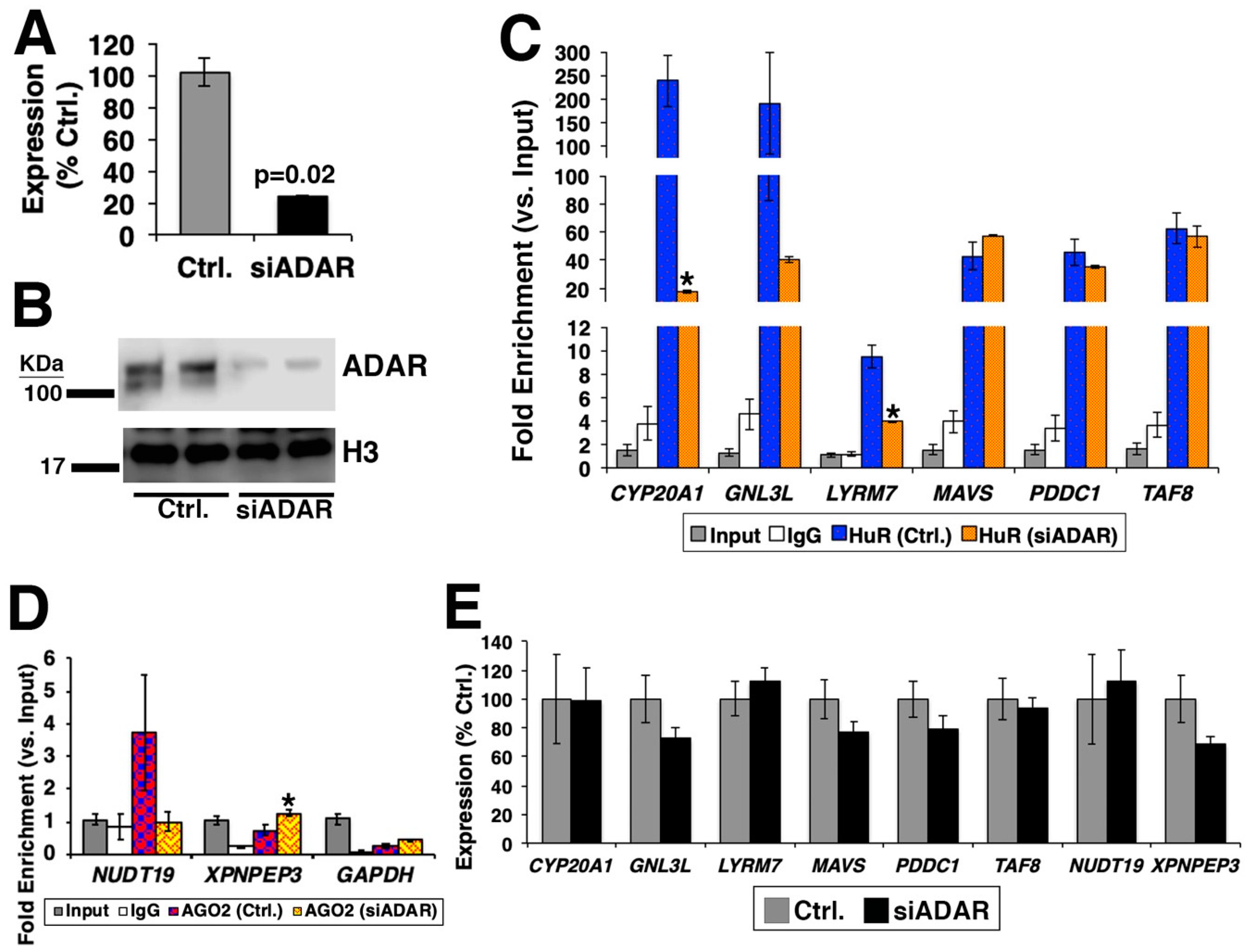
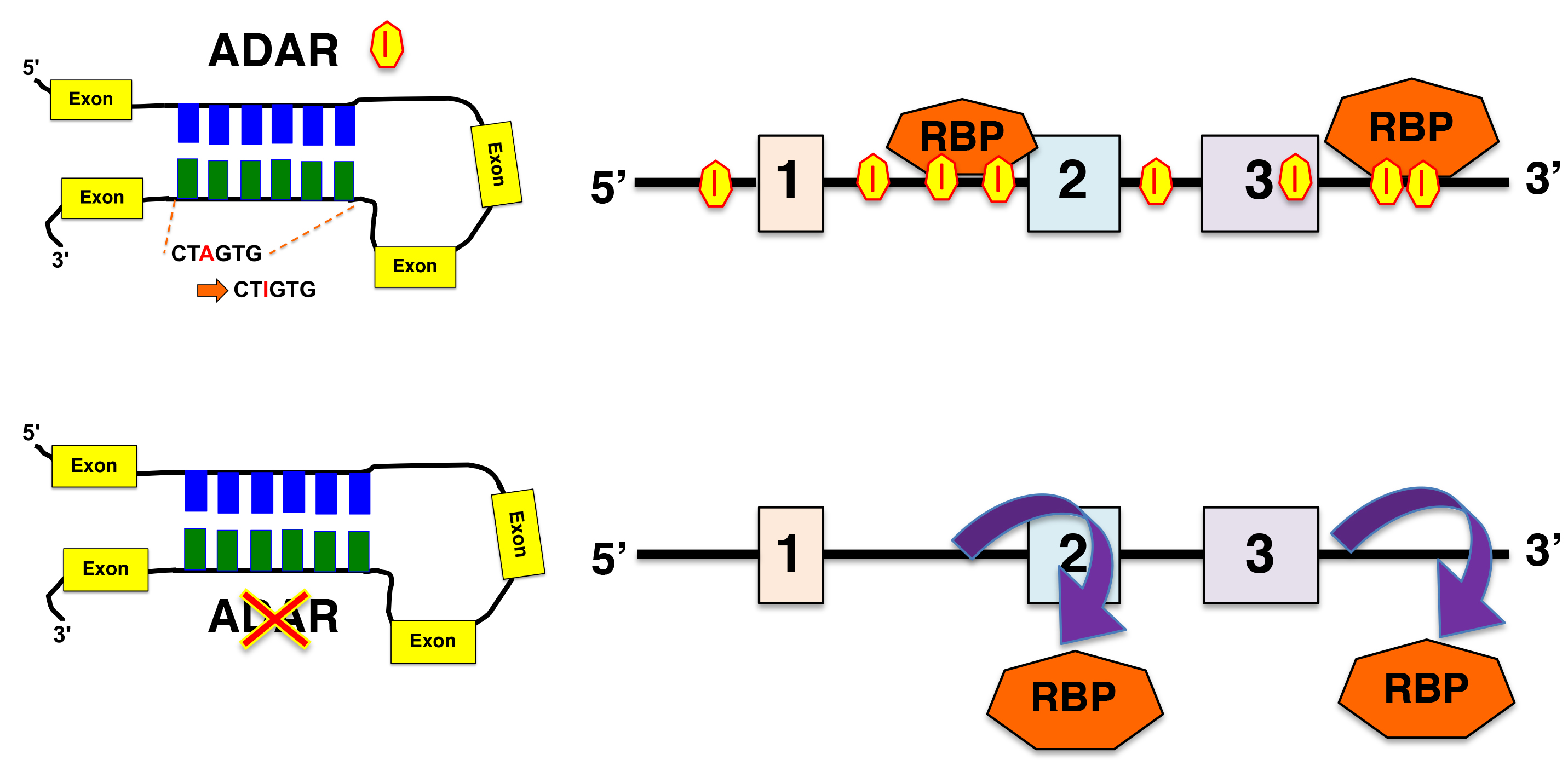
3.1. Presence of RNA Editing Sites in RBP-Bound Regions
3.2. Consequence of Loss of ADAR for the Binding of RBP
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cantara, W.A.; Crain, P.F.; Rozenski, J.; McCloskey, J.A.; Harris, K.A.; Zhang, X.; Vendeix, F.A.; Fabris, D.; Agris, P.F. The rna modification database, rnamdb: 2011 update. Nucleic Acids Res. 2011, 39, D195–D201. [Google Scholar] [CrossRef] [PubMed]
- He, C. Grand challenge commentary: RNA epigenetics? Nat. Chem. Biol. 2010, 6, 863–865. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Pan, T. RNA epigenetics. Trans. Res. 2015, 165, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Roundtree, I.A.; He, C. RNA epigenetics-chemical messages for posttranscriptional gene regulation. Curr. Opin. Chem. Biol. 2016, 30, 46–51. [Google Scholar] [CrossRef]
- Kadumuri, R.V.; Janga, S.C. Epitranscriptomic Code and Its Alterations in Human Disease. Trends Mol Med. 2018, 24, 886–903. [Google Scholar] [CrossRef]
- Diroma, M.A.; Ciaccia, L.; Pesole, G.; Picardi, E. Elucidating the editome: Bioinformatics approaches for rna editing detection. Brief. Bioinform. 2019, 20, 436–447. [Google Scholar] [CrossRef]
- Keegan, L.P.; Gallo, A.; O’Connell, M.A. The many roles of an rna editor. Nat. Rev. Genet. 2001, 2, 869–878. [Google Scholar] [CrossRef]
- Hideyama, T.; Kwak, S. When does als start? Adar2-glua2 hypothesis for the etiology of sporadic als. Front. Mol. Neurosci. 2011, 4, 33. [Google Scholar] [CrossRef]
- Savva, Y.A.; Rieder, L.E.; Reenan, R.A. The adar protein family. Genome Biol. 2012, 13, 252. [Google Scholar] [CrossRef]
- Chen, C.X.; Cho, D.S.; Wang, Q.; Lai, F.; Carter, K.C.; Nishikura, K. A third member of the rna-specific adenosine deaminase gene family, adar3, contains both single-and double-stranded rna binding domains. RNA 2000, 6, 755–767. [Google Scholar] [CrossRef]
- Wang, Q.; Miyakoda, M.; Yang, W.; Khillan, J.; Stachura, D.L.; Weiss, M.J.; Nishikura, K. Stress-induced apoptosis associated with null mutation of adar1 rna editing deaminase gene. J. Biol. Chem. 2004, 279, 4952–4961. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an ampa receptor gene rescues lethality in mice deficient in the rna-editing enzyme adar2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef]
