SNPs and Somatic Mutation on Long Non-Coding RNA: New Frontier in the Cancer Studies?



Abstract
:1. Introduction
1.1. Impact of SNPs on lncRNAs
1.2. LncRNAs Involved in Cancer
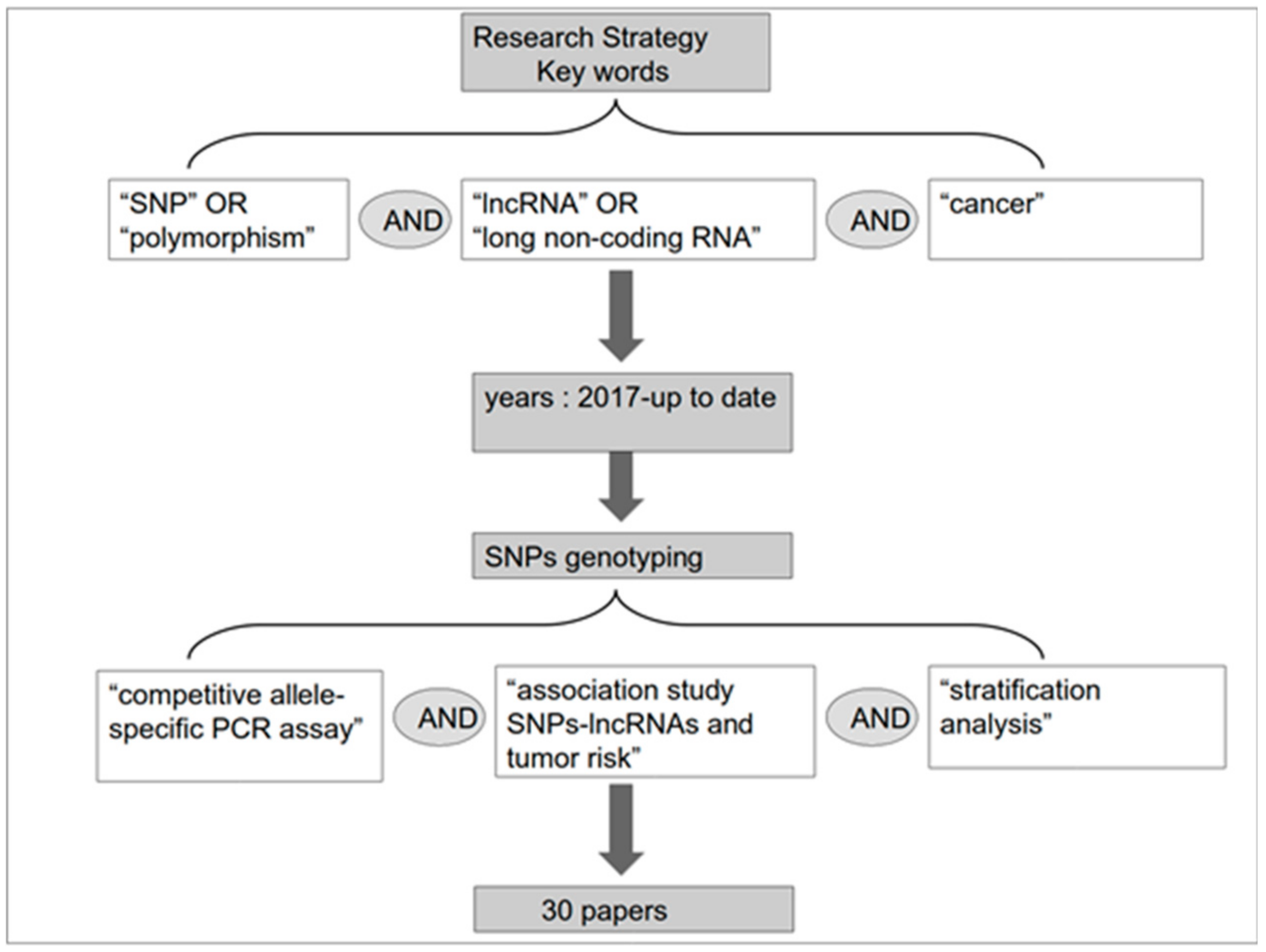
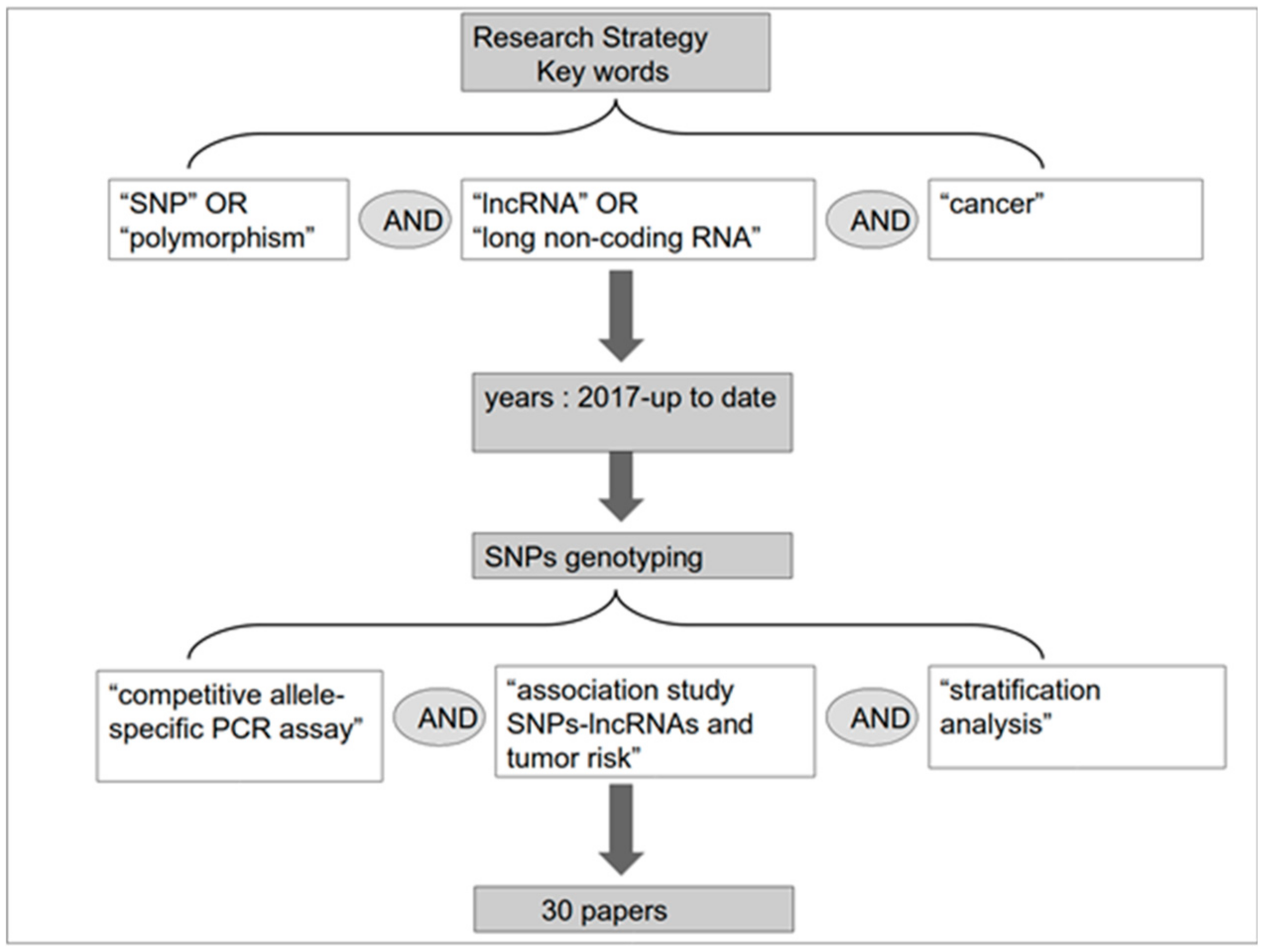
2. Methods for Detection of SNPs and Measurement of Association with Cancer Risk
2.1. SNPs on LncRNAs Involved in Cancer
2.2. Somatic Mutation of lncRNAs in Cancer
3. Discussion and Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- International HapMap Consortium; Frazer, K.A.; Ballinger, D.G.; Cox, D.R.; Hinds, D.A.; Stuve, L.L.; Gibbs, R.A.; Belmont, J.W.; Boudreau, A.; Hardenbol, P.; et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 2007, 449, 851–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachidanandam, R.; Weissman, D.; Schmidt, S.C.; Kakol, J.M.; Stein, L.D.; Marth, G.; Sherry, S.; Mullikin, J.C.; Mortimore, B.J.; Willey, D.L.; et al. International SNP Map Working Group A map of human genome sequence variation containing 1.42 million single nucleotide polymorphisms. Nature 2001, 409, 928–933. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Clements, J.A.; Batra, J. Single nucleotide polymorphisms in clinics: Fantasy or reality for cancer? Crit. Rev. Clin. Lab Sci. 2016, 53, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Westra, H.-J.; Karjalainen, J.; Zhernakova, D.V.; Esko, T.; Hrdlickova, B.; Almeida, R.; Zhernakova, A.; Reinmaa, E.; Võsa, U.; et al. Human disease-associated genetic variation impacts large intergenic non-coding RNA expression. PLoS Genet. 2013, 9, e1003201. [Google Scholar] [CrossRef] [PubMed]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucl. Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef] [PubMed]
- Sabarinathan, R.; Tafer, H.; Seemann, S.E.; Hofacker, I.L.; Stadler, P.F.; Gorodkin, J. The RNAsnp web server: Predicting SNP effects on local RNA secondary structure. Nucl. Acids Res. 2013, 41, W475–W479. [Google Scholar] [CrossRef] [PubMed]
- Bhartiya, D.; Jalali, S.; Ghosh, S.; Scaria, V. Distinct patterns of genetic variations in potential functional elements in long noncoding RNAs. Hum. Mutat. 2014, 35, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, L.; Chen, L.-L. Life without A tail: New formats of long noncoding RNAs. Int. J. Biochem. Cell Biol. 2014, 54, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Gutschner, T.; Diederichs, S. The hallmarks of cancer: A long non-coding RNA point of view. RNA Biol. 2012, 9, 703–719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhan, A.; Soleimani, M.; Mandal, S.S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 2017, 77, 3965–3981. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Chang, H.Y. Long Noncoding RNAs in Cancer Pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Bush, W.S.; Moore, J.H. Genome-Wide Association Studies. PLoS Comput. Biol. 2012, 8, e1002822. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Holme, J.; Anthony, J. SNP genotyping: The KASP assay. Methods Mol. Biol. 2014, 1145, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Tan, H. The association between gene SNPs and cancer predisposition: Correlation or causality? EBioMedicine 2017, 16, 8–9. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, L.W.; Ritchie, M.D.; Moore, J.H. Multifactor dimensionality reduction software for detecting gene-gene and gene-environment interactions. Bioinformatics 2003, 19, 376–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangiovanni, A.; Del Ninno, E.; Fasani, P.; De Fazio, C.; Ronchi, G.; Romeo, R.; Morabito, A.; De Franchis, R.; Colombo, M. Increased survival of cirrhotic patients with a hepatocellular carcinoma detected during surveillance. Gastroenterology 2004, 126, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Nahon, P.; Zucman-Rossi, J. Single nucleotide polymorphisms and risk of hepatocellular carcinoma in cirrhosis. J. Hepatol. 2012, 57, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Panzitt, K.; Tschernatsch, M.M.O.; Guelly, C.; Moustafa, T.; Stradner, M.; Strohmaier, H.M.; Buck, C.R.; Denk, H.; Schroeder, R.; Trauner, M.; et al. Characterization of HULC, a novel gene with striking up-regulation in hepatocellular carcinoma, as noncoding RNA. Gastroenterology 2007, 132, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Motsinger, A.A.; Ritchie, M.D. Multifactor dimensionality reduction: An analysis strategy for modelling and detecting gene-gene interactions in human genetics and pharmacogenomics studies. Hum. Genom. 2006, 2, 318–328. [Google Scholar]
- Wang, B.; Lv, Z.; Ding, H.; Fang, X.; Wen, J.; Xu, Q.; Yuan, Y. The association of lncRNA-HULC polymorphisms with hepatocellular cancer risk and prognosis. Gene 2018, 670, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.-G.; Xu, Q.; Lv, Z.; Fang, X.-X.; Ding, H.-X.; Wen, J.; Yuan, Y. Association of twelve polymorphisms in three onco-lncRNA genes with hepatocellular cancer risk and prognosis: A case-control study. World J. Gastroenterol. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-L.; Huang, Z.; Wang, Q.; Chen, H.-H.; Ma, S.-N.; Wu, R.; Cai, W.-S. The Association of Polymorphisms in lncRNA-H19 with Hepatocellular Cancer Risk and Prognosis. Biosci. Rep. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Thorat, M.A.; Andriole, G.; Brawley, O.W.; Brown, P.H.; Culig, Z.; Eeles, R.A.; Ford, L.G.; Hamdy, F.C.; Holmberg, L.; et al. Prevention and early detection of prostate cancer. Lancet Oncol. 2014, 15, e484–e492. [Google Scholar] [CrossRef] [Green Version]
