Impact of Genomics on Clarifying the Evolutionary Relationships amongst Mycobacteria: Identification of Molecular Signatures Specific for the Tuberculosis-Complex of Bacteria with Potential Applications for Novel Diagnostics and Therapeutics

Abstract
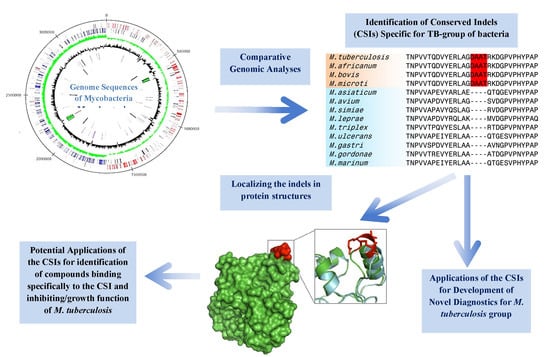
:
1. Introduction
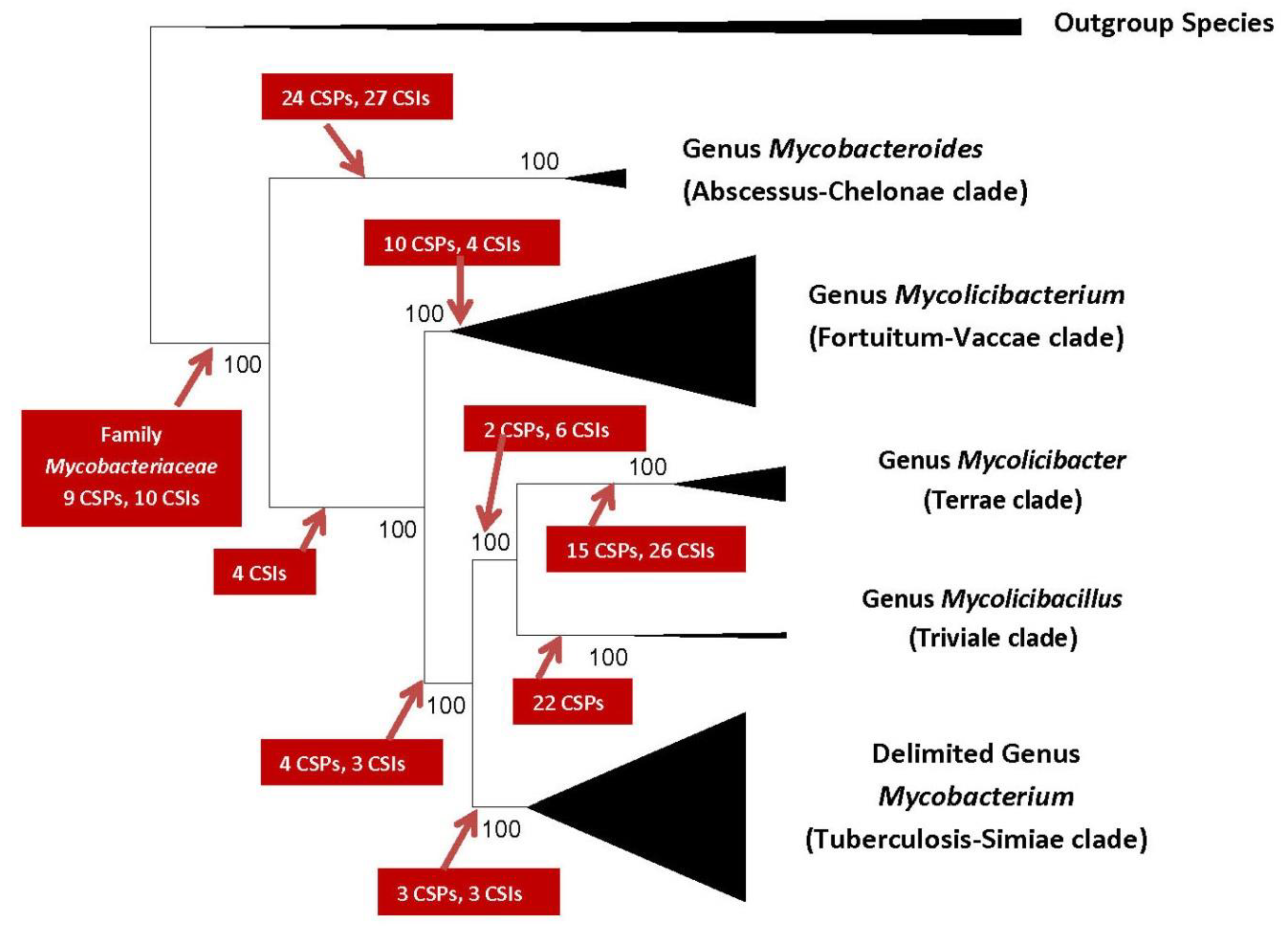
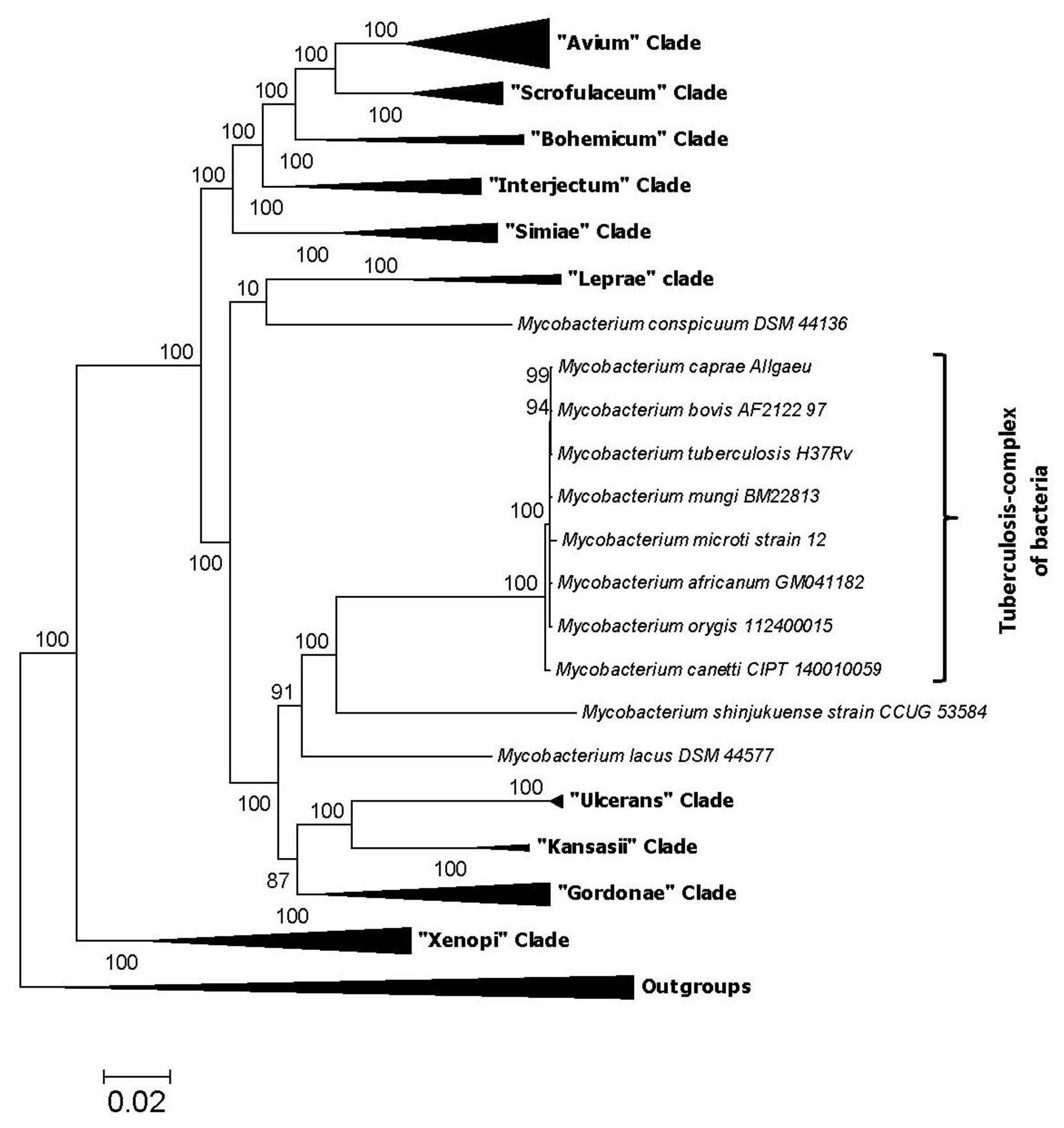
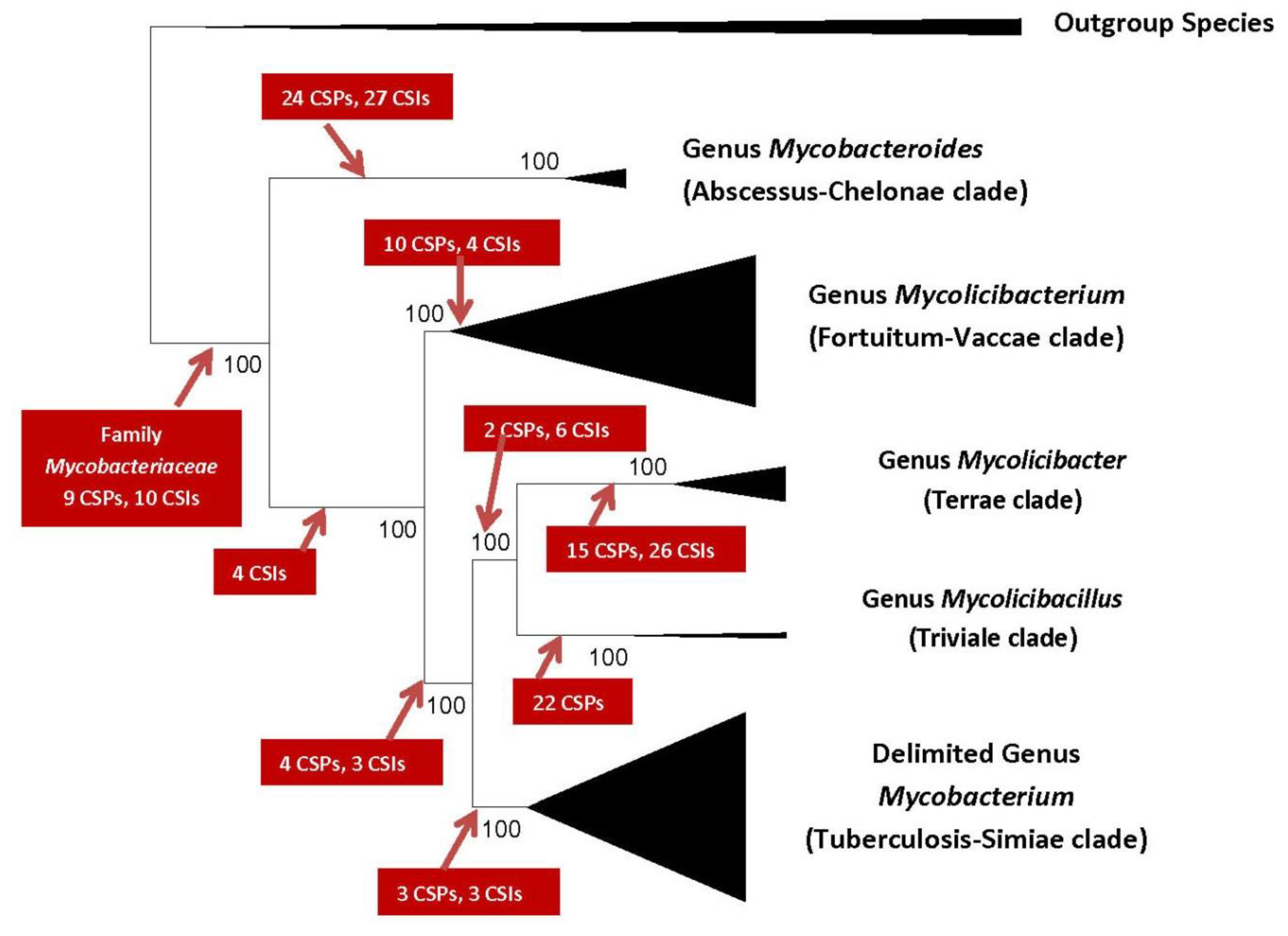
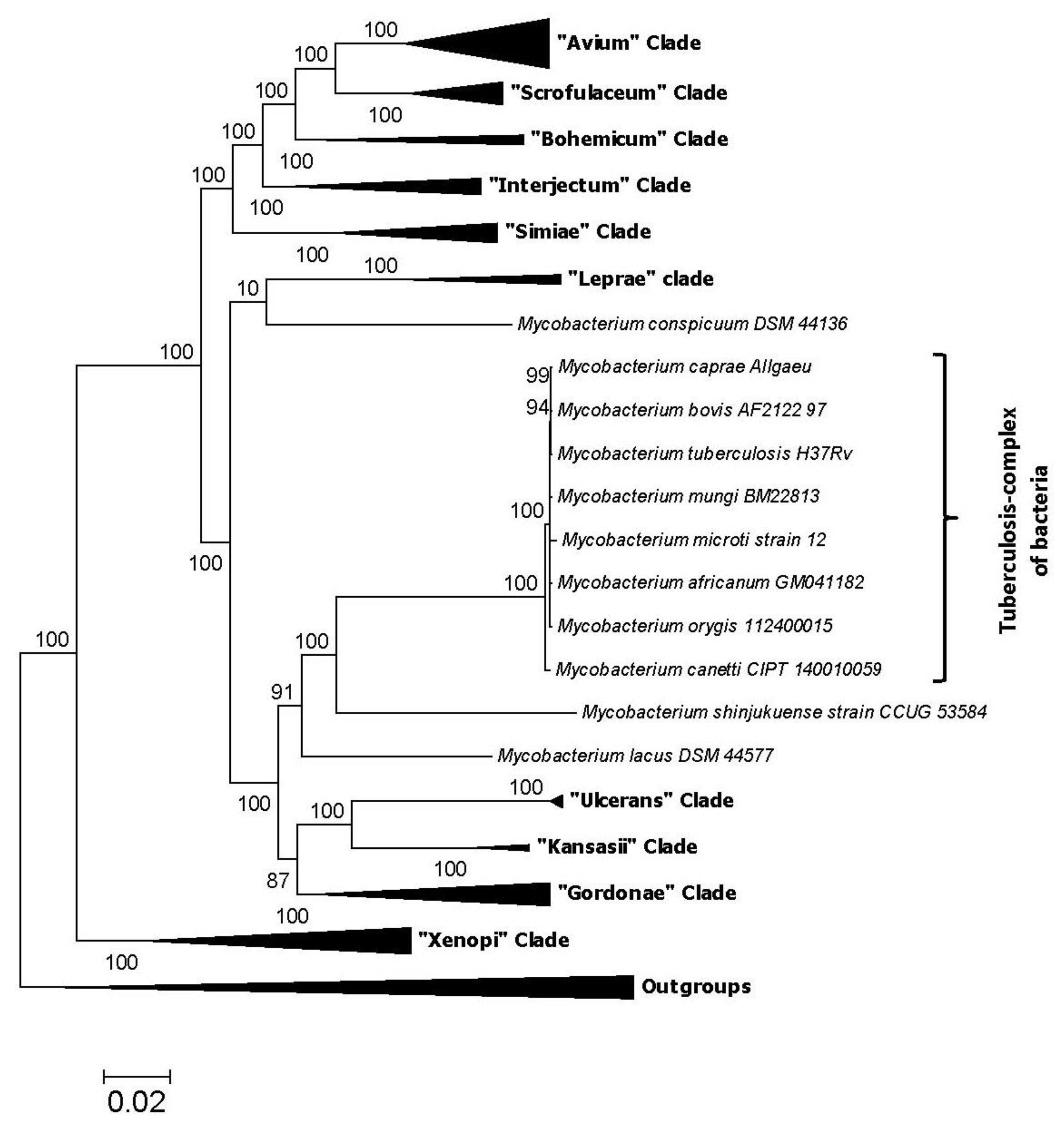
2. Impact of Genomics on Clarifying the Evolutionary Relationships amongst Mycobacteria
3. Genetic and Biological Significance of the Conserved Signature Indels
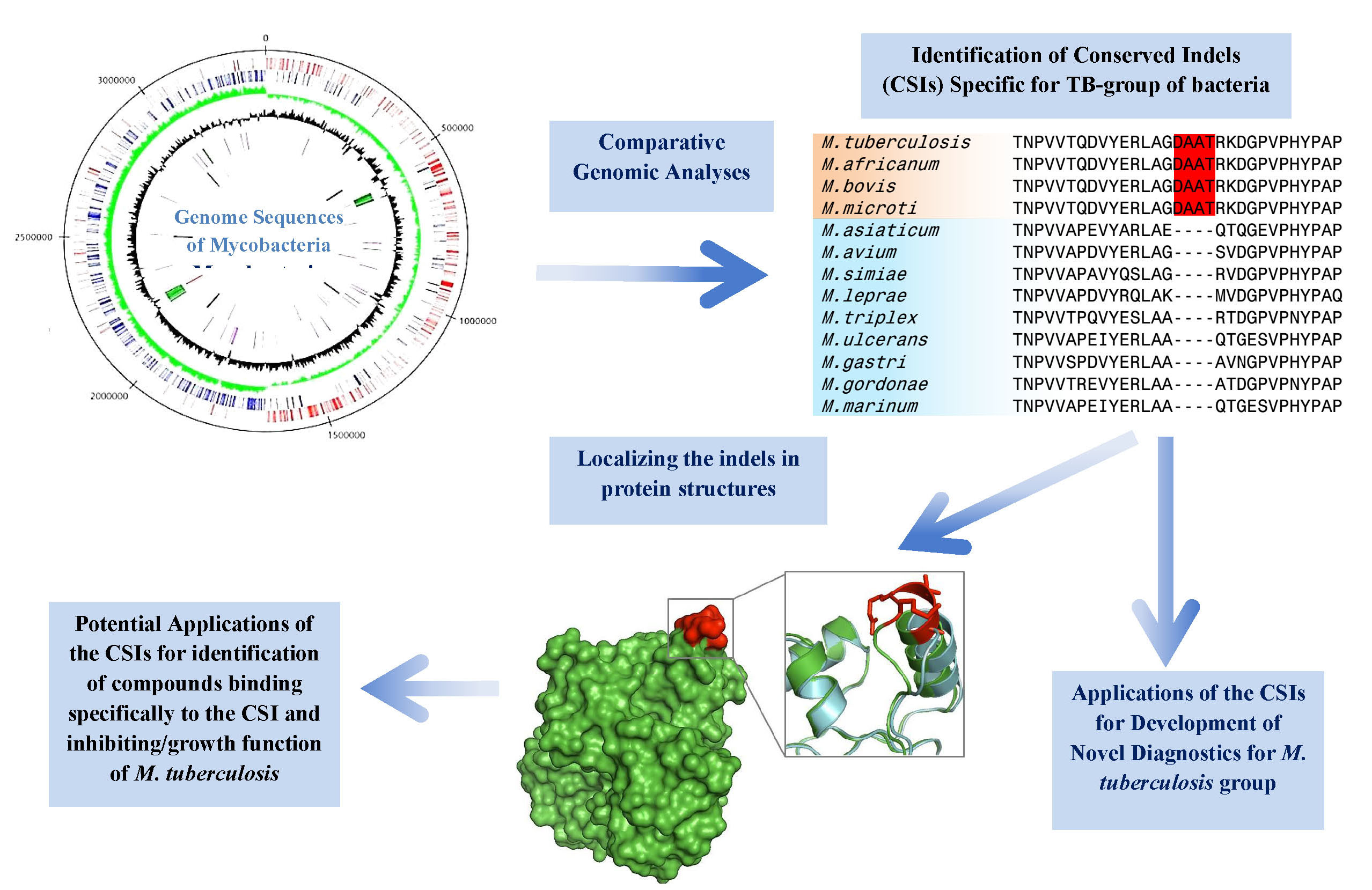
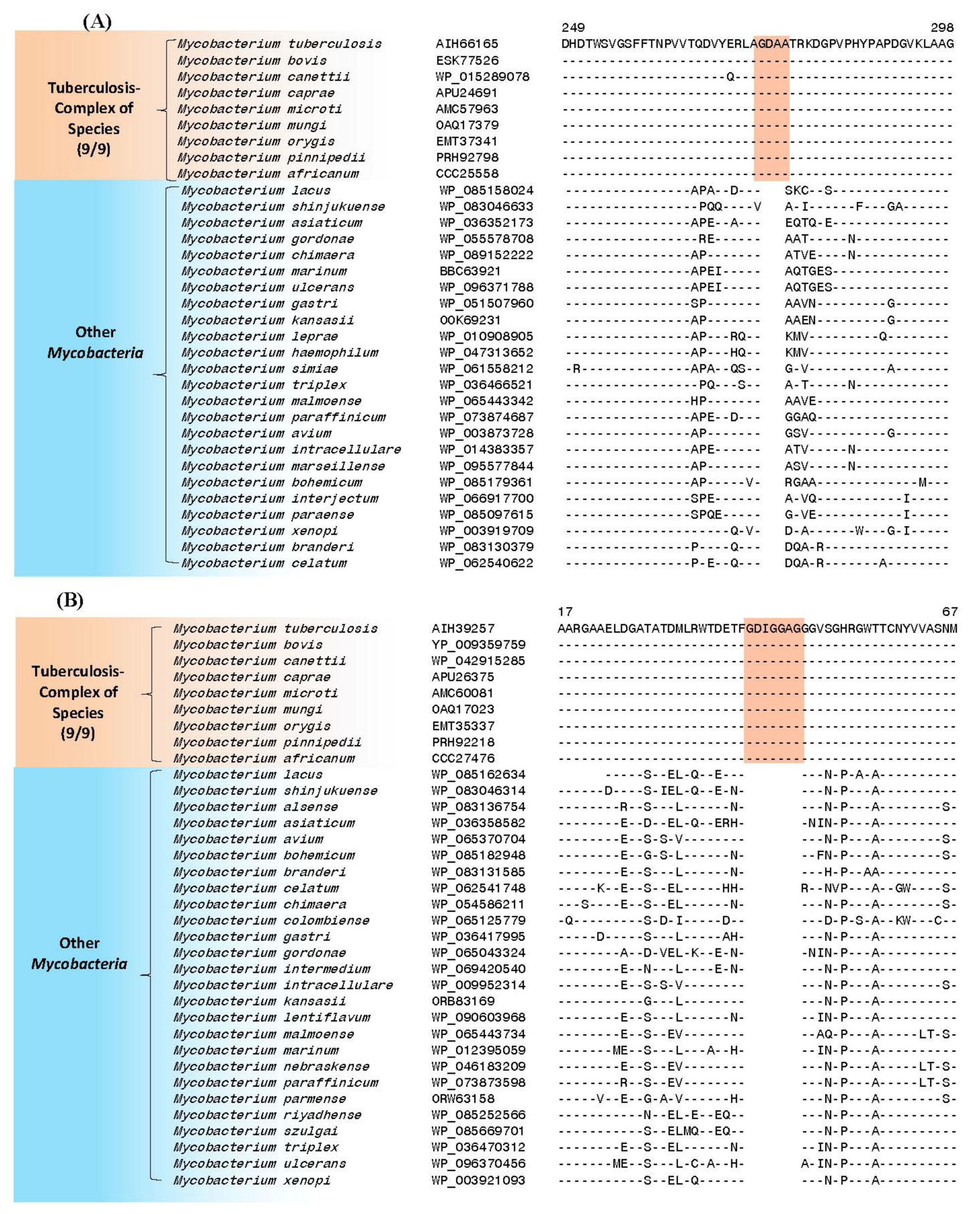
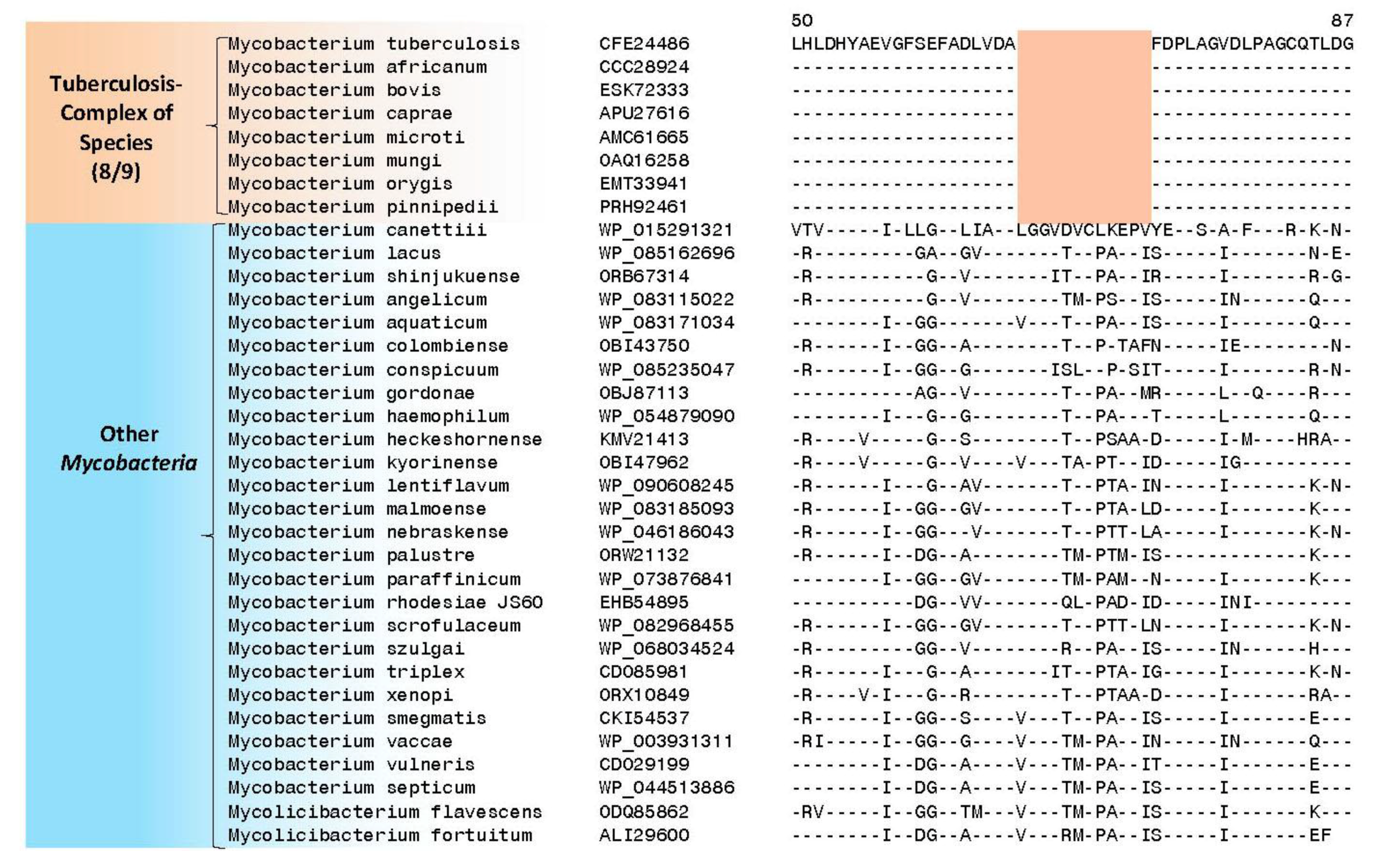
4. Conserved Signature Indels Specific for the M. tuberculosis Complex of Organisms
5. Significance and Applications of the Tuberculosis-Complex Specific Conserved Signature Indels for Development of Novel Diagnostics and Therapeutics
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Global Tuberculois Report 2016; World Health Organization: Geneva, Switzerland, 2017. [Google Scholar]
- Koch, A.; Cox, H.; Mizrahi, V. Drug-resistant tuberculosis: Challenges and opportunities for diagnosis and treatment. Curr. Opin. Pharmacol. 2018, 42, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Chesov, D.; Heyckendorf, J.; Leung, C.C.; Udwadia, Z.; Dheda, K. Drug-resistant tuberculosis: An update on disease burden, diagnosis and treatment. Respirology 2018, 23, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Hoagland, D.T.; Liu, J.; Lee, R.B.; Lee, R.E. New agents for the treatment of drug-resistant Mycobacterium tuberculosis. Adv. Drug Deliv. Rev. 2016, 102, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Bisht, D.; Khan, A.U. Potential Alternative Strategy against Drug Resistant Tuberculosis: A Proteomics Prospect. Proteomes 2018, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Patel, S.; Jain, R. New structural classes of antituberculosis agents. Med. Res. Rev. 2018, 38, 684–740. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.C.; Mizrahi, V. Priming the tuberculosis drug pipeline: New antimycobacterial targets and agents. Curr. Opin. Microbiol. 2018, 45, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Campanico, A.; Moreira, R.; Lopes, F. Drug discovery in tuberculosis. New drug targets and antimycobacterial agents. Eur. J. Med. Chem. 2018, 150, 525–545. [Google Scholar] [CrossRef] [PubMed]
- Tiberi, S.; du, P.N.; Walzl, G.; Vjecha, M.J.; Rao, M.; Ntoumi, F.; Mfinanga, S.; Kapata, N.; Mwaba, P.; McHugh, T.D.; et al. Tuberculosis: Progress and advances in development of new drugs, treatment regimens, and host-directed therapies. Lancet Infect. Dis. 2018, 18, e183–e198. [Google Scholar] [CrossRef]
- Magee, G.M.; Ward, A.C.; Genus, I. Mycobacterium Lehmann and Neumann 1896, 363AL. In Bergey’s Manual of Systematic Bacteriology, Vol. 5, Actinobacteria; Whitman, W., Goodfellow, M., Kampfer, P., Busse, H.-J., Trujillo, M.E., Ludwig, W., Suzuki, K., Parte, A., Eds.; Springer: New York, NY, USA, 2012; pp. 312–375. [Google Scholar]
- Parte, A.C. LPSN-list of prokaryotic names with standing in nomenclature. Nucl. Acids Res. 2014, 42, D613–D616. [Google Scholar] [CrossRef] [PubMed]
- Brzostek, A.; Pawelczyk, J.; Rumijowska-Galewicz, A.; Dziadek, B.; Dziadek, J. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J. Bacteriol. 2009, 191, 6584–6591. [Google Scholar] [CrossRef] [PubMed]
