In Silico Profiling of Clinical Phenotypes for Human Targets Using Adverse Event Data


Abstract
:1. Introduction
2. Materials and Methods
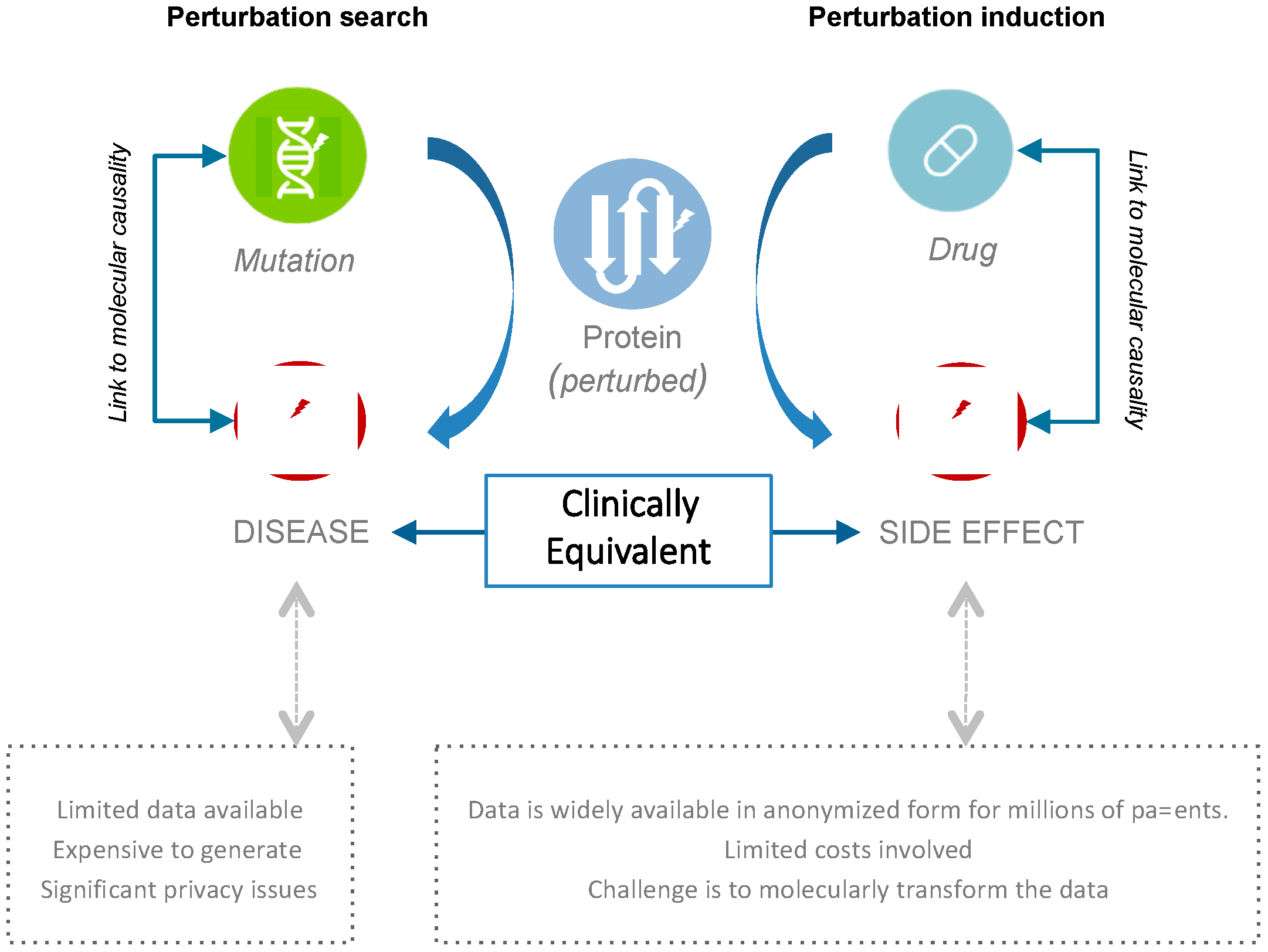
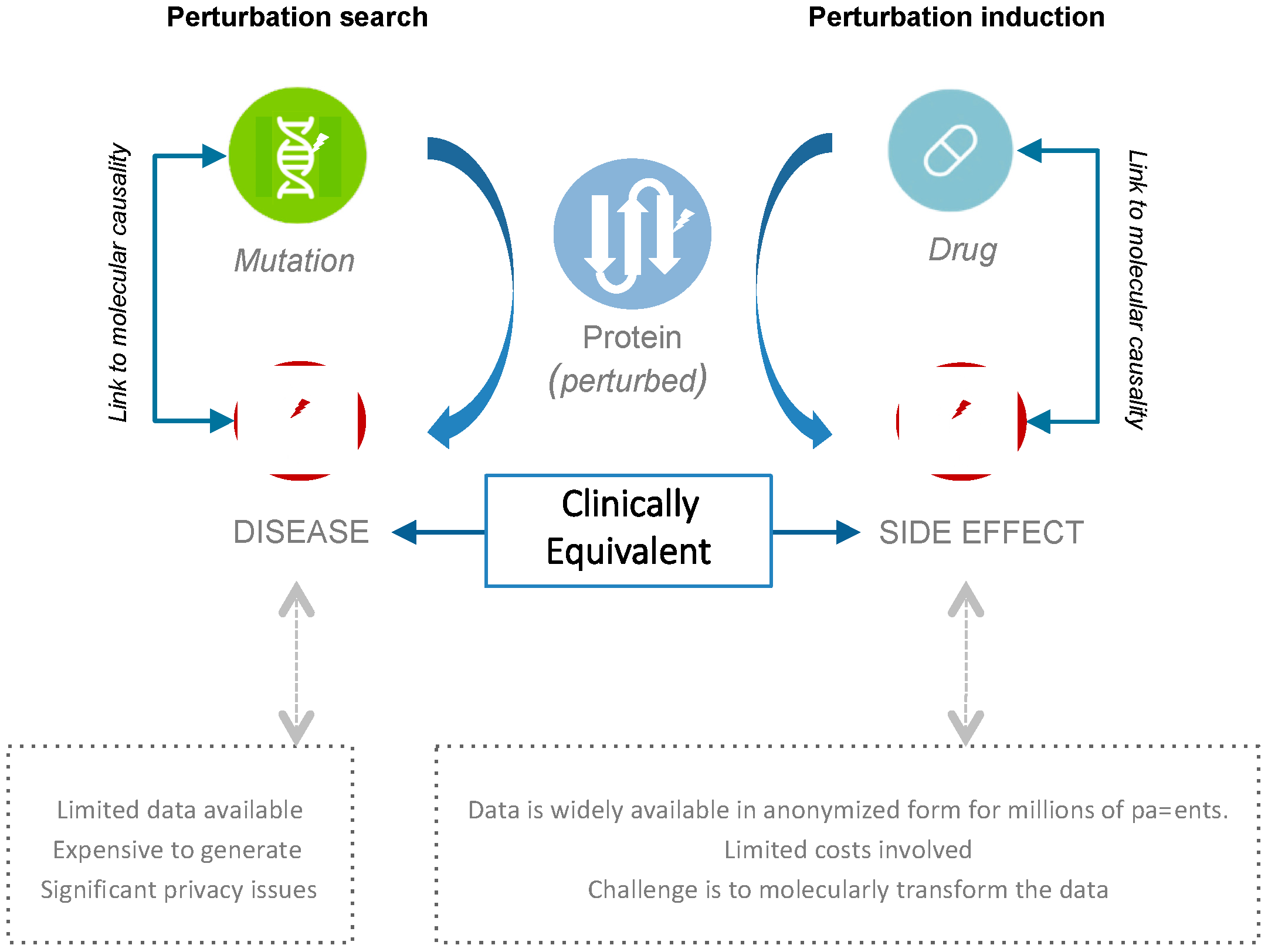
2.1. Connecting Clinical Phenotypes to Molecular Knowledge
