Pharmacogenomic Profiling of ADME Gene Variants: Current Challenges and Validation Perspectives



 ,
, 


Abstract
:1. Introduction
2. Pharmacogenomics Approaches for Germline SNP Identification
3. Pharmacogenomics Tools Currently Available
4. Candidate Biomarkers Discovery Process
5. Candidate Biomarkers Validation Process
6. Promise and Challenges of Pharmacogenomics Fallout in Clinical Practice
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- The International HapMap3 Consortium. Integrating common and rare genetic variation in diverse human populations. Nature 2010, 5, 52–58. [Google Scholar] [CrossRef]
- Olivier, M.; Taniere, P. Somatic mutations in cancer prognosis and prediction: Lessons from TP53 and EGFR genes. Curr. Opin. Oncol. 2011, 23, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Roden, D.M.; Wilke, R.A.; Kroemer, H.K.; Stein, C.M. Pharmacogenomics: The genetics of variable drug responses. Circulation 2011, 123, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Elmas, A.; OuYang, T.-H.; Wang, X.; Anastassiou, D. Discovering Genome-Wide tag SNPs based on the mutual information of the variants. PLoS ONE 2016, 11, e0167994. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; He, Y.; Wang, H.; Wang, Y.; Liu, Y.; Wang, Y.; Chu, X.; Wang, Y.; Xu, L.; Shen, Y.; et al. Linkage disequilibrium sharing and haplotype-tagged SNP portability between populations. Proc. Natl. Acad. Sci. USA 2006, 103, 1418–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, F.; Yanai, K.; Morii, T.; Ishinaga, Y.; Taniguchi-Yanai, K.; Nagano, S.; Kato, N. Linkage disequilibrium grouping of Single Nucleotide Polymorphisms (SNPs) reflecting haplotype phylogeny for efficient selection of tag SNPs. Genetics 2005, 170, 291–304. [Google Scholar] [CrossRef] [PubMed]
- Maggo, S.D.; Savage, R.L.; Kennedy, M.A. Impact of new genomic technologies on understanding adverse drug reactions. Clin. Pharmacokinet. 2016, 55, 419–436. [Google Scholar] [CrossRef] [PubMed]
- Stadler, Z.K.; Thom, P.; Robson, M.E.; Weitzel, J.N.; Kauff, N.D.; Hurley, K.E.; Devlin, V.; Gold, B.; Klein, R.J.; Offit, K. Genome-Wide Association Studies of Cancer. JCO 2010, 28, 4255–4267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunter, D.J.; Kraft, P. Drinking from the fire hose—Statistical issues in genomewide association studies. N. Engl. J. Med. 2007, 357, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Leusink, M.; Maitland-van der Zee, A.H.; Ding, B.; Drenos, F.; van Iperen, E.P.; Warren, H.R.; Caulfield, M.J.; Cupples, L.A.; Cushman, M.; Hingorani, A.D.; et al. A genetic risk score is associated with statin-induced low-density lipoprotein cholesterol lowering. Pharmacogenomics 2016, 17, 583–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jukema, J.W.; Trompet, S. Genetics: Genetic risk scores—new promises for drug evaluation. Nat. Rev. Cardiol. 2015, 12, 321–322. [Google Scholar] [CrossRef] [PubMed]
- Ciuculete, D.M.; Bandstein, M.; Benedict, C.; Waeber, G.; Vollenweider, P.; Lind, L.; Schiöth, H.B.; Mwinyi, J. A genetic risk score is significantly associated with statin therapy response in the elderly population. Clin. Genet. 2017, 91, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Tabor, H.K.; Risch, N.J.; Myers, R.M. Candidate-gene approaches for studying complex genetic traits: Practical considerations. Nat. Rev. Genet. 2002, 3, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Hardenbol, P.; Yu, F.; Belmont, J.; Mackenzie, J.; Bruckner, C.; Brundage, T.; Boudreau, A.; Chow, S.; Eberle, J.; Erbilgin, A.; et al. Highly multiplexed molecular inversion probe genotyping: Over 10,000 targeted SNPs genotyped in a single tube assay. Genome Res. 2005, 15, 269–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardenbol, P.; Baner, J.; Jain, M.; Nilsson, M.; Namsaraev, E.A.; Karlin-Neumann, G.A.; Fakhrai-Rad, H.; Ronaghi, M.; Willis, T.D.; Landegren, U.; et al. Multiplexed genotyping with sequence-tagged molecular inversion probes. Nat. Biotechnol. 2003, 21, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Burmester, J.K.; Sedova, M.; Shapero, M.H.; Mansfield, E. DMET microarray technology for pharmacogenomics-based personalized medicine. Methods Mol. Biol. 2010, 632, 99–124. [Google Scholar] [CrossRef] [PubMed]