- Tortoli, E. Phylogeny of the genus Mycobacterium: Many doubts, few certainties. Infect. Genet. Evol. 2012, 12, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Falkinham, J.O. Surrounded by mycobacteria: Nontuberculous mycobacteria in the human environment. J. Appl. Microbiol. 2009, 107, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Hartmans, S.; de Bont, J.A.M.; Stackebrandt, E. Chapter 1.1.18—The genus Mycobacterium—Nonmedical. In The Prokaryotes: Volume 3: Archaea. Bacteria: Firmicutes, Actinomycetes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.H., Stackebrandt, E., Eds.; Springer New York: New York, NY, USA, 2006; pp. 889–918. [Google Scholar]
- Gao, B.; Gupta, R.S. Phylogenetic framework and molecular signatures for the main clades of the phylum Actinobacteria. Microbiol. Mol. Biol. Rev. 2012, 76, 66–112. [Google Scholar] [CrossRef] [PubMed]
- Tortoli, E.; Fedrizzi, T.; Pecorari, M.; Giacobazzi, E.; De Sanctis, V.; Bertorelli, R.; Grottola, A.; Fabio, A.; Ferretti, P.; Di Leva, F.; et al. The new phylogenesis of the genus Mycobacterium. Int. J. Mycobacteriol. 2015, 4, 77. [Google Scholar] [CrossRef]
- Wang, J.; McIntosh, F.; Radomski, N.; Dewar, K.; Simeone, R.; Enninga, J.; Brosch, R.; Rocha, E.P.; Veyrier, F.J.; Behr, M.A. Insights on the emergence of Mycobacterium tuberculosis from the analysis of Mycobacterium kansasii. Genome Biol. Evol. 2015, 7, 856–870. [Google Scholar] [CrossRef] [PubMed]
- Fedrizzi, T.; Meehan, C.J.; Grottola, A.; Giacobazzi, E.; Fregni, S.G.; Tagliazucchi, S.; Fabio, A.; Bettua, C.; Bertorelli, R.; De Sanctis, V.; et al. Genomic characterization of Nontuberculous Mycobacteria. Sci. Rep. 2017, 7, 45258. [Google Scholar] [CrossRef] [PubMed]
- Wee, W.Y.; Dutta, A.; Choo, S.W. Comparative genome analyses of mycobacteria give better insights into their evolution. PLoS ONE 2017, 12, e0172831. [Google Scholar] [CrossRef] [PubMed]
- Naushad, H.S.; Lee, B.; Gupta, R.S. Conserved signature indels and signature proteins as novel tools for understanding microbial phylogeny and systematics: Identification of molecular signatures that are specific for the phytopathogenic genera Dickeya, Pectobacterium and Brenneria. Int. J. Syst. Evol. Microbiol. 2014, 64, 366–383. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Paramanathan, R.; Gupta, R.S. Signature proteins that are distinctive characteristics of Actinobacteria and their subgroups. Antonie Leeuwenhoek 2006, 90, 69–91. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Gupta, R.S. Conserved indels in protein sequences that are characteristic of the phylum Actinobacteria. Int. J. Syst. Evol. Microbiol. 2005, 55, 2401–2412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lory, S. The Family Mycobacteriaceae. In The Prokaryotes-Actinobacteria; Rosenberg, E., DeLong, E., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: New York, NY, USA, 2014; pp. 571–575. [Google Scholar]
- Gupta, R.S.; Lo, B.; Son, J. Phylogenomics and Comparative Genomic Studies Robustly Support Division of the Genus Mycobacterium into an Emended Genus Mycobacterium and Four Novel Genera. Front. Microbiol. 2018, 9, 67. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Bornigen, D.; Morgan, X.C.; Huttenhower, C. PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 2013, 4, 2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Hugenholtz, P.; Mavromatis, K.; Pukall, R.; Dalin, E.; Ivanova, N.N.; Kunin, V.; Goodwin, L.; Wu, M.; Tindall, B.J.; et al. A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 2009, 462, 1056–1060. [Google Scholar] [CrossRef] [PubMed]
- Adeolu, M.; Alnajar, S.; Naushad, S.; Gupta, S. Genome-based phylogeny and taxonomy of the ‘Enterobacteriales’: Proposal for Enterobacterales ord. nov. divided into the families Enterobacteriaceae, Erwiniaceae fam. nov., Pectobacteriaceae fam. nov., Yersiniaceae fam. nov., Hafniaceae fam. nov., Morganellaceae fam. nov., and Budviciaceae fam. nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 5575–5599. [Google Scholar] [PubMed]