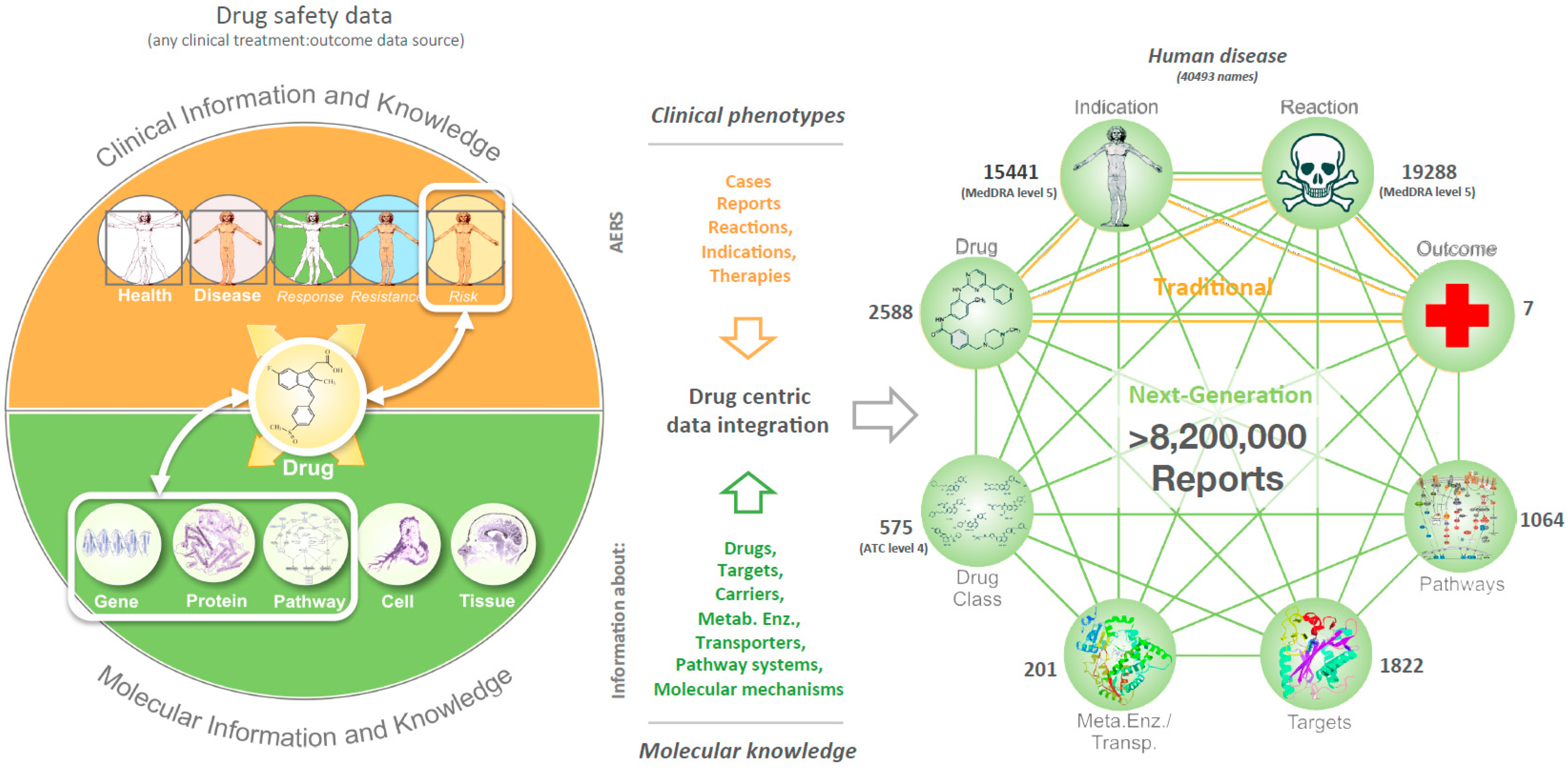
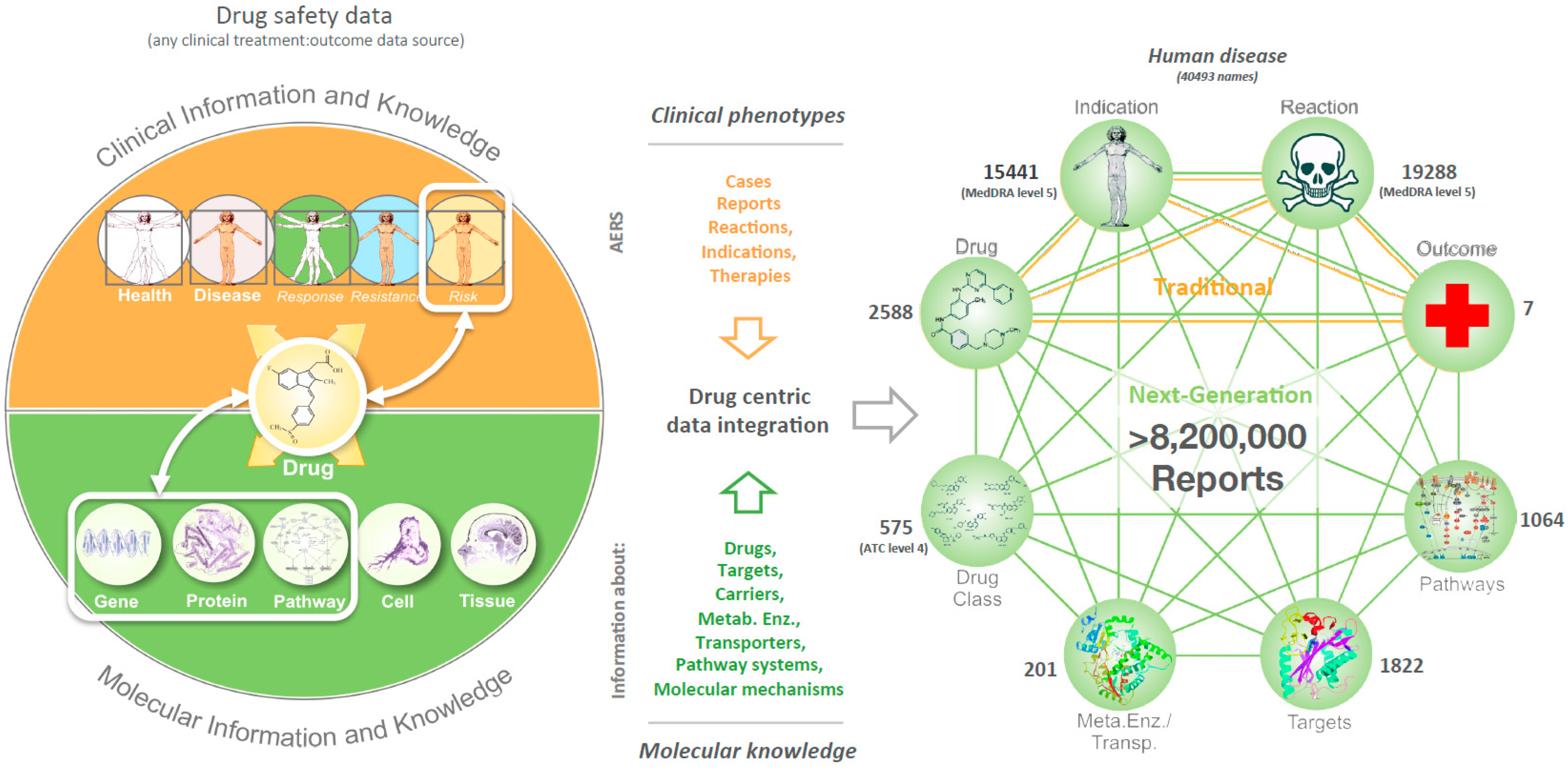
2.2. Data Integration
2.3. Characterization of Relationships
2.4. The Collection of Relationships
2.5. Presented Validation Examples
2.5.1. Prospective Prediction of Side Effects
2.5.2. Hypothesis Testing for Improved Use of Drugs
2.6. Benchmarking
- (a)
- To ensure specificity we primarily considered reactions described at MedDRA level 4. In many cases AEs mentioned exactly a certain term as reported in the original dataset (e.g., ‘Tachycardia’), whereas other times an effect could only be captured in AEs via higher MedDRA level descriptions (e.g., ‘salivation’ and ‘Salivary gland disorders NEC’). In some other cases this strategy led to the identification of reaction terms too specific and with scant occurrence in AEs (e.g., ‘Hypophosphatemia’ or ‘Hypocalcaemia’).