- Arbitrio, M.; Martino, M.T.D.; Scionti, F.; Agapito, G.; Guzzi, P.H.; Cannataro, M.; Tassone, P.; Tagliaferri, P. DMETTM (Drug Metabolism Enzymes and Transporters): A pharmacogenomic platform for precision medicine. Oncotarget 2016, 7, 54028–54050. [Google Scholar] [CrossRef] [PubMed]
- Guzzi, P.H.; Agapito, G.; Di Martino, M.T.; Arbitrio, M.; Tassone, P.; Tagliaferri, P.; Cannataro, M. DMET-Analyzer: Automaticanalysis of Affymetrix DMET Data. BMC Bioinform. 2012, 13, 258. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, M.T.; Arbitrio, M.; Leone, E.; Guzzi, P.H.; Rotundo, M.S.; Ciliberto, D.; Tomaino, V.; Fabiani, F.; Talarico, D.; Sperlongano, P.; et al. Single nucleotide polymorphisms of ABCC5 and ABCG1 transporter genes correlate to irinotecan-associated gastrointestinal toxicity in colorectal cancer patients: A DMET microarray profiling study. Cancer Biol. Ther. 2011, 12, 780–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Martino, M.T.; Arbitrio, M.; Guzzi, P.H.; Leone, E.; Baudi, F.; Piro, E.; Prantera, T.; Cucinotto, I.; Calimeri, T.; Rossi, M.; et al. A peroxisome proliferator-activated receptor gamma (PPARG) polymorphism is associated with zoledronic acid-related osteonecrosis of the jaw in multiple myeloma patients: Analysis by DMET microarray profiling. Br. J. Haematol. 2011, 154, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Arbitrio, M.; Di Martino, M.T.; Barbieri, V.; Agapito, G.; Guzzi, P.H.; Botta, C.; Iuliano, E.; Scionti, F.; Altomare, E.; Codispoti, S.; et al. Identification of polymorphic variants associated with erlotinib-related skin toxicity in advanced non-small cell lung cancer patients by DMET microarray analysis. Cancer Chemother. Pharmacol. 2016, 77, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Scionti, F.; Di Martino, M.T.; Sestito, S.; Nicoletti, A.; Falvo, F.; Roppa, K.; Arbitrio, M.; Guzzi, P.H.; Agapito, G.; Pisani, A.; et al. Genetic variants associated with Fabry disease progression despite enzyme replacement therapy. Oncotarget 2017, 8, 107558–107564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Martino, M.T.; Scionti, F.; Sestito, S.; Nicoletti, A.; Arbitrio, M.; Guzzi, H.P.; Talarico, V.; Altomare, F.; Sanseviero, M.T.; Agapito, G.; et al. Genetic variants associated with gastrointestinal symptoms in Fabry disease. Oncotarget 2016, 7, 85895–85904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, J.M.; Haidar, C.E.; Wilkinson, M.R.; Crews, K.R.; Baker, D.K.; Kornegay, N.M.; Yang, W.; Pui, C.H.; Reiss, U.M.; Gaur, A.H.; et al. PG4KDS: A model for the clinical implementation of pre-emptive pharmacogenetics. Am. J. Med. Genet. C. Semin. Med. Genet. 2014, 166, 45–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.H.; Yeakley, J.M.; McDaniel, T.K.; Shen, R. Medium to high-throughput SNP genotyping using Vera Code microbeads. Methods Mol. Biol. 2009, 496, 129–142. [Google Scholar] [PubMed]