- Sawana, A.; Adeolu, M.; Gupta, R.S. Molecular signatures and phylogenomic analysis of the genus Burkholderia: Proposal for division of this genus into the emended genus Burkholderia containing pathogenic organisms and a new genus Paraburkholderia gen. nov. harboring environmental species. Front. Genet. 2014, 5, 429. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S. Editorial: Applications of genome sequences for discovering characteristics that are unique to different groups of organisms and provide insights into evolutionary relationships. Front. Genet. 2016, 7, 27. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S. Chapter 8—Identification of conserved indels that are useful for classification and evolutionary studies. In Methods in Microbiology: New Approaches to Prokaryotic Systematics; Goodfellow, M., Sutcliffe, I., Chun, J., Eds.; Academic Press: Oxford, UK, 2014; pp. 153–182. [Google Scholar]
- Turenne, C.; Chedore, P.; Wolfe, J.; Jamieson, F.; Broukhanski, G.; May, K.; Kabani, A. Mycobacterium lacus sp. nov., a novel slowly growing, non-chromogenic clinical isolate. Int. J. Syst. Evol. Microbiol. 2002, 52, 2135–2140. [Google Scholar] [PubMed]
- Saito, H.; Iwamoto, T.; Ohkusu, K.; Otsuka, Y.; Akiyama, Y.; Sato, S.; Taguchi, O.; Sueyasu, Y.; Kawabe, Y.; Fujimoto, H.; et al. Mycobacterium shinjukuense sp. nov., a slowly growing, non-chromogenic species isolated from human clinical specimens. Int. J. Syst. Evol. Microbiol. 2011, 61, 1927–1932. [Google Scholar] [CrossRef] [PubMed]
- Rokas, A.; Holland, P.W. Rare genomic changes as a tool for phylogenetics. Trends Ecol. Evol. 2000, 15, 454–459. [Google Scholar] [CrossRef]
- Gupta, R.S. Impact of genomics on the understanding of microbial evolution and classification: The importance of Darwin’s views on classification. FEMS Microbiol. Rev. 2016, 40, 520–553. [Google Scholar] [CrossRef] [PubMed]
- Alnajar, S.; Gupta, R.S. Phylogenomics and comparative genomic studies delineate six main clades within the family Enterobacteriaceae and support the reclassification of several polyphyletic members of the family. Infect. Genet. Evol. 2017, 54, 108–127. [Google Scholar] [CrossRef] [PubMed]
- Adeolu, M.; Gupta, R.S. A phylogenomic and molecular marker based proposal for the division of the genus Borrelia into two genera: The emended genus Borrelia containing only the members of the relapsing fever Borrelia, and the genus Borreliella gen. nov. containing the members of the Lyme disease Borrelia (Borrelia burgdorferi sensu lato complex). Antonie Leeuwenhoek 2014, 105, 1049–1072. [Google Scholar] [PubMed]
- Barbour, A.G.; Adeolu, M.; Gupta, R.S. Division of the genus Borrelia into two genera (corresponding to Lyme disease and relapsing fever groups) reflects their genetic and phenotypic distinctiveness and will lead to a better understanding of these two groups of microbes. Int. J. Syst. Evol. Microbiol. 2017, 67, 2058–2067. [Google Scholar] [PubMed]
- Khadka, B.; Gupta, R.S. Identification of a conserved 8 aa insert in the PIP5K protein in the Saccharomycetaceae family of fungi and the molecular dynamics simulations and structural analysis to investigate its potential functional role. Proteins 2017, 85, 1454–1467. [Google Scholar] [CrossRef] [PubMed]