- Botti, G.; Collina, F.; Scognamiglio, G.; Aquino, G.; Cerrone, M.; Liguori, G.; Gigantino, V.; Malzone, M.G.; Cantile, M. LncRNA HOTAIR Polymorphisms Association with Cancer Susceptibility in Different Tumor Types. Curr. Drug Targets 2018, 19, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.I.; Xavier-Magalhães, A.; Moreira-Barbosa, C.; Magalhães, H.; Henrique, R.; Jerónimo, C.; Costa, B.M. Influence of HOTAIR rs920778 and rs12826786 genetic variants on prostate cancer risk and progression-free survival. Biomark. Med. 2018, 12, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Cui, H.; Lou, Z.; Wang, X.; Chen, L.; Xie, Z.; Hehir, M.; Yao, X.; Ren, Y.; Cen, D.; et al. Association of rs3787016 in Long Non-coding RNAs POLR2E and rs2910164 in MiRNA-146a with Prostate Cancer: A Systematic Review and Meta-analysis. Iran. J. Public Health 2018, 47, 623–632. [Google Scholar] [PubMed]
- Hua, J.T.; Ahmed, M.; Guo, H.; Zhang, Y.; Chen, S.; Soares, F.; Lu, J.; Zhou, S.; Wang, M.; Li, H.; et al. Risk SNP-Mediated Promoter-Enhancer Switching Drives Prostate Cancer through lncRNA PCAT19. Cell 2018, 174, 564.e18–575.e18. [Google Scholar] [CrossRef] [PubMed]
- Sattarifard, H.; Hashemi, M.; Hassanzarei, S.; Narouie, B.; Bahari, G. Association between genetic polymorphisms of long non-coding RNA PRNCR1 and prostate cancer risk in a sample of the Iranian population. Mol. Clin. Oncol. 2017, 7, 1152–1158. [Google Scholar] [CrossRef] [PubMed]
- Taheri, M.; Pouresmaeili, F.; Omrani, M.D.; Habibi, M.; Sarrafzadeh, S.; Noroozi, R.; Rakhshan, A.; Sayad, A.; Ghafouri-Fard, S. Association of ANRIL gene polymorphisms with prostate cancer and benign prostatic hyperplasia in an Iranian population. Biomark. Med. 2017, 11, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Harismendy, O.; Notani, D.; Song, X.; Rahim, N.G.; Tanasa, B.; Heintzman, N.; Ren, B.; Fu, X.-D.; Topol, E.J.; Rosenfeld, M.G.; et al. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature 2011, 470, 264–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef]
- Gong, W.-J.; Yin, J.-Y.; Li, X.-P.; Fang, C.; Xiao, D.; Zhang, W.; Zhou, H.-H.; Li, X.; Liu, Z.-Q. Association of well-characterized lung cancer lncRNA polymorphisms with lung cancer susceptibility and platinum-based chemotherapy response. Tumour Biol. 2016, 37, 8349–8358. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Chen, S.-H.; Lv, Q.-L.; Sun, B.; Qu, Q.; Qin, C.-Z.; Fan, L.; Guo, Y.; Cheng, L.; Zhou, H.-H. Clinical Significance of Long Non-Coding RNA CASC8 rs10505477 Polymorphism in Lung Cancer Susceptibility, Platinum-Based Chemotherapy Response, and Toxicity. Int. J. Environ. Res. Public Health 2016, 13, 545. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, H.; Lv, X.; Yang, Z.; Gao, M.; Bi, Y.; Zhang, Z.; Wang, S.; Cui, Z.; Zhou, B.; et al. Polymorphism in lncRNA AC016683.6 and its interaction with smoking exposure on the susceptibility of lung cancer. Cancer Cell Int. 2018, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Li, H.; Li, J.; Lv, X.; Gao, M.; Bi, Y.; Zhang, Z.; Wang, S.; Li, S.; Li, N.; et al. Association Between Long Noncoding RNA MEG3 Polymorphisms and Lung Cancer Susceptibility in Chinese Northeast Population. DNA Cell Biol. 2018, 37, 812–820. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Cui, Z.; Li, H.; Li, J.; Yang, Z.; Bi, Y.; Gao, M.; Zhou, B.; Yin, Z. Polymorphism in lncRNA AC008392.1 and its interaction with smoking on the risk of lung cancer in a Chinese population. Cancer Manag. Res. 2018, 10, 1377–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadaş, E.; Aydın, M. Effect of HOTAIR rs12826786 and rs1899663 polymorphisms on lung cancer susceptibility and clinicopathological characteristics in a Turkish population: A hospital-based case-control study. Cell. Mol. Biol. 2018, 64, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Gurney, J.G.; Ross, J.A.; Wall, D.A.; Bleyer, W.A.; Severson, R.K.; Robison, L.L. Infant cancer in the U.S.: Histology-specific incidence and trends, 1973 to 1992. J. Pediatr. Hematol. Oncol. 1997, 19, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, Z.-J.; Zhang, R.; Zhang, J.; Zhu, J.; Yang, T.; Zou, Y.; He, J.; Xia, H. Associations between lncRNA MEG3 polymorphisms and neuroblastoma risk in Chinese children. Aging 2018, 10, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; He, J.; Chang, Y.; Luo, A.; Luo, A.; Zhang, J.; Zhang, R.; Xia, H.; Xu, L. HOTAIR gene polymorphisms contribute to increased neuroblastoma susceptibility in Chinese children. Cancer 2018, 124, 2599–2606. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chang, Y.; Jia, W.; Zhang, J.; Zhang, R.; Zhu, J.; Yang, T.; Xia, H.; Zou, Y.; He, J. LINC00673 rs11655237 C>T confers neuroblastoma susceptibility in Chinese population. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhall, G. Medulloblastoma. J. Child Neurol. 2009, 24, 1418–1430. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-D.; Zhang, N.; Qiu, X.-G.; Yuan, J.; Yang, M. LncRNA CDKN2BAS rs2157719 genetic variant contributes to medulloblastoma predisposition. J. Gene Med. 2018, 20. [Google Scholar] [CrossRef] [PubMed]
- Haggar, F.A.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Hu, Y.; Jing, F.; Li, Y.; Gu, S.; Jiang, X.; Mao, Y.; Li, Q.; Jin, M.; Chen, K. A novel SNP in promoter region of RP11-3N2.1 is associated with reduced risk of colorectal cancer. J. Hum. Genet. 2018, 63, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bao, C.; Gu, S.; Ye, D.; Jing, F.; Fan, C.; Jin, M.; Chen, K. Associations between novel genetic variants in the promoter region of MALAT1 and risk of colorectal cancer. Oncotarget 2017, 8, 92604–92614. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Gu, S.; Ye, D.; Li, Y.; Jing, F.; Li, Q.; Chen, K. Association between genetic variants in the promoter region of a novel antisense long noncoding RNA RP11-392P7.6 and colorectal cancer risk. Environ. Mol. Mutagen. 2017, 58, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jing, F.; Ding, Y.; He, Q.; Zhong, Y.; Fan, C. Long noncoding RNA CCAT1 polymorphisms are associated with the risk of colorectal cancer. Cancer Genet. 