- Pulley, J.M.; Denny, J.C.; Petersonm, J.F.; Bernard, G.R.; Vnencak-Jones, C.L.; Ramirez, A.H.; Delaney, J.T.; Bowton, E.; Brothers, K.; Johnson, K.; et al. Operational implementation of prospective genotyping for personalized medicine: The design of the Vanderbilt PREDICT project. Clin. Pharmacol. Ther. 2012, 92, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Tremaine, L.; Brian, W.; DelMonte, T.; Francke, S.; Groenen, P.; Johnson, K.; Li, L.; Pearson, K.; Marshall, J.C. The role of ADME pharmacogenomics in early clinical trials: Perspective of the Industry Pharmacogenomics Working Group (I-PWG). Pharmacogenomics 2015, 16, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Mukerjee, G.; Huston, A.; Kabakchiev, B.; Piquette-Miller, M.; van Schaik, R.; Dorfman, R. User considerations in assessing pharmacogenomic tests and their clinical support tools. NPJ Genom. Med. 2018, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, S.; Barone, J.; Malone, B. Warfarin pharmacogenomics. Curr. Opin. Mol. Ther. 2009, 34, 422–427. [Google Scholar]
- Available online: https://www.pharmgkb.org/ (accessed on 3 October 2018).
- Available online: https://cpicpgx.org/ (accessed on 27 September 2018).
- Available online: https://www.ncbi.nlm.nih.gov/snp (accessed on 27 September 2018).
- Committee on the Review of Omics-Based Tests for Predicting Patient Outcomes in Clinical Trials; Board on Health Care Services; Board on Health Sciences Policy; Institute of Medicine; Micheel, C.M.; Nass, S.J.; Omenn, G.S. (Eds.) Evolution of Translational Omics: Lessons Learned and the Path Forward; National Academies Press (US): Washington, DC, USA, 2012. [Google Scholar]
- Jennings, L.; Van Deerlin, V.M.; Gulley, M.L. Recommended principles and practices for validating clinical molecular pathology tests. Arch. Pathol. Lab. Med. 2009, 133, 743–755. [Google Scholar] [PubMed]
- McShane, L.M.; Cavenagh, M.M.; Lively, T.G.; Eberhard, D.A.; Bigbee, W.L.; Williams, P.M.; Mesirov, J.P.; Polley, M.Y.; Kim, K.Y.; Tricoli, J.V.; et al. Criteria for the use of omics-based predictors in clinical trials. Nature 2013, 502, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Mattocks, C.J.; Morris, M.A.; Matthijs, G.; Swinnen, E.; Corveleyn, A.; Dequeker, E.; Muller, C.R.; Pratt, V.; Wallace, A.; EuroGentest Validation Group. A standardized framework for the validation and verification of clinical molecular genetic tests. Eur. J. Hum. Genet. 2010, 18, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Ortega, V.E.; Meyers, D.A. Pharmacogenetics: Implications of race and ethnicity on defining genetic profiles for personalized medicine. J. Allergy ClinImmunol. 2014, 133, 16–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelman-Sundberg, M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): Clinical consequences, evolutionary aspects and functional diversity. Pharmacogenom. J. 2005, 5, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindeman, N.I.; Cagle, P.T.; Beasley, M.B.; Chitale, D.A.; Dacic, S.; Giaccone, G.; Jenkins, R.B.; Kwiatkowski, D.J.; Saldivar, J.S.; Squire, J.; et al. Molecular testing guideline for selection of lung cancer patients for EGFR and ALK tyrosine kinase inhibitors: Guideline from the College of American Pathologists, International Association for the Study of Lung Cancer, and Association for Molecular Pathology. J. Thorac. Oncol. 2013, 8, 823–859. [Google Scholar] [PubMed]
- Rakha, E.A.; Starczynski, J.; Lee, A.H.; Ellis, I.O. The updated ASCO/CAP guideline recommendations for HER2 testing in the management of invasive breast cancer: A critical review of their implications for routine practice. Histopathology 2014, 64, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Benson, A.B., 3rd; Venook, A.P.; Cederquist, L.; Chan, E.; Chen, Y.J.; Cooper, H.S.; Deming, D.; Engstrom, P.F.; Enzinger, P.C.; Fichera, A.; et al. Colon Cancer, Version 1.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2017, 15, 370–398. [Google Scholar] [CrossRef] [Green Version]
- Bode, A.M.; Dong, Z. Recent advances in precision oncology research. NPJ Precis. Oncol. 2018, 16, 11. [Google Scholar] [CrossRef] [PubMed]
- Annotation of FDA Label for Irinotecan and UGT1A1. Available online: https://www.pharmgkb.org/ (accessed on 12 September 2018).