- (b)
- We searched only for reactions: The physiological effects expected to occur when compounds hit the listed targets of [15] may be characterized with increased PRRs in AEs, however sometimes they may only reflect a condition and not an adverse reaction.
- (c)
- We searched for up to two synonyms per case: Sometimes the same effect may have been declared via its synonyms or in similar spelling (e.g., ‘anemia’ and ‘anaemia’). In other cases, alternative names were searched to describe generic phrases of the original dataset (e.g., both ‘hypertension’ and ‘hypotension’ were searched for the originally termed effect ‘blood pressure changes’).
- (d)
- We also did not consider effects of proteins that were not targets or had no drugs reported in AEs.
2.7. Comparison of Target-Reaction Signals
2.8. Data Availability
3. Results
3.1. Phenotypic Profiling of Human Target Proteins
3.2. Using the Approach: Examples and Perspectives of Analytical Strategies
3.3. Validation Results
- Prospective prediction of drug side effects. We asked whether it might be possible to predict AE-profiles of novel drugs. Examining the reaction profiles of tyrosine kinase inhibitors (TKIs) revealed that Sorafenib is more strongly associated with dermatological reactions than Sunitinib (Supplementary Table S2). Indeed, skin-related AEs are a common form of side-effects observed in patients treated with TKIs [16,17,21,22]. However, while these two TKIs share a common target-inhibition profile [23,24,25,26], Sorafenib additionally inhibits BRAF. We capitalized then on the Sorafenib-specific side-effect difference to hone in on the BRAF-specific clinical effects and hypothesized that the reaction profiles of other BRAF inhibitors may also include skin-related AEs. To test whether we could have predicted this relationship for the BRAF-specific inhibitor Vemurafenib, we used a data slice of all patient-cases reported prior to Vemurafenib’s FDA-approval in August 2011. Through this prospective-retrospective analysis we found that BRAF perturbation was strongly related to dermatologic reactions (Supplementary Figure S2)—thus predicting the primary side-effects observed in Vemurafenib’s phase-3 trial [27] (NCT01006980). Indeed, predicted skin-related AEs are consistent with BRAF pathway findings from recent studies and other independent work [28,29,30,31]. Importantly, this clinical effect is also included in Vemurafenib’s FDA-label [32].
- Rational prediction of combination therapies. We also assessed whether we could identify combinations of targets that may improve therapeutic outcomes in patients. We reasoned that AE outcomes and in particular “death rate”, might be used as surrogate marker for treatment efficacy. Using prior knowledge about the association between biobehavioral stress and tumorigenesis [12,13], we investigated whether perturbation of beta-adrenergic receptor (BAR) function in cancer patients might result in lower patient mortality. We examined the phenotypic consequences of BAR activity modulation by comparing two similar cohorts of skin cancer patients—one arm with and one without inhibition of BAR activity (Supplementary Figure S3). In absence of BAR antagonism, reported deaths occurred in 23.6% cases, whereas with BAR inhibition deaths were reported in 18.4% cases. These results suggested that co-medication of BAR blockers may be associated with reduced mortality of skin cancer patients, and were supported by subsequent in vitro and in vivo studies which demonstrated the role of BAR in tumor growth and stress-response signaling via SRC activation in cancer cells. Further analysis revealed that mortality was reduced across major cancer types in patients where BAR signaling is inhibited with beta-blockers and identified BAR inhibition as a potential combinatorial route to anti-cancer treatment [33]. In support of these observations, a recent phase-2 pilot-study (NCT01265576) examining clinical effects of beta-blocker usage in hepatocellular carcinoma (HCC) patients with VT-122 (combination of the non-selective beta-blocker Propranolol and the COX2-selective Etodolac) demonstrated potential anticancer effects when co-administered with Sorafenib by increasing therapy duration and overall survival, as compared to HCC patients treated with Sorafenib alone [34]. Our example study emphasizes the importance of clinical phenotype profiling at the level of target perturbations, but also provides a potentially novel approach to drug repositioning, via rational design of combination therapies.