- Bahn, J.H.; Lee, J.H.; Li, G.; Greer, C.; Peng, G.; Xiao, X. Accurate identification of a-to-i rna editing in human by transcriptome sequencing. Genome Res. 2012, 22, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Zhang, W.; Liang, Y.; Hu, X.; Tan, X.; et al. Comprehensive analysis of rna-seq data reveals extensive rna editing in a human transcriptome. Nat. Biotechnol. 2012, 30, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Ramaswami, G.; Lin, W.; Piskol, R.; Tan, M.H.; Davis, C.; Li, J.B. Accurate identification of human alu and non-alu rna editing sites. Nat. Methods 2012, 9, 579–581. [Google Scholar] [CrossRef]
- Park, E.; Williams, B.; Wold, B.J.; Mortazavi, A. Rna editing in the human encode rna-seq data. Genome Res. 2012, 22, 1626–1633. [Google Scholar] [CrossRef]
- Ramaswami, G.; Zhang, R.; Piskol, R.; Keegan, L.P.; Deng, P.; O’Connell, M.A.; Li, J.B. Identifying rna editing sites using rna sequencing data alone. Nat. Methods 2013, 10, 128–132. [Google Scholar] [CrossRef]
- Solomon, O.; Oren, S.; Safran, M.; Deshet-Unger, N.; Akiva, P.; Jacob-Hirsch, J.; Cesarkas, K.; Kabesa, R.; Amariglio, N.; Unger, R.; et al. Global regulation of alternative splicing by adenosine deaminase acting on rna (adar). RNA 2013, 19, 591–604. [Google Scholar] [CrossRef]
- Stellos, K.; Gatsiou, A.; Stamatelopoulos, K.; Perisic Matic, L.; John, D.; Lunella, F.F.; Jae, N.; Rossbach, O.; Amrhein, C.; Sigala, F.; et al. Adenosine-to-inosine rna editing controls cathepsin s expression in atherosclerosis by enabling hur-mediated post-transcriptional regulation. Nat. Med. 2016, 22, 1140–1150. [Google Scholar] [CrossRef]
- Picardi, E.; D’Antonio, M.; Carrabino, D.; Castrignano, T.; Pesole, G. Expedit: A webserver to explore human rna editing in rna-seq experiments. Bioinform. (Oxf. Engl.) 2011, 27, 1311–1312. [Google Scholar] [CrossRef]
- D’Antonio, M.; Picardi, E.; Castrignano, T.; D’Erchia, A.M.; Pesole, G. Exploring the rna editing potential of rna-seq data by expedit. Methods Mol. Biol. 2015, 1269, 327–338. [Google Scholar] [PubMed]
- Zhang, Q.; Xiao, X. Genome sequence-independent identification of rna editing sites. Nat. Methods 2015, 12, 347–350. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Xiao, X. Raser: Reads aligner for snps and editing sites of rna. Bioinform. (Oxf. Engl.) 2015, 31, 3906–3913. [Google Scholar] [CrossRef] [PubMed]
- Picardi, E.; Pesole, G. Reditools: High-throughput rna editing detection made easy. Bioinform. (Oxf. Engl.) 2013, 29, 1813–1814. [Google Scholar] [CrossRef]
- Picardi, E.; D’Erchia, A.M.; Montalvo, A.; Pesole, G. Using reditools to detect rna editing events in ngs datasets. Curr. Protoc. Bioinform. 2015, 49, 12.12.1–12.12.15. [Google Scholar] [CrossRef]