- Alnajar, S.; Khadka, B.; Gupta, R.S. Ribonucleotide reductases from Bifidobacteria contain multiple conserved indels distinguishing them from all other organisms: In silico analysis of the possible role of a 43 aa Bifidobacteria-specific insert in the Class III RNR homolog. Front. Microbiol. 2017, 8, 1409. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Gupta, R.S. Conserved inserts in the Hsp60 (GroEL) and Hsp70 (DnaK) proteins are essential for cellular growth. Mol. Genet. Genom. 2009, 281, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Schoeffler, A.J.; May, A.P.; Berger, J.M. A domain insertion in Escherichia coli GyrB adopts a novel fold that plays a critical role in gyrase function. Nucl. Acids Res. 2010, 38, 7830–7844. [Google Scholar] [CrossRef] [PubMed]
- Barry, C.E.; Crick, D.C.; McNeil, M.R. Targeting the formation of the cell wall core of M. tuberculosis. Infect. Disord. Drug Targets 2007, 7, 182–202. [Google Scholar] [CrossRef] [PubMed]
- Moraes, G.L.; Gomes, G.C.; Monteiro de Sousa, P.R.; Alves, C.N.; Govender, T.; Kruger, H.G.; Maguire, G.E.M.; Lamichhane, G.; Lameira, J. Structural and functional features of enzymes of Mycobacterium tuberculosis peptidoglycan biosynthesis as targets for drug development. Tuberculosis 2015, 95, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.E.; Gawronski, J.D.; Dejesus, M.A.; Ioerger, T.R.; Akerley, B.J.; Sassetti, C.M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251. [Google Scholar] [CrossRef] [PubMed]
- Dejesus, M.A.; Gerrick, E.R.; Xu, W.; Park, S.W.; Long, J.E.; Boutte, C.C.; Rubin, E.J.; Schnappinger, D.; Ehrt, S.; Fortune, S.M.; et al. Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Cosconati, S.; Hong, J.A.; Novellino, E.; Carroll, K.S.; Goodsell, D.S.; Olson, A.J. Structure-based virtual screening and biological evaluation of Mycobacterium tuberculosis adenosine 5′-phosphosulfate reductase inhibitors. J. Med. Chem. 2008, 51, 6627–6630. [Google Scholar] [CrossRef] [PubMed]
- Senaratne, R.H.; De Silva, A.D.; Williams, S.J.; Mougous, J.D.; Reader, J.R.; Zhang, T.; Chan, S.; Sidders, B.; Lee, D.H.; Chan, J.; et al. 5′-Adenosinephosphosulphate reductase (CysH) protects Mycobacterium tuberculosis against free radicals during chronic infection phase in mice. Mol. Microbiol. 2006, 59, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Duckworth, B.P.; Nelson, K.M.; Aldrich, C.C. Adenylating enzymes in Mycobacterium tuberculosis as drug targets. Curr. Top Med. Chem. 2012, 12, 766–796. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhang, H.; Tonge, P.J.; Tan, D.S. Mechanism-based inhibitors of MenE, an acyl-CoA synthetase involved in bacterial menaquinone biosynthesis. Bioorg. Med. Chem. Lett. 2008, 18, 5963–5966. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, R.S.; Tonkin, L.M.; Green, A.J.; Doherty, A.J. Domain structure of a NHEJ DNA repair ligase from Mycobacterium tuberculosis. J. Mol. Biol. 2005, 351, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Dubey, V.S.; Sirakova, T.D.; Cynamon, M.H.; Kolattukudy, P.E. Biochemical function of msl5 (pks8 plus pks17) in Mycobacterium tuberculosis H37Rv: Biosynthesis of monomethyl branched unsaturated fatty acids. J. Bacteriol. 2003, 185, 4620–4625. [Google Scholar] [CrossRef] [PubMed]
- Portevin, D.; Sousa-D’Auria, C.; Houssin, C.; Grimaldi, C.; Chami, M.; Daffe, M.; Guilhot, C. A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc. Natl. Acad. Sci. USA 2004, 101, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Bellinzoni, M.; Buroni, S.; Pasca, M.R.; Guglierame, P.; Arcesi, F.; De Rossi, E.; Riccardi, G. Glutamine amidotransferase activity of NAD+ synthetase from Mycobacterium tuberculosis depends on an amino-terminal nitrilase domain. Res. Microbiol. 2005, 156, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Kime, L.; Vincent, H.A.; Gendoo, D.M.; Jourdan, S.S.; Fishwick, C.W.; Callaghan, A.J.; McDowall, K.J. The first small-molecule inhibitors of members of the ribonuclease E family. Sci. Rep. 2015, 5, 8028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, S.; Gruber, T.; Barry, C.E.; Boshoff, H.I., III; Rhee, K.Y. Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 2013, 339, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Wang, X.D.; Erber, L.N.; Luo, M.; Guo, A.Z.; Yang, S.S.; Gu, J.; Turman, B.J.; Gao, Y.; Li, D.; et al. Binding pocket alterations in dihydrofolate synthase confer resistance to para-aminosalicylic acid in clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Minato, Y.; Thiede, J.M.; Kordus, S.L.; McKlveen, E.J.; Turman, B.J.; Baughn, A.D. Mycobacterium tuberculosis folate metabolism and the mechanistic basis for para-aminosalicylic acid susceptibility and resistance. Antimicrob. Agents Chemther. 2015, 59, 5097–5106. [Google Scholar] [CrossRef] [PubMed]
- Rashid, A.M.; Batey, S.F.; Syson, K.; Koliwer-Brandl, H.; Miah, F.; Findlay, K.C.; Nartowski, K.P.; Kalscheuer, R.; Bornemann, S. Assembly of α-Glucan by GlgE and GlgB in Mycobacteria and Streptomycetes. Biochemistry 2016, 55, 3270–3284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.W.; Melgar, M.M.; Kurth, D.; Swamidass, S.J.; Purdon, J.; Tseng, T.; Gago, G.; Baldi, P.; Gramajo, H.; Tsai, S.-C. Structure-based inhibitor design of AccD5, an essential acyl-CoA carboxylase carboxyltransferase domain of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2006, 103, 3072–3077. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Wang, C.; Besra, G.S. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin. Microbiol. Rev. 2005, 18, 81–101. [Google Scholar] [CrossRef] [PubMed]
- Sidorenko, V.S.; Rot, M.A.; Filipenko, M.L.; Nevinsky, G.A.; Zharkov, D.O. Novel DNA glycosylases from Mycobacterium tuberculosis. Biochemistry 2008, 73, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; He, Z.G. Characterization of an interplay between a Mycobacterium tuberculosis MazF homolog, Rv1495 and its sole DNA topoisomerase I. Nucl. Acids Res. 2010, 38, 8219–8230. [Google Scholar] [CrossRef] [PubMed]
- Goude, R.; Amin, A.G.; Chatterjee, D.; Parish, T. The arabinosyltransferase EmbC is inhibited by ethambutol in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2009, 53, 4138–4146. [Google Scholar] [CrossRef] [PubMed]
- Alderwick, L.J.; Birch, H.L.; Mishra, A.K.; Eggeling, L.; Besra, G.S. Structure, function and biosynthesis of the Mycobacterium tuberculosis cell wall: Arabinogalactan and lipoarabinomannan assembly with a view to discovering new drug targets. Biochem. Soc. Trans. 2007, 35, 1325–1328. [Google Scholar] [CrossRef] [PubMed]
- Meena, L.S.; Chopra, P.; Bedwal, R.S.; Singh, Y. Cloning and characterization of GTP-binding proteins of Mycobacterium tuberculosis H37Rv. Enzyme Microb. Technol. 2008, 42, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Hassan, F.M.N.; Gupta, R.S. Novel Sequence Features of DNA Repair Genes/Proteins from Deinococcus Species Implicated in Protection from Oxidatively Generated Damage. Genes 2018, 9, 149. [Google Scholar] [CrossRef] [PubMed]
- Akiva, E.; Itzhaki, Z.; Margalit, H. Built-in loops allow versatility in domain-domain interactions: Lessons from self-interacting domains. Proc. Natl. Acad. Sci. USA 2008, 105, 13292–13297. [Google Scholar] [CrossRef] [PubMed]