2018, 222–223, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yang, C.; Tong, S.; Ding, Y.; Deng, W.; Song, D.; Xiao, K. Genetic variation of long non-coding RNA TINCR contribute to the susceptibility and progression of colorectal cancer. Oncotarget 2017, 8, 33536–33543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkali, Z.; Chan, T.; Manoharan, M.; Algaba, F.; Busch, C.; Cheng, L.; Kiemeney, L.; Kriegmair, M.; Montironi, R.; Murphy, W.M.; et al. Bladder cancer: Epidemiology, staging and grading, and diagnosis. Urology 2005, 66, 4–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Zhang, Q.; Gu, D.; Zhang, K.; Ge, Y.; Chu, H.; Du, M.; Xu, B.; Wang, M.; et al. Tagging SNPs in the HOTAIR gene are associated with bladder cancer risk in a Chinese population. Gene 2018, 664, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Shah, R.; Rosso, K.; Nathanson, S.D. Pathogenesis, prevention, diagnosis and treatment of breast cancer. World J. Clin. Oncol. 2014, 5, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Hassanzarei, S.; Hashemi, M.; Sattarifard, H.; Hashemi, S.M.; Bahari, G.; Ghavami, S. Genetic polymorphisms of HOTAIR gene are associated with the risk of breast cancer in a sample of southeast Iranian population. Tumour Biol. 2017, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markopoulos, A.K. Current Aspects on Oral Squamous Cell Carcinoma. Open Dent. J. 2012, 6, 126. [Google Scholar] [CrossRef] [PubMed]
- Gorlick, R.; Khanna, C. Osteosarcoma. J. Bone Miner. Res. 2010, 25, 683–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Q.-Y.; Wang, H.; Wang, Y. LncRNA H19 polymorphisms associated with the risk of OSCC in Chinese population. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3770–3774. [Google Scholar] [PubMed]
- He, T.-D.; Xu, D.; Sui, T.; Zhu, J.-K.; Wei, Z.-X.; Wang, Y.-M. Association between H19 polymorphisms and osteosarcoma risk. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3775–3780. [Google Scholar] [PubMed]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Qiao, O.; Wang, J.; Li, J.; Jin, H.; Li, Z.; Jin, Y. rs1859168 A > C polymorphism regulates HOTTIP expression and reduces risk of pancreatic cancer in a Chinese population. World J. Surg. Oncol. 2017, 15, 155. [Google Scholar] [CrossRef] [PubMed]
- Zali, H.; Rezaei-Tavirani, M.; Azodi, M. Gastric cancer: Prevention, risk factors and treatment. Gastroenterol. Hepatol. Bed Bench 2011, 4, 175–185. [Google Scholar] [PubMed]
- Duan, F.; Jiang, J.; Song, C.; Wang, P.; Ye, H.; Dai, L.; Zhang, J.; Wang, K. Functional long non-coding RNAs associated with gastric cancer susceptibility and evaluation of the epidemiological efficacy in a central Chinese population. Gene 2018, 646, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Ching, T.; Garmire, L.X. Pan-cancer analysis of expressed somatic nucleotide variants in long intergenic non-coding RNA. Pac. Symp. Biocomput. 2018, 23, 512–523. [Google Scholar] [PubMed]