- Kiran, A.; Baranov, P.V. Darned: A database of rna editing in humans. Bioinform. (Oxf. Engl.) 2010, 26, 1772–1776. [Google Scholar] [CrossRef]
- Ramaswami, G.; Li, J.B. Radar: A rigorously annotated database of a-to-i rna editing. Nucleic Acids Res. 2014, 42, D109–D113. Available online: http://rnaedit.com/download/ (accessed on 4 September 2017). [CrossRef]
- Weirick, T.; Militello, G.; Ponomareva, Y.; John, D.; Doring, C.; Dimmeler, S.; Uchida, S. Logic programming to infer complex rna expression patterns from rna-seq data. Brief. Bioinform. 2016, 19, 199–209. [Google Scholar] [CrossRef]
- Brinegar, A.E.; Cooper, T.A. Roles for rna-binding proteins in development and disease. Brain Res. 2016, 1647, 1–8. [Google Scholar] [CrossRef]
- Calabretta, S.; Richard, S. Emerging roles of disordered sequences in rna-binding proteins. Trends. Biochem. Sci. 2015, 40, 662–672. [Google Scholar] [CrossRef]
- Cookson, M.R. Rna-binding proteins implicated in neurodegenerative diseases. Wiley Interdiscip. Rev. RNA 2017, 8, e1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, S.; Siddam, A.D.; Barnum, C.E.; Janga, S.C.; Lachke, S.A. Rna-binding proteins in eye development and disease: Implication of conserved rna granule components. Wiley Interdiscip. Rev. RNA 2016, 7, 527–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Bruin, R.G.; Rabelink, T.J.; van Zonneveld, A.J.; van der Veer, E.P. Emerging roles for rna-binding proteins as effectors and regulators of cardiovascular disease. Eur. Heart J. 2016, 38, 1380–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fredericks, A.M.; Cygan, K.J.; Brown, B.A.; Fairbrother, W.G. Rna-binding proteins: Splicing factors and disease. Biomolecules 2015, 5, 893–909. [Google Scholar] [CrossRef] [Green Version]
- Sephton, C.F.; Yu, G. The function of rna-binding proteins at the synapse: Implications for neurodegeneration. Cell. Mol. Life Sci. 2015, 72, 3621–3635. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated rnas by rip-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Wessels, H.H.; Hirsekorn, A.; Ohler, U.; Mukherjee, N. Identifying rbp targets with rip-seq. Methods Mol. Biol. (Clifton N. J.) 2016, 1358, 141–152. [Google Scholar]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. Clip identifies nova-regulated rna networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M., Jr.; Jungkamp, A.C.; Munschauer, M.; et al. Transcriptome-wide identification of rna-binding protein and microrna target sites by par-clip. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Konig, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. Iclip reveals the function of hnrnp particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010, 17, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Kayikci, M.; Briese, M.; Zarnack, K.; Luscombe, N.M.; Rot, G.; Zupan, B.; Curk, T.; Ule, J. Iclip predicts the dual splicing effects of tia-rna interactions. PLoS Biol. 2010, 8, e1000530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.C.; Di, C.; Hu, B.; Zhou, M.; Liu, Y.; Song, N.; Li, Y.; Umetsu, J.; Lu, Z.J. Clipdb: A clip-seq database for protein-rna interactions. BMC Genom. 2015, 16, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.W.; Xie, C.; Yang, J.R.; Li, J.H.; Yang, J.H.; Zheng, L. Mtibase: A database for decoding microrna target sites located within cds and 5’utr regions from clip-seq and expression profile datasets. Database J. Biol. Databases Curation 2015. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Yang, Y.T.; Huang, Y.; Zhu, Y.; Lu, Z.J. Postar: A platform for exploring post-transcriptional regulation coordinated by rna-binding proteins. Nucleic Acids Res. 2017, 45, D104–D114. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Liu, S.; Zhou, H.; Qu, L.H.; Yang, J.H. Starbase v2.0: Decoding mirna-cerna, mirna-ncrna and protein-rna interaction networks from large-scale clip-seq data. Nucleic Acids Res. 2014, 42, D92–D97. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.H.; Li, J.H.; Shao, P.; Zhou, H.; Chen, Y.Q.; Qu, L.H. Starbase: A database for exploring microrna-mrna interaction maps from argonaute clip-seq and degradome-seq data. Nucleic Acids Res. 2011, 39, D202–D209. Available online: http://starbase.sysu.edu.cn/download.php (accessed on 4 September 2017). [CrossRef] [Green Version]
- Gaisler-Salomon, I.; Kravitz, E.; Feiler, Y.; Safran, M.; Biegon, A.; Amariglio, N.; Rechavi, G. Hippocampus-specific deficiency in rna editing of glua2 in alzheimer’s disease. Neurobiol. Aging 2014, 35, 1785–1791. [Google Scholar] [CrossRef]
- Xiang, J.F.; Yin, Q.F.; Chen, T.; Zhang, Y.; Zhang, X.O.; Wu, Z.; Zhang, S.; Wang, H.B.; Ge, J.; Lu, X.; et al. Human colorectal cancer-specific ccat1-l lncrna regulates long-range chromatin interactions at the myc locus. Cell Res. 2014, 24, 513–531. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Kwak, S. The molecular link between inefficient glua2 q/r site-rna editing and tdp-43 pathology in motor neurons of sporadic amyotrophic lateral sclerosis patients. Brain Res. 2014, 1584, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Hui, H.; Guo, Z.; Zhang, W.; Hu, Y.; He, T.; Tai, Y.; Peng, P.; Wang, L. Adar1 regulates arhgap26 gene expression through rna editing by disrupting mir-30b-3p and mir-573 binding. RNA 2013, 19, 1525–1536. [Google Scholar] [CrossRef] [Green Version]