3.4. Benchmarcking and Comparison
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Horrobin, D.F. Modern biomedical research: An internally self-consistent universe with little contact with medical reality? Nat Rev. Drug Discov. 2003, 2, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [Green Version]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Shrivastava, S.; Hassanali, M.; Stothard, P.; Chang, Z.; Woolsey, J. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, D668–D672. [Google Scholar] [CrossRef] [PubMed]
- MedDRA. Available online: http://meddra.org/ (accessed on 21 March 2017).
- Soldatos, T.G.; Perdigão, N.; Brown, N.P.; Sabir, K.S.; O’Donoghue, S.I. How to learn about gene function: Text-mining or ontologies? Methods 2015, 74, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Milacic, M.; Haw, R.; Rothfels, K.; Wu, G.; Croft, D.; Hermjakob, H.; D’Eustachio, P.; Stein, L. Annotating cancer variants and anti-cancer therapeutics in reactome. Cancers 2012, 4, 1180–1211. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, C.F.; Anthony, K.; Krupa, S.; Buchoff, J.; Day, M.; Hannay, T.; Buetow, K.H. PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37, D674–D679. [Google Scholar] [CrossRef] [PubMed]
- WHOCC—Structure and Principles. Available online: https://www.whocc.no/atc/structure_and_principles/ (accessed on 21 March 2017).
- Van Puijenbroek, E.P.; Bate, A.; Leufkens, H.G.; Lindquist, M.; Orre, R.; Egberts, A.C. A comparison of measures of disproportionality for signal detection in spontaneous reporting systems for adverse drug reactions. Pharmacoepidemiol. Drug Saf. 2002, 11, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaker, P.H.; Han, L.Y.; Kamat, A.A.; Arevalo, J.M.; Takahashi, R.; Lu, C.; Jennings, N.B.; Armaiz-Pena, G.; Bankson, J.A.; Ravoori, M.; et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat. Med. 2006, 12, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Antoni, M.H.; Lutgendorf, S.K.; Cole, S.W.; Dhabhar, F.S.; Sephton, S.E.; McDonald, P.G.; Stefanek, M.; Sood, A.K. The influence of bio-behavioural factors on tumour biology: Pathways and mechanisms. Nat. Rev. Cancer 2006, 6, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Al Banchaabouchi, M.; Campillos, M.; Jensen, L.J.; Gross, C.; Gavin, A.C.; Bork, P. Systematic identification of proteins that elicit drug side effects. Mol. Syst. Biol. 2013, 9, 663. [Google Scholar] [CrossRef] [PubMed]
- Whitebread, S.; Hamon, J.; Bojanic, D.; Urban, L. Keynote review: In vitro safety pharmacology profiling: An essential tool for successful drug development. Drug Discov. Today 2005, 10, 1421–1433. [Google Scholar] [CrossRef]
- Perez-Soler, R. Rash as a surrogate marker for efficacy of epidermal growth factor receptor inhibitors in lung cancer. Clin. Lung Cancer 2006, 8 (Suppl. 1), S7–S14. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wu, Y.; Lv, T.F.; Yao, Y.W.; Xiao, Y.Y.; Yuan, D.M.; Song, Y. Skin Rash could Predict the Response to EGFR Tyrosine Kinase Inhibitor and the Prognosis for Patients with Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2013, 8, e55128. [Google Scholar] [CrossRef] [PubMed]