- Hormozdiari, F.; Salari, R.; Hsing, M.; Schonhuth, A.; Chan, S.K.; Sahinalp, S.C.; Cherkasov, A. The effect of insertions and deletions on wirings in protein-protein interaction networks: A large-scale study. J. Comput. Biol. 2009, 16, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Baugh, L.; Phan, I.; Begley, D.W.; Clifton, M.C.; Armour, B.; Dranow, D.M.; Taylor, B.M.; Muruthi, M.M.; Abendroth, J.; Fairman, J.W.; et al. Increasing the structural coverage of tuberculosis drug targets. Tuberculosis 2015, 95, 142–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Amzel, L.M. Tuberculosis drug targets. Curr. Drug Targets 2002, 3, 131–154. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Chartron, J.; Carroll, K.S.; Shiau, C.; Gao, H.; Leary, J.A.; Bertozzi, C.R.; Stout, C.D. Substrate recognition, protein dynamics, and iron-sulfur cluster in Pseudomonas aeruginosa adenosine 5′-phosphosulfate reductase. J. Mol. Biol. 2006, 364, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Eniyan, K.; Dharavath, S.; Vijayan, R.; Bajpai, U.; Gourinath, S. Crystal structure of UDP-N-acetylglucosamine-enolpyruvate reductase (MurB) from Mycobacterium tuberculosis. Biochim. Biophys. Acta 2018, 1866, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.S.; Nanda, A.; Khadka, B. Novel molecular, structural and evolutionary characteristics of the phosphoketolases from bifidobacteria and Coriobacteriales. PLoS ONE 2017, 12, e0172176. [Google Scholar] [CrossRef] [PubMed]
- Ahmod, N.Z.; Gupta, R.S.; Shah, H.N. Identification of a Bacillus anthracis specific indel in the yeaC gene and development of a rapid pyrosequencing assay for distinguishing B. anthracis from the B. cereus group. J. Microbiol. Methods 2011, 87, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Y.; Paschos, A.; Gupta, R.S.; Schellhorn, H.E. Insertion/deletion-based approach for the detection of Escherichia coli O157:H7 in freshwater environments. Environ. Sci. Technol. 2014, 48, 11462–11470. [Google Scholar] [CrossRef] [PubMed]
- Eddabra, R.; Ait, B.H. Rapid molecular assays for detection of tuberculosis. Pneumonia 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walzl, G.; McNerney, R.; du, P.N.; Bates, M.; McHugh, T.D.; Chegou, N.N.; Zumla, A. Tuberculosis: Advances and challenges in development of new diagnostics and biomarkers. Lancet Infe. Dis. 2018, 18, e199–e210. [Google Scholar] [CrossRef]
- Lamichhane, G.; Zignol, M.; Blades, N.J.; Geiman, D.E.; Dougherty, A.; Grosset, J.; Broman, K.W.; Bishai, W.R. A postgenomic method for predicting essential genes at subsaturation levels of mutagenesis: Application to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2003, 100, 7213–7218. [Google Scholar] [CrossRef] [PubMed]
- Keiser, T.L.; Purdy, G.E. Killing Mycobacterium tuberculosis In Vitro: What Model Systems Can Teach Us. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brotz-Oesterhelt, H.; Sass, P. Postgenomic strategies in antibacterial drug discovery. Future Microbiol. 2010, 5, 1553–1579. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Chopra, P.; Singh, Y. Recent advances towards identification of new drug targets for Mycobacterium tuberculosis. Expert Opin. Ther. Targets 2004, 8, 79–93. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Chen, Y.P. Structure-based drug design to augment hit discovery. Drug Discov. Today 2011, 16, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Mizrahi, V. Identification and validation of novel drug targets in Mycobacterium tuberculosis. Drug Discov. Today 2017, 22, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Nandan, D.; Lopez, M.; Ban, F.; Huang, M.; Li, Y.; Reiner, N.E.; Cherkasov, A. Indel-based targeting of essential proteins in human pathogens that have close host orthologue(s): Discovery of selective inhibitors for Leishmania donovani elongation factor-1α. Proteins 2007, 67, 53–64. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Gene Number (M. tuberculosis H37Rv) | Figure Number | Ins/Del | Location | Mutational Results # |
|---|---|---|---|---|---|
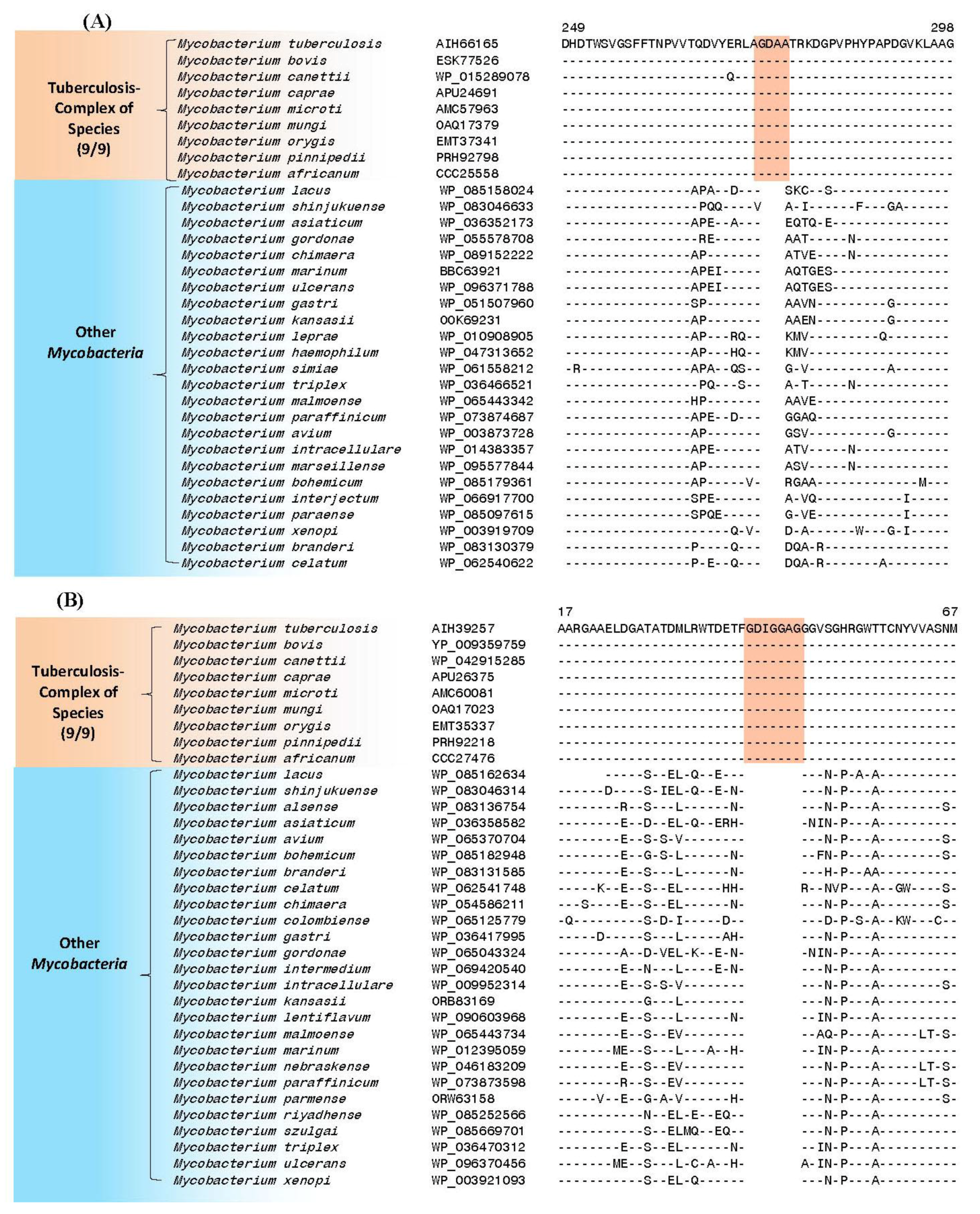
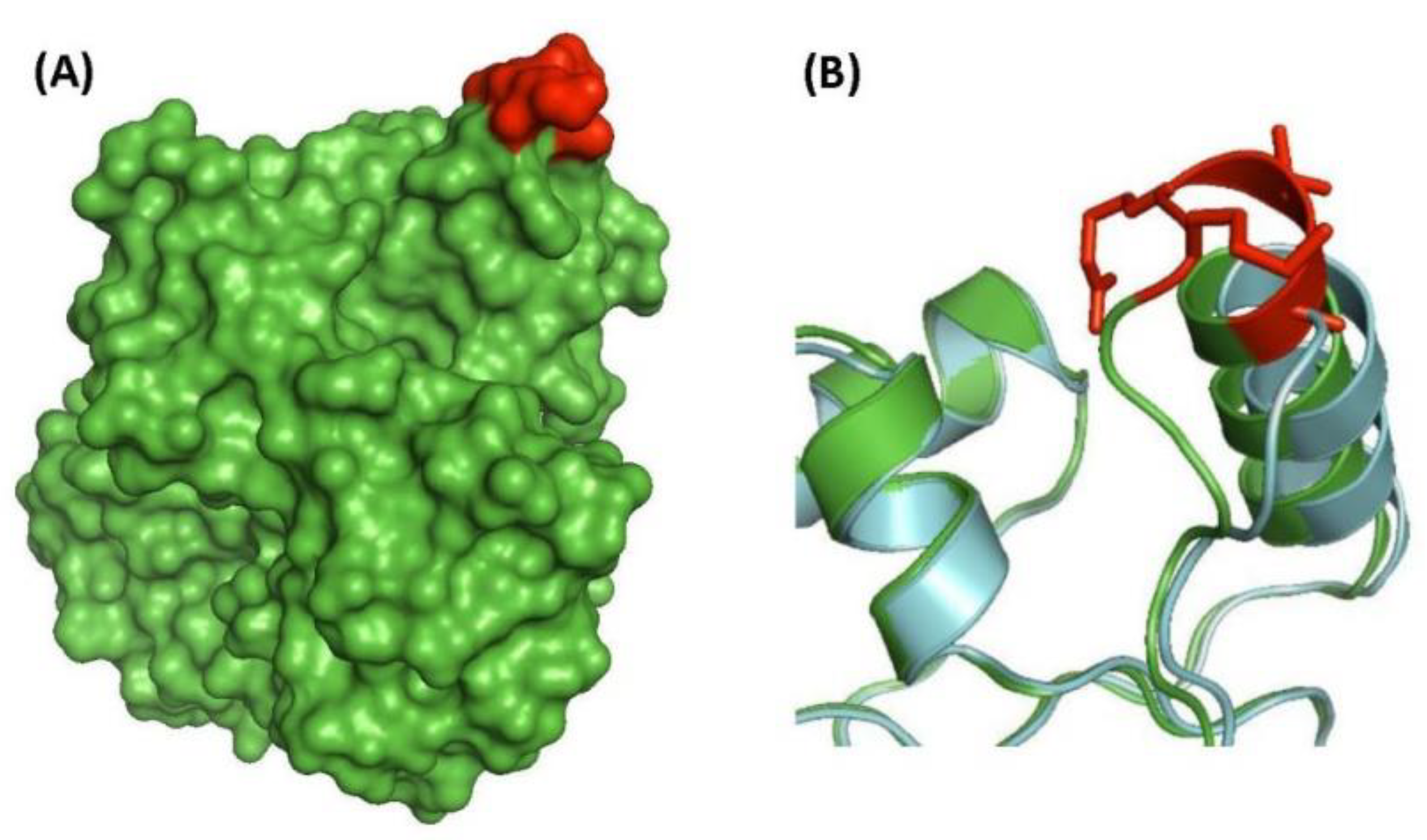