- Quinlan, A.R.; Hall, I.M. Bedtools: A flexible suite of utilities for comparing genomic features. Bioinform. (Oxf. Engl.) 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Militello, G.; Weirick, T.; John, D.; Doring, C.; Dimmeler, S.; Uchida, S. Screening and validation of lncrnas and circrnas as mirna sponges. Brief. Bioinform. 2016, 18, 780–788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, D.; Weirick, T.; Dimmeler, S.; Uchida, S. Rnaeditor: Easy detection of rna editing events and the introduction of editing islands. Brief. Bioinform. 2016, 18, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.Y.; Ramos, A. The double-stranded rna-binding motif, a versatile macromolecular docking platform. FEBS J. 2005, 272, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Varani, G. Protein families and rna recognition. FEBS J. 2005, 272, 2088–2097. [Google Scholar] [CrossRef] [PubMed]
- Masliah, G.; Barraud, P.; Allain, F.H. Rna recognition by double-stranded rna binding domains: A matter of shape and sequence. Cell. Mol. Life Sci. 2013, 70, 1875–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefl, R.; Skrisovska, L.; Allain, F.H. Rna sequence- and shape-dependent recognition by proteins in the ribonucleoprotein particle. EMBO Rep. 2005, 6, 33–38. [Google Scholar] [CrossRef]
- Wang, I.X.; So, E.; Devlin, J.L.; Zhao, Y.; Wu, M.; Cheung, V.G. Adar regulates rna editing, transcript stability, and gene expression. Cell Rep. 2013, 5, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Peng, S.S.; Chen, C.Y.; Xu, N.; Shyu, A.B. Rna stabilization by the au-rich element binding protein, hur, an elav protein. EMBO J. 1998, 17, 3461–3470. [Google Scholar] [CrossRef]
- Fan, X.C.; Steitz, J.A. Overexpression of hur, a nuclear-cytoplasmic shuttling protein, increases the in vivo stability of are-containing mrnas. EMBO J. 1998, 17, 3448–3460. [Google Scholar] [CrossRef] [Green Version]
- Levy, N.S.; Chung, S.; Furneaux, H.; Levy, A.P. Hypoxic stabilization of vascular endothelial growth factor mrna by the rna-binding protein hur. J. Biol. Chem. 1998, 273, 6417–6423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anantharaman, A.; Tripathi, V.; Khan, A.; Yoon, J.H.; Singh, D.K.; Gholamalamdari, O.; Guang, S.; Ohlson, J.; Wahlstedt, H.; Ohman, M.; et al. Adar2 regulates rna stability by modifying access of decay-promoting rna-binding proteins. Nucleic Acids Res. 2017, 45, 