- Camacho, N.; Van Loo, P.; Edwards, S.; Kay, J.D.; Matthews, L.; Haase, K.; Clark, J.; Dennis, N.; Thomas, S.; Kremeyer, B.; et al. Appraising the relevance of DNA copy number loss and gain in prostate cancer using whole genome DNA sequence data. PLoS Genet. 2017, 13, e1007001. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Trincado, J.L.; Tatlow, P.J.; Piccolo, S.R.; Eyras, E. Genome Sequencing and RNA-Motif Analysis Reveal Novel Damaging Noncoding Mutations in Human Tumors. Mol. Cancer Res. 2018, 16, 1112–1124. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhou, L.; Ge, M.; Zhang, B.; Yang, X.; Xiong, X.; Fu, G.; Zhang, J.; Nie, X.; Li, H.; et al. Whole exome sequencing identifies lncRNA GAS8-AS1 and LPAR4 as novel papillary thyroid carcinoma driver alternations. Hum. Mol. Genet. 2016, 25, 1875–1884. [Google Scholar] [CrossRef] [PubMed]
- Saka, E.; Harrison, B.J.; West, K.; Petruska, J.C.; Rouchka, E.C. Framework for reanalysis of publicly available Affymetrix® GeneChip® data sets based on functional regions of interest. BMC Genom. 2017, 18, 875. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, A.; Furuta, M.; Totoki, Y.; Tsunoda, T.; Kato, M.; Shiraishi, Y.; Tanaka, H.; Taniguchi, H.; Kawakami, Y.; Ueno, M.; et al. Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet. 2016, 48, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Puente, X.S.; Beà, S.; Valdés-Mas, R.; Villamor, N.; Gutiérrez-Abril, J.; Martín-Subero, J.I.; Munar, M.; Rubio-Pérez, C.; Jares, P.; Aymerich, M.; et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature 2015, 526, 519–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, R.M.; Musunuru, K. Mapping Novel Pathways in Cardiovascular Disease Using eQTL Data: The Past, Present, and Future of Gene Expression Analysis. Front. Genet. 2012, 3, 232. [Google Scholar] [CrossRef] [PubMed]
- Low, J.T.; Weeks, K.M. SHAPE-directed RNA secondary structure prediction. Methods 2010, 52, 150–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xie, Y.; Li, L.; He, Y.; Zheng, D.; Yu, P.; Yu, L.; Tang, L.; Wang, Z. EZH2 RIP-seq Identifies Tissue-specific Long Non-coding RNAs. Curr. Gene Ther. 2018, 18, 275. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; An, G.; Zhao, C.; Ouyang, Z.; Bo, X.; Shu, W. Lnc2Catlas: An atlas of long noncoding RNAs associated with risk of cancers. Sci. Rep. 2018, 8, 1909. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.R.; Liu, W.; Zhang, Q.; Guo, A.Y. lncRNASNP2: An updated database of functional SNPs and mutations in human and mouse lncRNAs. Nucl. Acids Res. 2017, 46, D276–D280. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.S.; Roy, C.R.; Mondal, N.R.; Chakravarty, B.; Chatterjee, T.; Roy, S.; Sengupta, S. Identification of genetic variation in the lncRNA HOTAIR associated with HPV16-related cervical cancer pathogenesis. Cell. Oncol. 2016, 39, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Liu, H.; Liu, Z.; Owzar, K.; Han, Y.; Su, L.; Wei, Y.; Hung, R.J.; McLaughlin, J.; Brhane, Y.; et al. A Novel Genetic Variant in Long Non-coding RNA Gene NEXN-AS1 is Associated with Risk of Lung Cancer. Sci. Rep. 2016, 6, 34234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
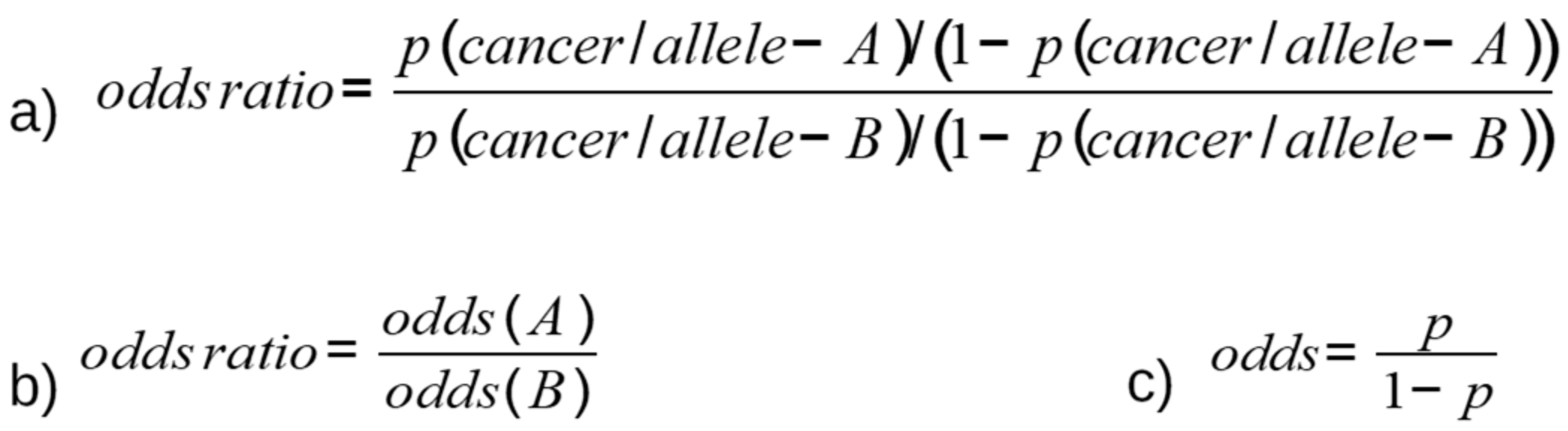

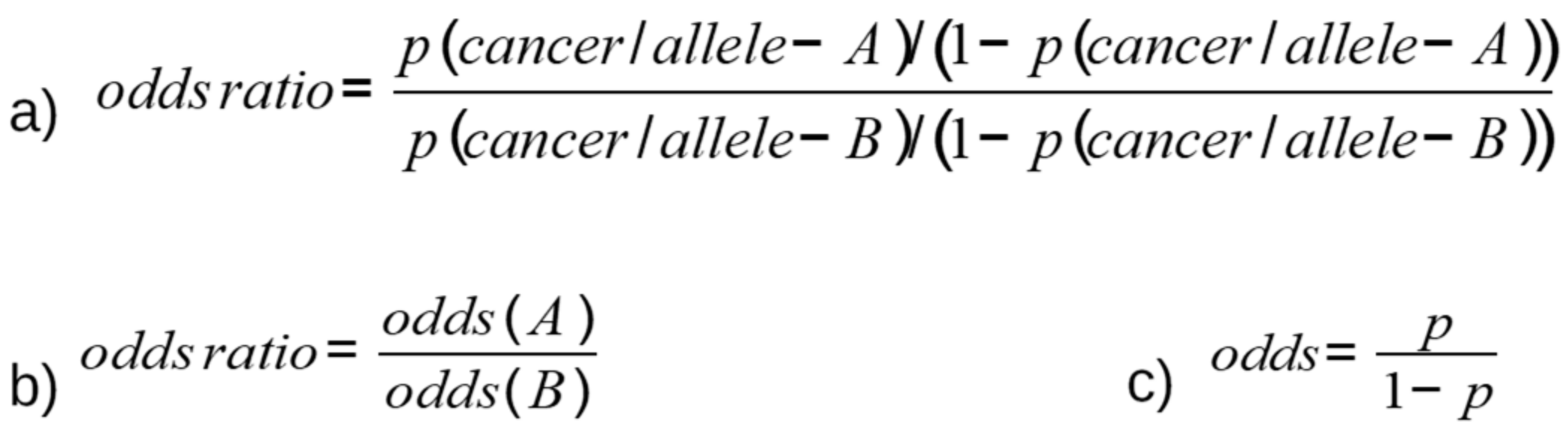{kind=link}
{kind=link}