| putative UDP-N-acetylenolpyruvoyl-glucosamine reductase (MurB) | Rv0482 | Figure 3A, Figure S1 | 4aa Ins | 249–298 | Essential |
| putative 3′-phosphoadenosine 5′-phosphosulfate reductase (CysH) (PAPS reductase, thioredoxin dep) | Rv2392 | Figure 3B, Figure S2 | 7aa Ins | 17–71 | Essential (growth defect) |
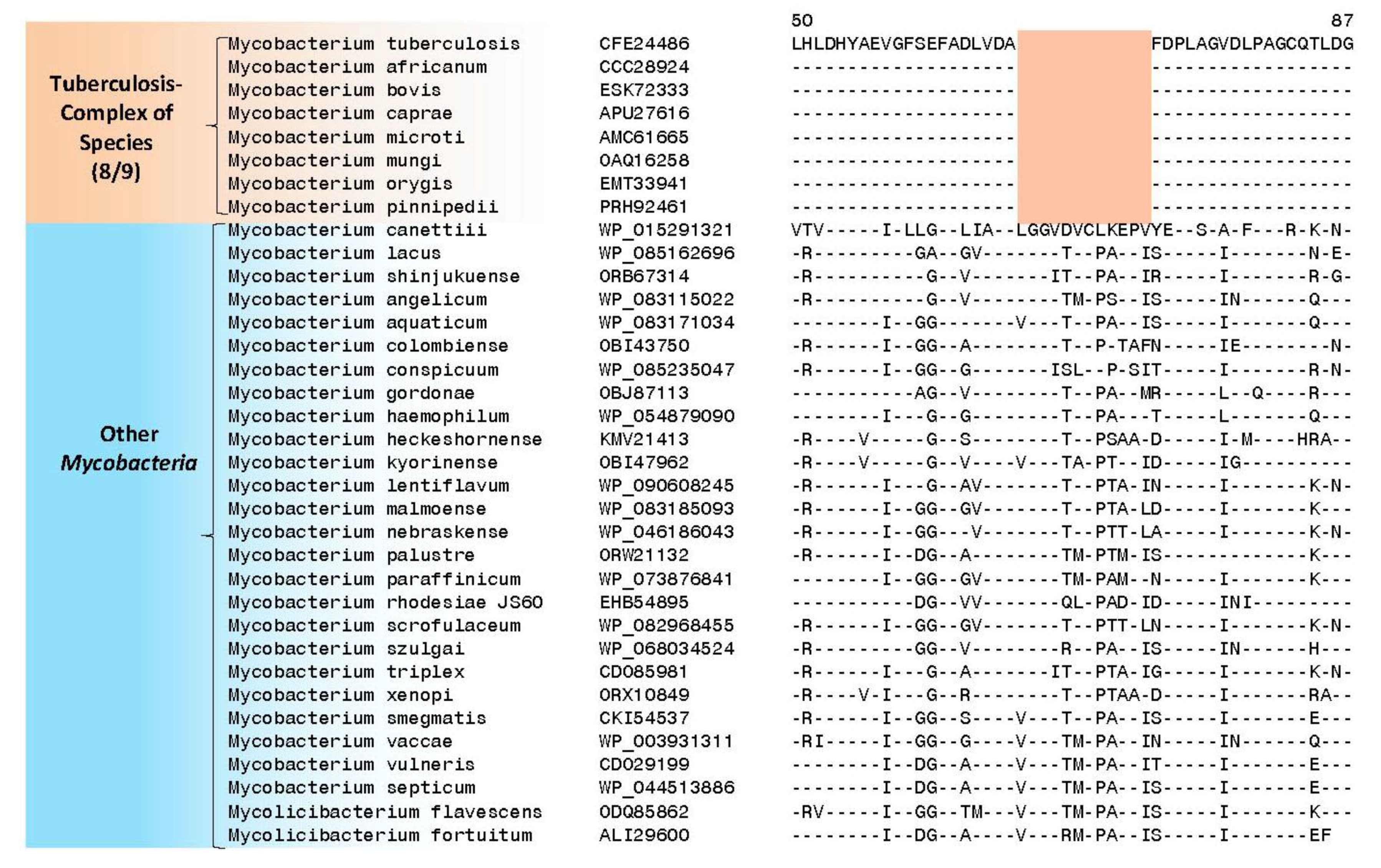
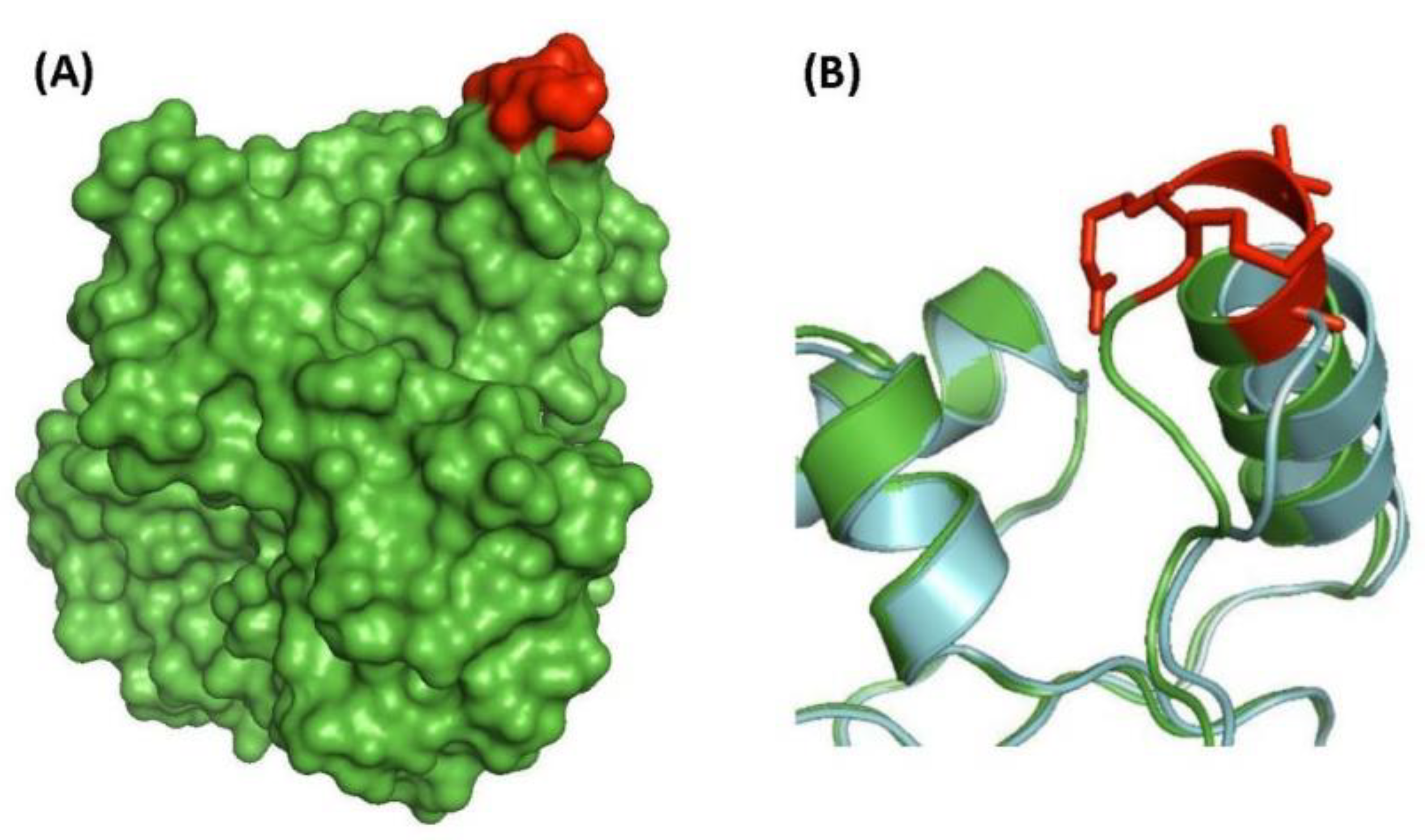
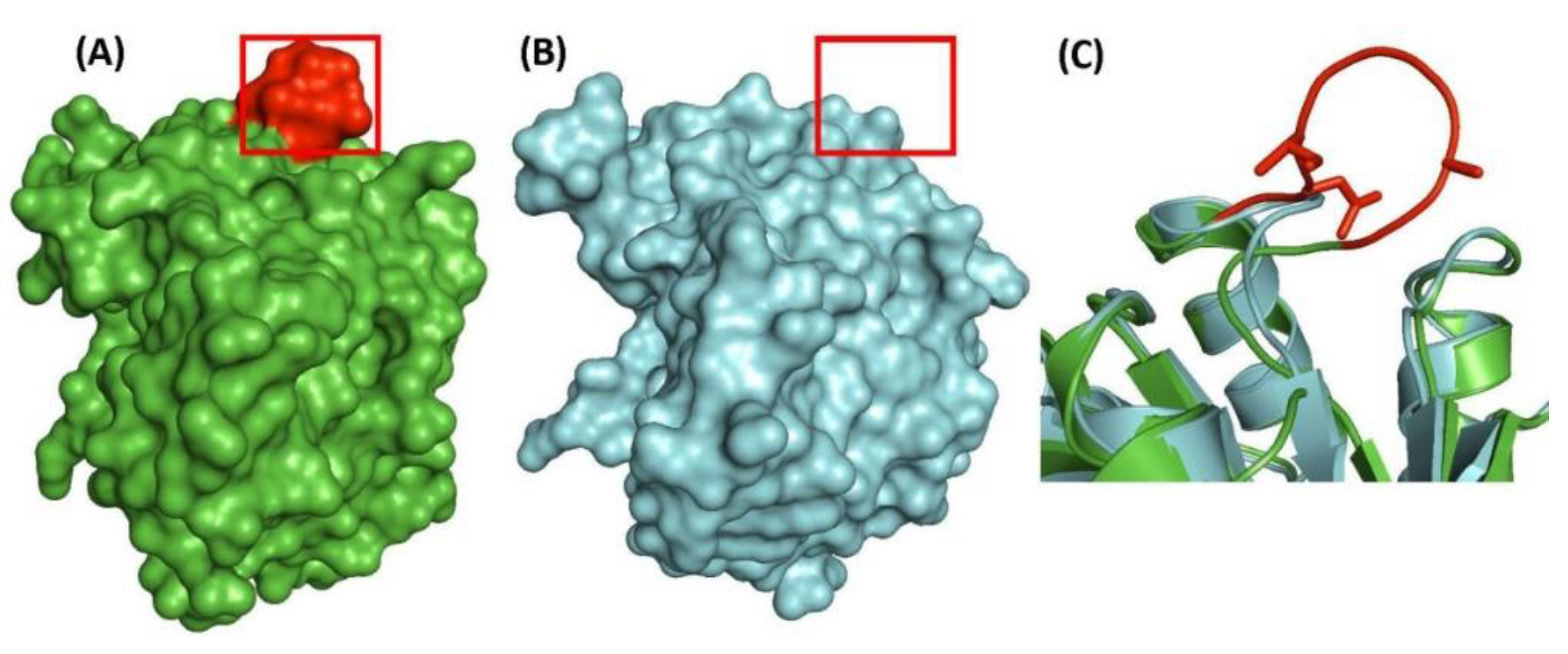
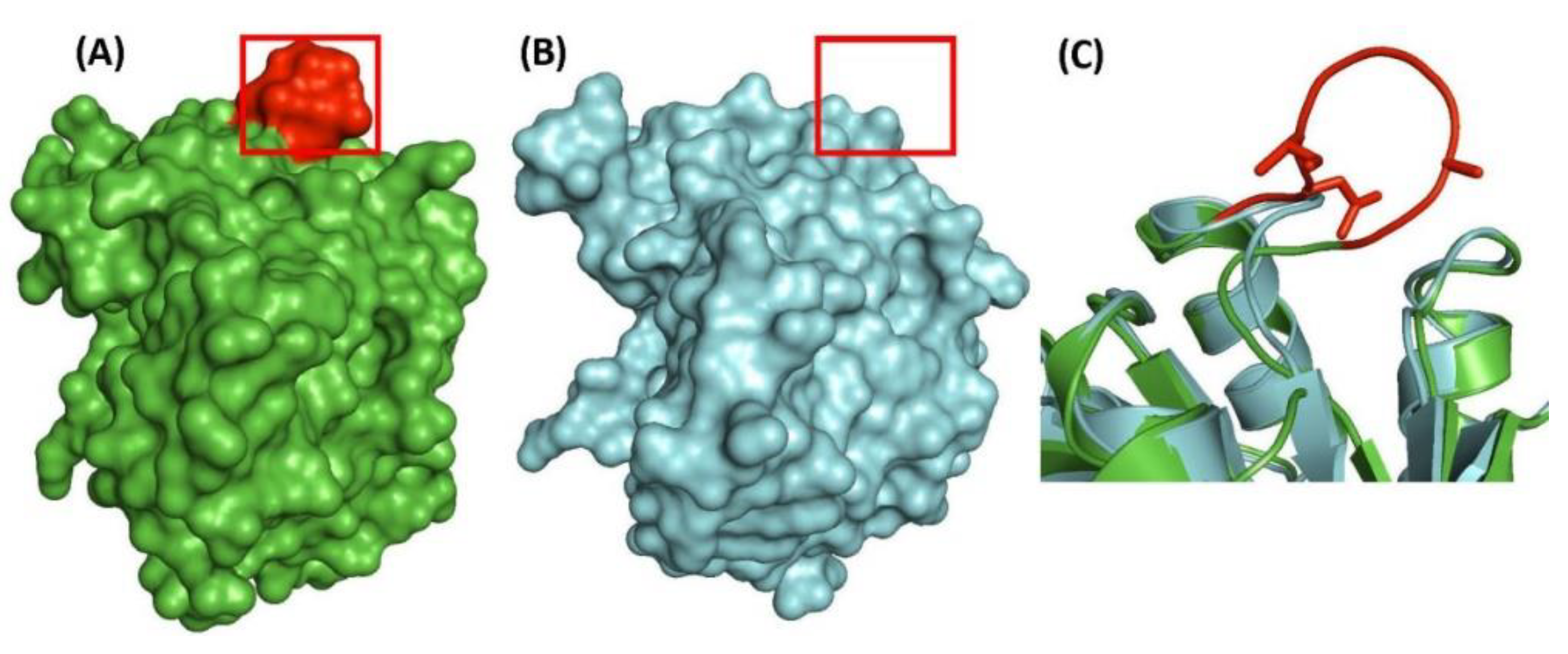
| transcriptional regulator, LytR family | Rv3840 | Figure 4, Figure S3 | 12aa Del | 50–87 | Non-essential |
| putative propionyl-CoA carboxylase beta chain 5 ACCD5 (PCCASE) | Rv3280 | Figure S4 | 1aa Del | 172–220 | Essential |
| O-succinylbenzoic acid-CoA ligase MenE * | Rv0542c | Figure S5 | 2aa Ins | 41–95 | Essential |
| ligase * | Rv3712 | Figure S6 | 4aa Ins | 180–234 | Essential |
| arabinosyltransferase EmbB * | Rv3795 | Figure S7 | 3aa Ins | 747–795 | Essential |
| GTPase Era | Rv2364c | Figure S8 | 1aa Ins | 225–283 | Essential (growth defect) |
| primosome assembly protein PriA | Rv1402 | Figure S9 | 3aa Ins | 609–655 | Essential |
| putative phospho-sugar mutase/MRSA homolog * | Rv3441c | Figure S10 | 3aa Ins | 43–102 | Essential |
| polyketide synthase Pks8 | Rv1662 | Figure S11 | 1aa Del | 539–586 | Non-essential |
| Glutamine-dependent NAD(+) synthetase | Rv2438c | Figure S12 | 1aa Del | 584–641 | Essential |
| ribonuclease E | Rv2444c | Figure S13 | 3aa Ins | 219–269 | Essential |
| putative folylpolyglutamate synthase protein (FolC) | Rv2447c | Figure S14 | 3aa Ins | 111–170 | Essential |
| DNA topoisomerase I TOPA (omega-protein) | Rv3646c | Figure S15 | 3aa Ins | 392–440 | Essential |
| metal cation transporting ATPase H | Rv0425c | Figure S16 | 1aa Del | 963–1014 | Non-essential |
| Acyltransferase * | Rv1565c | Figure S17 | 4aa Ins | 162–220 | Non-essential |
| α-amylase | Rv2471 | Figure S18 | 1aa Del | 428–477 | Non-essential |
| hypothetical protein IQ48_14915, partial | Rv0897c | Figure S19 | 3aa Ins | 257–306 | Non-essential |
| hypothetical protein CAB90_01059 * | Rv0938 | Figure S20 | 3aa Ins | 422–469 | Non-essential |
| transcriptional regulator * | Rv1186c | Figure S21 | 1aa Del | 406–457 | Non-essential |
| hypothetical protein IU12_21070 | Rv0008c | Figure S22 | 4aa Ins | 10–59 | Non-essential |
| hypothetical protein IU14_19860 | Rv0029 | Figure S23 | 2aa Del | 194–250 | Non-essential |
| membrane protein | Rv0051 | Figure S24 | 8aa Ins | 470–522 | Non-essential |
| hypothetical protein RN11_1864 * | Rv0094c | Figure S25 | 8aa Ins | 18–67 | Non-essential |
| transmembrane protein | Rv0188 | Figure S26 | 3aa Del | 18–55 | Non-essential |
| hypothetical protein ERS181347_00724 | Rv0209 | Figure S27 | 3aa Ins | 195–242 | Non-essential |
| conserved membrane protein | Rv0210 | Figure S28 | 3aa Ins | 10–59 | Non-essential |
| fructose-bisphosphate aldolase * | Rv0365c | Figure S29 | 4aa Ins | 144–193 | Non-essential |
| anti-sigma K factor | Rv0444c | Figure S30 | 1aa Ins | 147–206 | Non-essential |
| conserved protein of uncharacterised function % 2C possibly exported | Rv0518 | Figure S31 | 3aa Ins | 36–84 | Non-essential |
| exonuclease V subunit α | Rv0629c | Figure S32 | 2aa Del | 109–159 | Non-essential |
| multidrug resistance protein EmrB | Rv0783c | Figure S33 | 3aa Del | 320–366 | Non-essential |
| Hypothetical protein ERS024213_05484 | Rv0789c | Figure S34 | 1aa Del | 39–91 | Non-essential |
| LuxR family transcriptional regulator | RVBD_0890c | Figure S35 | 1aa Del | 290–328 | Non-essential |
| polyprenyl-diphosphate synthase GrcC | Rv0989c | Figure S36 | 3aa Del | 205–251 | Non-essential |
| polyprenyl-diphosphate synthase GrcC | Rv0989c | Figure S37 | 1aa Del | 94–141 | Non-essential |