4189–4201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anantharaman, A.; Gholamalamdari, O.; Khan, A.; Yoon, J.H.; Jantsch, M.F.; Hartner, J.C.; Gorospe, M.; Prasanth, S.G.; Prasanth, K.V. Rna-editing enzymes adar1 and adar2 coordinately regulate the editing and expression of ctn rna. FEBS Lett. 2017, 591, 2890–2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacobi, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [Green Version]
- Cartolano, M.; Huettel, B.; Hartwig, B.; Reinhardt, R.; Schneeberger, K. Cdna library enrichment of full length transcripts for smrt long read sequencing. PLoS ONE 2016, 11, e0157779. [Google Scholar] [CrossRef]
- Gonzalez-Ibeas, D.; Martinez-Garcia, P.J.; Famula, R.A.; Delfino-Mix, A.; Stevens, K.A.; Loopstra, C.A.; Langley, C.H.; Neale, D.B.; Wegrzyn, J.L. Assessing the gene content of the megagenome: Sugar pine (pinus lambertiana). G3 (Bethesda Md.) 2016, 6, 3787–3802. [Google Scholar] [CrossRef] [Green Version]
- Hoang, N.V.; Furtado, A.; Mason, P.J.; Marquardt, A.; Kasirajan, L.; Thirugnanasambandam, P.P.; Botha, F.C.; Henry, R.J. A survey of the complex transcriptome from the highly polyploid sugarcane genome using full-length isoform sequencing and de novo assembly from short read sequencing. BMC Genom. 2017, 18, 395. [Google Scholar] [CrossRef]
- Jiang, X.; Hall, A.B.; Biedler, J.K.; Tu, Z. Single molecule rna sequencing uncovers trans-splicing and improves annotations in anopheles stephensi. Insect Mol. Biol. 2017, 26, 298–307. [Google Scholar] [CrossRef]
- Kim, M.A.; Rhee, J.S.; Kim, T.H.; Lee, J.S.; Choi, A.Y.; Choi, B.S.; Choi, I.Y.; Sohn, Y.C. Alternative splicing profile and sex-preferential gene expression in the female and male pacific abalone haliotis discus hannai. Genes 2017, 8, 99. [Google Scholar] [CrossRef] [Green Version]
- Kuo, R.I.; Tseng, E.; Eory, L.; Paton, I.R.; Archibald, A.L.; Burt, D.W. Normalized long read rna sequencing in chicken reveals transcriptome complexity similar to human. BMC Genom. 2017, 18, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Mei, W.; Soltis, P.S.; Soltis, D.E.; Barbazuk, W.B. Detecting alternatively spliced transcript isoforms from single-molecule long-read sequences without a reference genome. Mol. Ecol. Resour. 2017, 7, 1243–1256. [Google Scholar] [CrossRef] [PubMed]