- Kozuki, T. Skin problems and EGFR-tyrosine kinase inhibitor. Jpn. J. Clin. Oncol. 2016, 46, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Racz, R.; Soldatos, T.G.; Jackson, D.; Burkhart, K. Association between Serotonin Syndrome and Second-Generation Antipsychotics via Pharmacological Target-Adverse Event Analysis. Clin. Transl. Sci. 2018. [Google Scholar] [CrossRef] [PubMed]
- Soldatos, T.G.; Dimitrakopoulou-Strauss, A.; Larribere, L.; Hassel, J.C.; Sachpekidis, C. Retrospective Side Effect Profiling of the Metastatic Melanoma Combination Therapy Ipilimumab-Nivolumab Using Adverse Event Data. Diagnostics 2018, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Poprach, A.; Pavlik, T.; Melichar, B.; Puzanov, I.; Dusek, L.; Bortlicek, Z.; Vyzula, R.; Abrahamova, J.; Buchler, T. Skin toxicity and efficacy of sunitinib and sorafenib in metastatic renal cell carcinoma: A national registry-based study. Ann. Oncol. 2012, 23, 3137–3143. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Wang, F.; Wu, G.; Zhang, L.; Wang, Y.; Wang, Z.; Chen, P.; Wang, Q.; Lu, J.; Wang, Y.; et al. The Relationship between the Adverse Events and Efficacy of Sorafenib in Patients with Metastatic Renal Cell Carcinoma. Medicine 2015, 94, e2222. [Google Scholar] [CrossRef] [PubMed]
- Van Erp, N.P.; Gelderblom, H.; Guchelaar, H.J. Clinical pharmacokinetics of tyrosine kinase inhibitors. Cancer Treat. Rev. 2009, 35, 692–706. [Google Scholar] [CrossRef] [PubMed]
- Di Gion, P.; Kanefendt, F.; Lindauer, A.; Scheffler, M.; Doroshyenko, O.; Fuhr, U.; Wolf, J.; Jaehde, U. Clinical pharmacokinetics of tyrosine kinase inhibitors: Focus on pyrimidines, pyridines and pyrroles. Clin. Pharmacokinet. 2011, 50, 551–603. [Google Scholar] [CrossRef] [PubMed]
- Sorafenib: FDA Label—NEXAVAR (in DailyMed). Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=b50667e4-5ebc-4968-a646-d605058dbef0 (accessed on 29 April 2016).
- Sunitinib: FDA Label—SUTENT (in DailyMed). Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=43a4d7f8-48ae-4a63-9108-2fa8e3ea9d9c (accessed on 29 April 2016).
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Blank, C.U.; Larkin, J.; Arance, A.M.; Hauschild, A.; Queirolo, P.; Del Vecchio, M.; Ascierto, P.A.; Krajsova, I.; Schachter, J.; Neyns, B.; et al. Open-label, multicentre safety study of vemurafenib in 3219 patients with BRAFV600 mutation-positive metastatic melanoma: 2-year follow-up data and long-term responders’ analysis. Eur. J. Cancer 2017, 79, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Kopetz, S.; Desai, J.; Chan, E.; Hecht, J.R.; O’Dwyer, P.J.; Maru, D.; Morris, V.; Janku, F.; Dasari, A.; Chung, W.; et al. Phase II Pilot Study of Vemurafenib in Patients with Metastatic BRAF-Mutated Colorectal Cancer. J. Clin. Oncol. 2015, 33, 4032–4038. [Google Scholar] [CrossRef] [PubMed]
- Huang, V.; Hepper, D.; Anadkat, M.; Cornelius, L. Cutaneous toxic effects associated with vemurafenib and inhibition of the BRAF pathway. Arch. Dermatol. 2012, 148, 628–633. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. RAS mutations in cutaneous squamous-cell carcinomas in patients treated with BRAF inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Vemurafenib: FDA Label—ZELBORAF (in DailyMed). Available online: https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=38eea320-7e0c-485a-bc30-98c3c45e2763 (accessed on 14 April 2016).