- Pirmohamed, M. Acceptance of biomarker-based tests for application in clinical practice: Criteria and obstacles. Clin. Pharmacol. Ther. 2010, 88, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Vijverberg, S.J.; Pieters, T.; Cornel, M.C. Ethical and social issues in pharmacogenomics testing. Curr. Pharm. Des. 2010, 16, 245–252. [Google Scholar] [CrossRef] [PubMed]
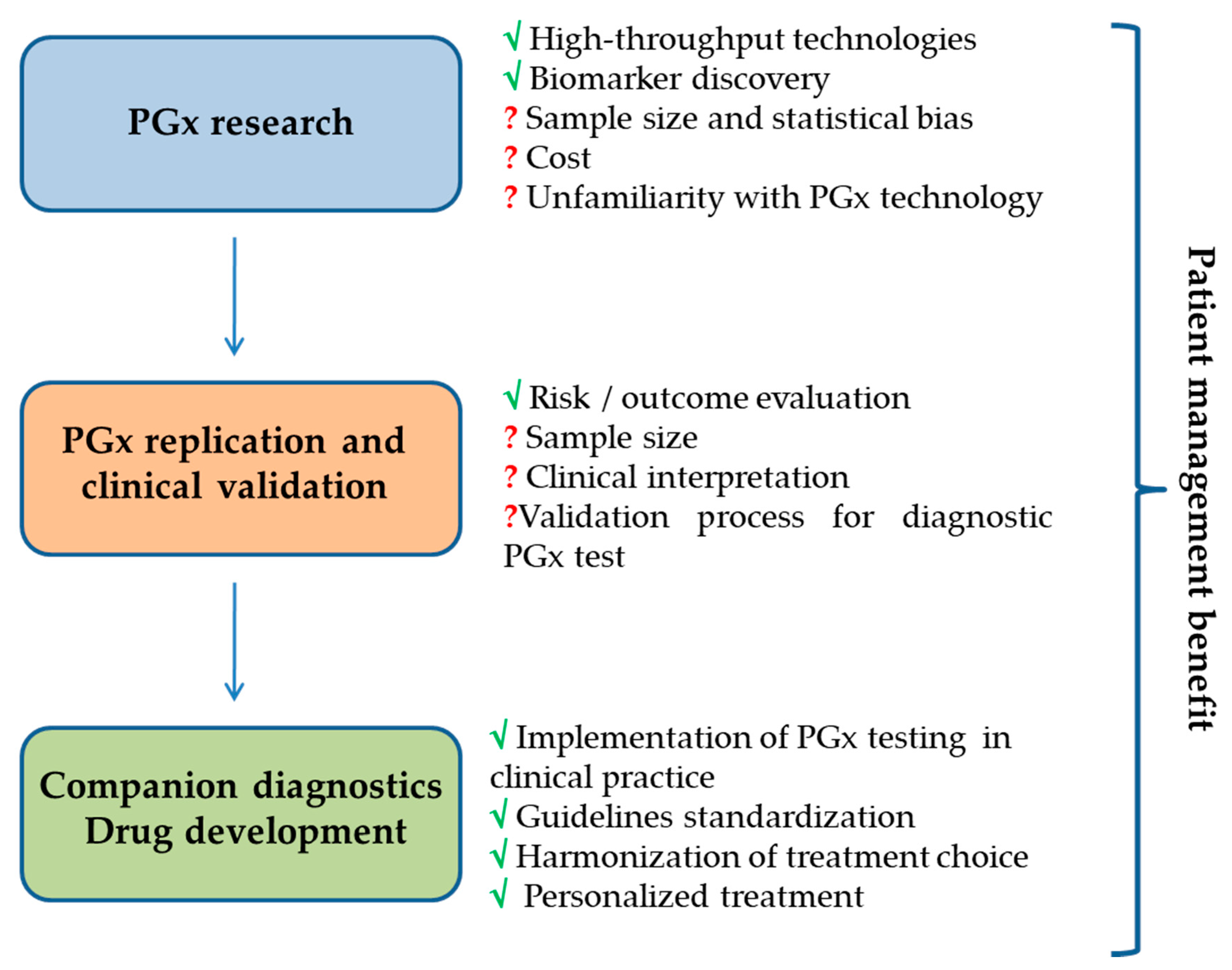

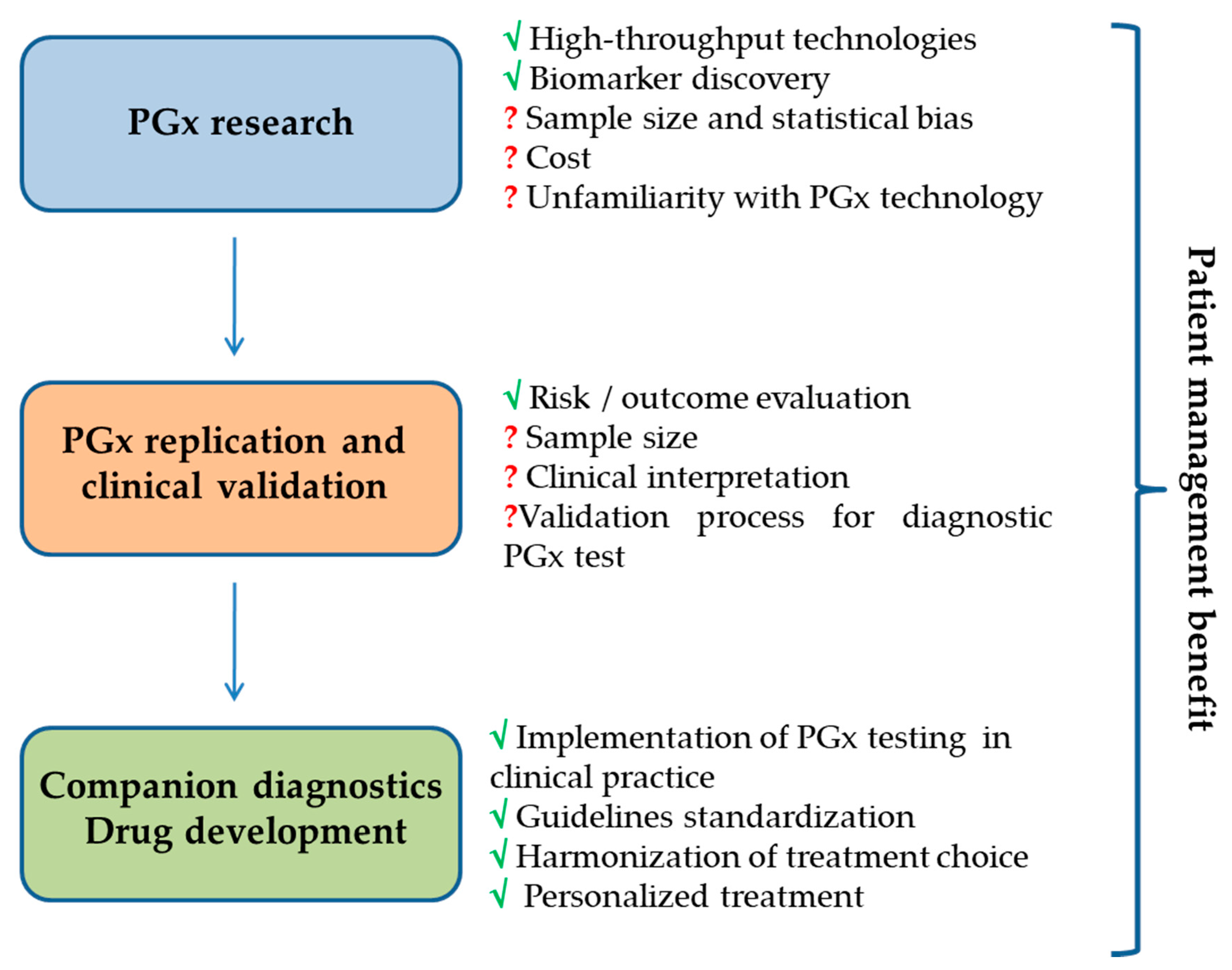{kind=link}
| PGx Approach | GWAS | SNPs Panel | Candidate SNP |
|---|---|---|---|
| Sample size | Tailored for large populations | Tailored for small populations | Tailored for small populations |
| Number of investigated markers | Larger numbers | 1–2 thousand | Smaller number |
| Hypothesis | Hypothesis-free and hypothesis generating | Hypothesis-free and hypothesis generating/PK and PD coverage | Selected on a priori knowledge |
| Study Design | Exploratory | Confirmatory/Exploratory | Confirmatory |
| Limitations | False Negative/control for multiple testing | Coverage of limited genes | False positive/non-replication of results/low genetic coverage |