| lncRNA | SNP | Allele (Major/Minor) | Cancer Risk/Model | Environmental Risk | Subgroup Risk | Cancer | PMID | n. Cases Test | n. Controls Tests |
|---|---|---|---|---|---|---|---|---|---|
| AC008392.1 | rs7248320 | A/G | ↓ recessive | ↓ recessive > 60 years | lung NSCLC | 29881308 | 512 | 588 | |
| AC016683.6 | rs4848320 | C/G/T | ↑ dominant smoking | lung | 29997452 | 434 | 593 | ||
| AC016683.6 | rs111083 | A/C/G/T | ↑ homozygous smoking | lung | 29997452 | 434 | 593 | ||
| AMFR-1:1 | rs4784659 | C/T | ↓ dominant and additive | gastric cancer | 29305976 | 470 | 470 | ||
| ANRIL | rs4977574 | A/G/T | ↑ allelic | ↑ BPH | prostate | 28621612 | 125 | 220 | |
| ANRIL | rs1333048 | A/C | ↑ allelic | ↑ BPH | prostate | 28621612 | 125 | 220 | |
| ANRIL | rs10757278 | A/G | ↑ allelic | ↑ BPH | prostate | 28621612 | 125 | 220 | |
| CASC8 | rs10505477 | C/T | ↓ dominant platinum-based chemotherapy resistance | ↑ recessive male and adenocarcinoma | lung | 27249003 | 498 | 213 | |
| CCAT1 | rs7013433 | A/C/T | ↑ dominant late clinical stage | colon rectal cancer | 29666003 | 507 | 503 | ||
| CCAT1 | rs67085638 | C/T | ↑ dominant | colon rectal cancer | 29666003 | 507 | 503 | ||
| CDKN2BAS | rs2157719 | T/C | ↑ dominant | ↑ pediatric and males | medulloblastoma | 29314442 | 160 | 443 | |
| EVX1-3:3 | rs1859168 | A/C/G/T | ↓ dominant and recessive | gastric cancer | 29305976 | 470 | 470 | ||
| H19 | rs2839698 | C/T/A | ↑ dominant | ↑ ever smoking | ↑ <60 years | hepatocellular carcinoma | 29511035 | 472 | 472 |
| H19 | rs3024270 | C/G | ↑ <60 years | hepatocellular carcinoma | 29511035 | 472 | 472 | ||
| H19 | rs217727 | C/T | ↑ additive | oral squamous cell carcinoma | 28975993 | 362 | 741 | ||
| H19 | rs217727 | G/A | ↑ dominant | osteosarcoma | 28975992 | 193 | 393 | ||
| HOTAIR | rs874945 | G/A | ↑ dominant | ↑ >60 years never smoking | bladder | 29673865 | 1050 | 1407 | |
| HOTAIR | rs920778 | C/T | ↑ dominant | breast | 29022495 | 220 | 231 | ||
| HOTAIR | rs12826786 | C/T | ↑ dominant | breast | 29022495 | 220 | 231 | ||
| HOTAIR | rs1899663 | G/T | ↓ dominant | breast | 29022495 | 220 | 231 | ||
| HOTAIR | rs12826786 | C/T | ↑ dominant | ↑ female | infant neuroblastoma | 29603181 | 393 | 812 | |
| HOTAIR | rs874945 | C/T | ↑ dominant | ↑ female | infant neuroblastoma | 29603181 | 393 | 812 | |
| HOTAIR | rs1899663 | C/A | ↑ dominant | ↑ female | infant neuroblastoma | 29603181 | 393 | 812 | |
| HOTAIR | rs12826786 | C/T | ↑ combination | lung | 29974853 | 87 | 93 | ||
| HOTAIR | rs1899663 | G/T | ↑ combination | lung | 29974853 | 87 | 93 | ||
| HOTAIR | rs12826786 | C/T | ↑ shorter biochemical recurrence-free survival in pT3-stage | prostate | 29436234 | 151 | 180 | ||
| HOTTIP | rs17501292 | T/C/G | ↑ allelic | hepatocellular carcinoma | 29930469 | 521 | 817 | ||
| HOTTIP | rs2067087 | G/C | ↑ allelic | hepatocellular carcinoma | 29930469 | 521 | 817 | ||
| HOTTIP | rs17427960 | C/A | ↑ allelic | hepatocellular carcinoma | 29930469 | 521 | 817 | ||