- Armaiz-Pena, G.N.; Allen, J.K.; Cruz, A.; Stone, R.L.; Nick, A.M.; Lin, Y.G.; Han, L.Y.; Mangala, L.S.; Villares, G.J.; Vivas-Mejia, P.; et al. Src activation by β-adrenoreceptors is a key switch for tumour metastasis. Nat. Commun. 2013, 4, 1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Torre, A.N.; Castaneda, I.; Hezel, A.F.; Bascomb, N.F.; Bhattacharyya, G.S.; Abou-Alfa, G.K. Effect of coadministration of propranolol and etodolac (VT-122) plus sorafenib for patients with advanced hepatocellular carcinoma (HCC). J. Clin. Oncol. 2015, 33. [Google Scholar] [CrossRef]
- Luo, Y.; Zhao, X.; Zhou, J.; Yang, J.; Zhang, Y.; Kuang, W.; Peng, J.; Chen, L.; Zeng, J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat. Commun. 2017, 8, 573. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Campillos, M.; González, P.; Jensen, L.J.; Bork, P. Large-scale prediction of drug-target relationships. FEBS Lett. 2008, 582, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Campillos, M.; Kuhn, M.; Gavin, A.C.; Jensen, L.J.; Bork, P. Drug target identification using side-effect similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Letunic, I.; Jensen, L.J.; Bork, P. The SIDER database of drugs and side effects. Nucleic Acids Res. 2016, 44, D1075–D1079. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Campillos, M.; Letunic, I.; Jensen, L.J.; Bork, P. A side effect resource to capture phenotypic effects of drugs. Mol. Syst. Biol. 2010, 6, 343. [Google Scholar] [CrossRef] [PubMed]
- Banda, J.M.; Evans, L.; Vanguri, R.S.; Tatonetti, N.P.; Ryan, P.B.; Shah, N.H. A curated and standardized adverse drug event resource to accelerate drug safety research. Sci. Data 2016, 3, 160026. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.R.; Jacunski, A.; Lorberbaum, T.; Romano, J.D.; Moskovitch, R.; Tatonetti, N.P. Systems biology approaches for identifying adverse drug reactions and elucidating their underlying biological mechanisms. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 104–122. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Clark, N.R.; Ma’ayan, A. Drug-induced adverse events prediction with the LINCS L1000 data. Bioinformatics 2016, 32, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Duggirala, H.J.; Tonning, J.M.; Smith, E.; Bright, R.A.; Baker, J.D.; Ball, R.; Bell, C.; Bright-Ponte, S.J.; Botsis, T.; Bouri, K.; et al. Use of data mining at the Food and Drug Administration. J. Am. Med. Inform. Assoc. 2016, 23, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Health Informatics at FDA > Data Mining at FDA—White Paper ‘Summary of Past and Present Data Mining Activities at the Food and Drug Administration’. Available online: http://www.fda.gov/scienceresearch/healthinformatics/ucm446239.htm (accessed on 21 January 2016).

{kind=link}
{kind=link}
| AE Cases | Event (E) | Not E | Totals |
|---|---|---|---|
| D (T) | a | b | A + b |
| Not D (T) | c | d | C + d |
| Totals | a + c | b + d | N = a + b + c + d |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Soldatos, T.G.; Taglang, G.; Jackson, D.B. In Silico Profiling of Clinical Phenotypes for Human Targets Using Adverse Event Data. High-Throughput 2018, 7, 37. https://doi.org/10.3390/ht7040037
Soldatos TG, Taglang G, Jackson DB. In Silico Profiling of Clinical Phenotypes for Human Targets Using Adverse Event Data. High-Throughput. 2018; 7(4):37. https://doi.org/10.3390/ht7040037
Chicago/Turabian StyleSoldatos, Theodoros G., Guillaume Taglang, and David B. Jackson. 2018. "In Silico Profiling of Clinical Phenotypes for Human Targets Using Adverse Event Data" High-Throughput 7, no. 4: 37. https://doi.org/10.3390/ht7040037
APA StyleSoldatos, T. G., Taglang, G., & Jackson, D. B. (2018). In Silico Profiling of Clinical Phenotypes for Human Targets Using Adverse Event Data. High-Throughput, 7(4), 37. https://doi.org/10.3390/ht7040037