| cold-shock protein | Rv1253 | Figure S38 | 2aa Del | 220–275 | Non-essential |
| transcriptional regulator | Rv1358 | Figure S39 | 1aa Ins | 94–152 | Non-essential |
| hypothetical protein IQ40_04435 | Rv1359 | Figure S40 | 4aa Ins | 150–208 | Non-essential |
| esterase | Rv1497 | Figure S41 | 1aa Ins | 296–344 | Non-essential |
| hypothetical protein RN11_1864 * | Rv0094c | Figure S42 | 8aa Ins | 165–213 | Non-essential |
| DEAD/DEAH box helicase | Rv2092c | Figure S43 | 1aa Del | 579–624 | Non-essential |
| phosphoglycerate mutase | Rv2135c | Figure S44 | 1aa Del | 01–48 | Non-essential |
| hypothetical protein CAB90_02390 | Rv2137c | Figure S45 | 2aa Ins | 10–54 | Non-essential |
| putative glycerol-3-phosphate dehydrogenase * | Rv2249c | Figure S46 | 4aa Ins | 333–380 | Non-essential |
| GTP-binding protein LepA | Rv2404c | Figure S47 | 3aa Ins | 298–355 | Non-essential |
| type I restriction/modification system specificity determinant HsdS | Rv2761c | Figure S48 | 4aa Ins | 10–58 | Non-essential |
| hypothetical protein IQ38_12515, partial | Rv2762c | Figure S49 | 2aa Del | 42–81 | Non-essential |
| polyketide synthase * | Rv2940c | Figure S50 | 3aa Ins | 1311–1359 | Non-essential |
| lipase | Rv2970c | Figure S51 | 1aa Ins | 170–225 | Non-essential |
| secreted protein | Rv3054c | Figure S52 | 1aa Del | 13–61 | Non-essential |
| DNA polymerase IV * | Rv3056 | Figure S53 | 1aa Del | 208–254 | Non-essential |
| ATP-dependent DNA helicase * | Rv3202c | Figure S54 | 1aa Del | 378–426 | Non-essential |
| membrane protein | Rv3207c | Figure S55 | 1aa Ins | 207–256 | Non-essential |
| ATPase | Rv3220c | Figure S56 | 1aa Ins | 215–269 | Non-essential |
| DNA glycosylase * | Rv3297 | Figure S57 | 4aa Ins | 80–132 | Non-essential |
| hypothetical protein IQ47_16905, partial * | Rv3394c | Figure S58 | 3aa Ins | 78–123 | Non-essential |
| hydrolase * | Rv3400 | Figure S59 | 3aa Ins | 141–186 | Non-essential |
| hypothetical protein RN11_1864 * | Rv0094c | Figure S60 | 8aa Ins | 19–68 | Non-essential |
| acyl-CoA dehydrogenase FadE27 | Rv3505 | Figure S61 | 8aa Ins | 162–211 | Non-essential |
| oxidoreductase * | Rv3742c | Figure S62 | 11aa Del | 25–65 | Non-essential |
| hypothetical protein IQ42_20035 * | Rv3912 | Figure S63 | 2aa Del | 113–159 | Non-essential |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gupta, R.S. Impact of Genomics on Clarifying the Evolutionary Relationships amongst Mycobacteria: Identification of Molecular Signatures Specific for the Tuberculosis-Complex of Bacteria with Potential Applications for Novel Diagnostics and Therapeutics. High-Throughput 2018, 7, 31. https://doi.org/10.3390/ht7040031
Gupta RS. Impact of Genomics on Clarifying the Evolutionary Relationships amongst Mycobacteria: Identification of Molecular Signatures Specific for the Tuberculosis-Complex of Bacteria with Potential Applications for Novel Diagnostics and Therapeutics. High-Throughput. 2018; 7(4):31. https://doi.org/10.3390/ht7040031
Chicago/Turabian StyleGupta, Radhey S. 2018. "Impact of Genomics on Clarifying the Evolutionary Relationships amongst Mycobacteria: Identification of Molecular Signatures Specific for the Tuberculosis-Complex of Bacteria with Potential Applications for Novel Diagnostics and Therapeutics" High-Throughput 7, no. 4: 31. https://doi.org/10.3390/ht7040031
APA StyleGupta, R. S. (2018). Impact of Genomics on Clarifying the Evolutionary Relationships amongst Mycobacteria: Identification of Molecular Signatures Specific for the Tuberculosis-Complex of Bacteria with Potential Applications for Novel Diagnostics and Therapeutics. High-Throughput, 7(4), 31. https://doi.org/10.3390/ht7040031