- O’Grady, T.; Wang, X.; Honer Zu Bentrup, K.; Baddoo, M.; Concha, M.; Flemington, E.K. Global transcript structure resolution of high gene density genomes through multi-platform data integration. Nucleic Acids Res. 2016, 44, e145. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Sahu, D.K.; Chowdhry, R.; Mishra, A.; Goel, M.M.; Faheem, M.; Srivastava, C.; Ojha, B.K.; Gupta, D.K.; Kant, R. Isoseq analysis and functional annotation of the infratentorial ependymoma tumor tissue on pacbio rsii platform. Meta Gene 2016, 7, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Tombacz, D.; Balazs, Z.; Csabai, Z.; Moldovan, N.; Szucs, A.; Sharon, D.; Snyder, M.; Boldogkoi, Z. Characterization of the dynamic transcriptome of a herpesvirus with long-read single molecule real-time sequencing. Sci. Rep. 2017, 7, 43751. [Google Scholar] [CrossRef] [PubMed]
- Zulkapli, M.M.; Rosli, M.A.F.; Salleh, F.I.M.; Mohd Noor, N.; Aizat, W.M.; Goh, H.H. Iso-seq analysis of nepenthes ampullaria, nepenthes rafflesiana and nepenthes x hookeriana for hybridisation study in pitcher plants. Genom. Data 2017, 12, 130–131. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Genic Location | All Sites | Alu | Non-Alu |
|---|---|---|---|
| 5′-UTR | 6775 | 6131 | 644 |
| Exon | 4405 | 2082 | 2323 |
| Intron | 1,936,801 | 1,870,644 | 66,157 |
| 3′-UTR | 85,169 | 79,434 | 5735 |
| Intergenic | 516,714 | 479,480 | 37,234 |
| ncRNA | 26,595 | 24,184 | 2411 |
| Genic Location | Count |
|---|---|
| 5′-UTR | 50 |
| 5′-UTR;Intron | 10 |
| Exon | 20 |
| Exon;Intron | 9 |
| Intron;Exon | 17 |
| Intron;5′-UTR | 4 |
| Intron | 16,707 |
| Intron;3′-UTR | 14 |
| Intron;ncRNA | 33 |
| 3′-UTR | 590 |
| 3′-UTR;Intergenic | 2 |
| 3′-UTR;Intron | 4 |
| Intergenic;5′-UTR | 1 |
| Intergenic | 3376 |
| Intergenic;3′-UTR | 2 |
| Intergenic;ncRNA | 3 |
| ncRNA | 258 |
| ncRNA;Intron | 18 |
| ncRNA;Intergenic | 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weirick, T.; Militello, G.; Hosen, M.R.; John, D.; Moore, J.B., IV; Uchida, S. Investigation of RNA Editing Sites within Bound Regions of RNA-Binding Proteins. High-Throughput 2019, 8, 19. https://doi.org/10.3390/ht8040019
Weirick T, Militello G, Hosen MR, John D, Moore JB IV, Uchida S. Investigation of RNA Editing Sites within Bound Regions of RNA-Binding Proteins. High-Throughput. 2019; 8(4):19. https://doi.org/10.3390/ht8040019
Chicago/Turabian StyleWeirick, Tyler, Giuseppe Militello, Mohammed Rabiul Hosen, David John, Joseph B. Moore, IV, and Shizuka Uchida. 2019. "Investigation of RNA Editing Sites within Bound Regions of RNA-Binding Proteins" High-Throughput 8, no. 4: 19. https://doi.org/10.3390/ht8040019
APA StyleWeirick, T., Militello, G., Hosen, M. R., John, D., Moore, J. B., IV, & Uchida, S. (2019). Investigation of RNA Editing Sites within Bound Regions of RNA-Binding Proteins. High-Throughput, 8(4), 19. https://doi.org/10.3390/ht8040019