| HOTTIP | rs3807598 | C/G | ↓ HBV-negative | hepatocellular carcinoma | 29930469 | 521 | 817 | ||
| HOTTIP | rs1859168 | A/C | ↓ dominant | pancreatic | 28818070 | 416 | 146 | ||
| HULC | rs1041279 | C/G | ↑ recessive | n.d | ↑ male | hepatocellular carcinoma | 29803923 | 517 | 810 |
| HULC | rs2038540 | C/G | n.d | ↑ smokers-drinkers | n.d | hepatocellular carcinoma | 29803923 | 517 | 810 |
| LINC00673 | rs11655237 | C/T | ↑ dominant | ↑ patients with tumor originating from the adrenal gland | infant neuroblastoma | 29339420 | 393 | 812 | |
| MALAT1 | rs1194338 | C/A | ↓ dominant | n.d | n.d | colon rectal cancer | 29190941 | 320 | 319 |
| MALAT1 | rs4102217 | G/C | ↑ dominant | hepatocellular carcinoma | 29930469 | 521 | 817 | ||
| MALAT1 | rs591291 | C/T | ↓ HBV-negative and female | hepatocellular carcinoma | 29930469 | 521 | 817 | ||
| MEG3 | rs7158663 | G/A | ↑ combination | infant neuroblastoma | 29615542 | 392 | 783 | ||
| MEG3 | rs4081134 | G/A | ↑ recessive > 18 month | infant neuroblastoma | 29615542 | 392 | 783 | ||
| MEG3 | rs4081134 | A/G | ↑ dominant | lung | 30113224 | 526 | 526 | ||
| PCAT19 | rs11672691 | A/G | ↑ bifunctional | prostate | 30033362 | ||||
| POLR2E | rs3787016 | C/T | ↑ all genotype | prostate | 29922603 | 5 | 5 | ||
| PRNCR1 | rs13252298 | A/G | ↑ recessive | prostate | 29285392 | 178 | 180 | ||
| PRNCR1 | rs1456315 | G/A | ↑ allelic | prostate | 29285392 | 178 | 180 | ||
| PRNCR1 | rs7841060 | T/G | ↑ allelic | prostate | 29285392 | 178 | 180 | ||
| RP11-392P7.6 | rs10845671 | A/C/T | ↑ dominant | colon rectal cancer | 28612367 | 320 | 319 | ||
| RP11-3N2.1 | rs13230517 | G/A | ↓ dominant | ↓ non-drinkers | n.d | colon rectal cancer | 29167551 | 320 | 319 |
| TINCR | rs2288947 | A/G | ↓ dominant | colon rectal cancer | 28418933 | 1400 | 1400 | ||
| TINCR | rs8105637 | A/G | ↑ dominant | colon rectal cancer | 28418933 | 1400 | 1400 | ||
| ZNF33B-2:1 | rs579501 | A/C | ↓ dominant and additive | gastric cancer | 29305976 | 470 | 470 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minotti, L.; Agnoletto, C.; Baldassari, F.; Corrà, F.; Volinia, S. SNPs and Somatic Mutation on Long Non-Coding RNA: New Frontier in the Cancer Studies? High-Throughput 2018, 7, 34. https://doi.org/10.3390/ht7040034
Minotti L, Agnoletto C, Baldassari F, Corrà F, Volinia S. SNPs and Somatic Mutation on Long Non-Coding RNA: New Frontier in the Cancer Studies? High-Throughput. 2018; 7(4):34. https://doi.org/10.3390/ht7040034
Chicago/Turabian StyleMinotti, Linda, Chiara Agnoletto, Federica Baldassari, Fabio Corrà, and Stefano Volinia. 2018. "SNPs and Somatic Mutation on Long Non-Coding RNA: New Frontier in the Cancer Studies?" High-Throughput 7, no. 4: 34. https://doi.org/10.3390/ht7040034
APA StyleMinotti, L., Agnoletto, C., Baldassari, F., Corrà, F., & Volinia, S. (2018). SNPs and Somatic Mutation on Long Non-Coding RNA: New Frontier in the Cancer Studies? High-Throughput, 7(4), 34. https://doi.org/10.3390/ht7040034