| Platform | TaqMan Open Array PGx Express Panel (Thermo Fisher Scientific) | DMET Plus (Thermo Fisher Scientific) | PharmacoScan (Thermo Fisher Scientific) | Ion AmpliSeqPGx (Thermo Fisher Scientific) | iPLEX ADME PGx (Sequenom) |
|---|---|---|---|---|---|
| Markers (SNP/indels/CNV) | 60 | 1936 | 4627 | 141 | 192 |
| Genes | 14 | 231 | 1191 | 40 | 38 |
| Sample per assay | 46 | 48 | 22, 94 | 48 | 3, 12, 48 |
| DNA input | 10 ng | 60 ng | 50 ng | 10 ng | 80 ng |
| Technology | Real-Time PCR | Microarray | Microarray | Next-generation sequencing | Mass spectrometry |
| Turnaround time | ~1 day | ~3 days | ~5 days | ~1.5 days | ~8 h |
| Average Call Rate | >99.8% | >99.8% | >99.0% | 99.8% | >99.0% |
| Concordance to reference | ≥99.5% | ≥99.5% | ≥99.5% | 99.9% | 98.9% |
| Reproducibility | ≥99.8% | ≥99.8% | ≥99.8% | 99.7% | >99.7% |
| For research only | Yes | Yes | Yes | Yes | Yes |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arbitrio, M.; Di Martino, M.T.; Scionti, F.; Barbieri, V.; Pensabene, L.; Tagliaferri, P. Pharmacogenomic Profiling of ADME Gene Variants: Current Challenges and Validation Perspectives. High-Throughput 2018, 7, 40. https://doi.org/10.3390/ht7040040
Arbitrio M, Di Martino MT, Scionti F, Barbieri V, Pensabene L, Tagliaferri P. Pharmacogenomic Profiling of ADME Gene Variants: Current Challenges and Validation Perspectives. High-Throughput. 2018; 7(4):40. https://doi.org/10.3390/ht7040040
Chicago/Turabian StyleArbitrio, Mariamena, Maria Teresa Di Martino, Francesca Scionti, Vito Barbieri, Licia Pensabene, and Pierosandro Tagliaferri. 2018. "Pharmacogenomic Profiling of ADME Gene Variants: Current Challenges and Validation Perspectives" High-Throughput 7, no. 4: 40. https://doi.org/10.3390/ht7040040
APA StyleArbitrio, M., Di Martino, M. T., Scionti, F., Barbieri, V., Pensabene, L., & Tagliaferri, P. (2018). Pharmacogenomic Profiling of ADME Gene Variants: Current Challenges and Validation Perspectives. High-Throughput, 7(4), 40. https://doi.org/10.3390/ht7040040