Recent Advances in Targeted and Untargeted Metabolomics by NMR and MS/NMR Methods


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Targeted Metabolomics
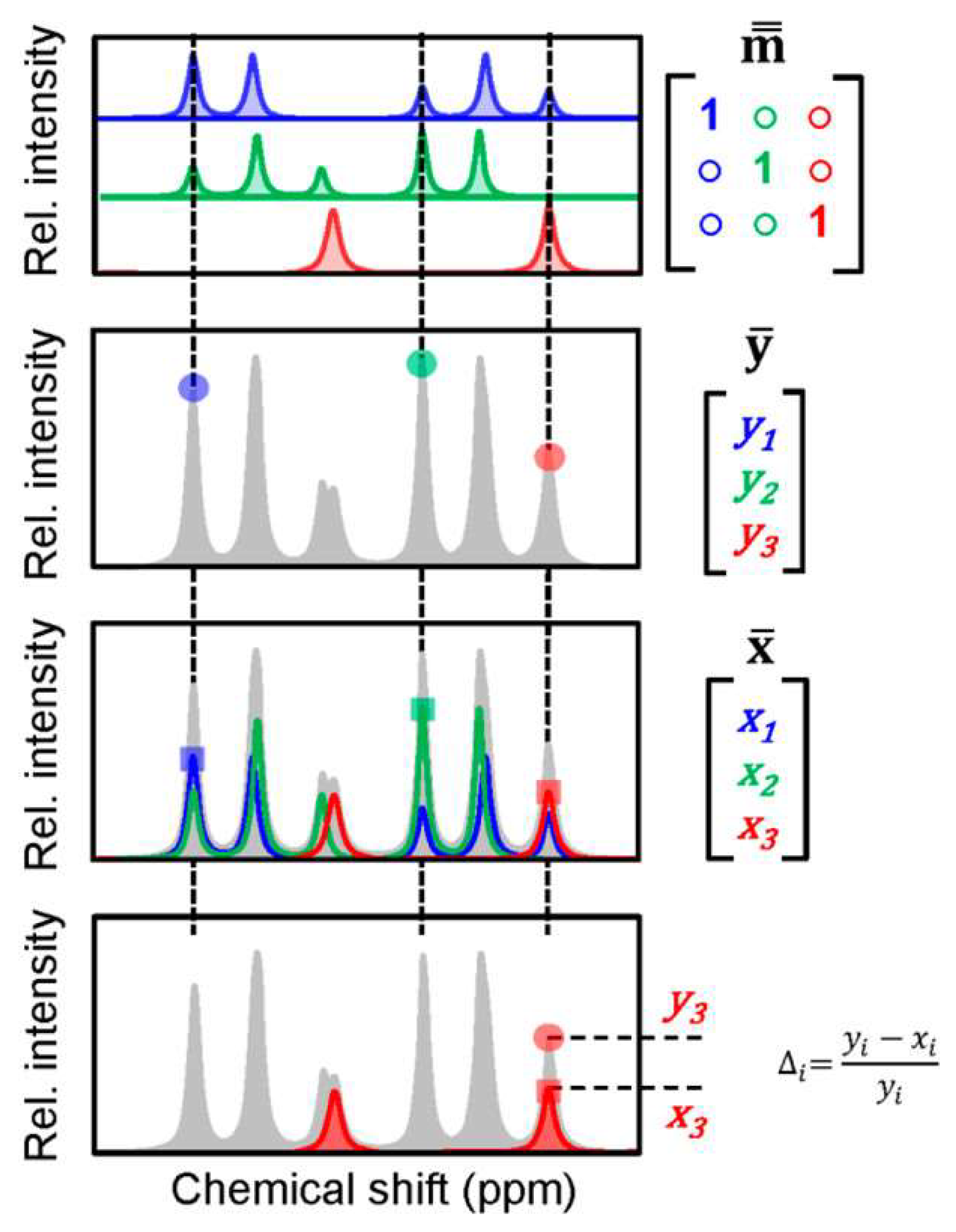
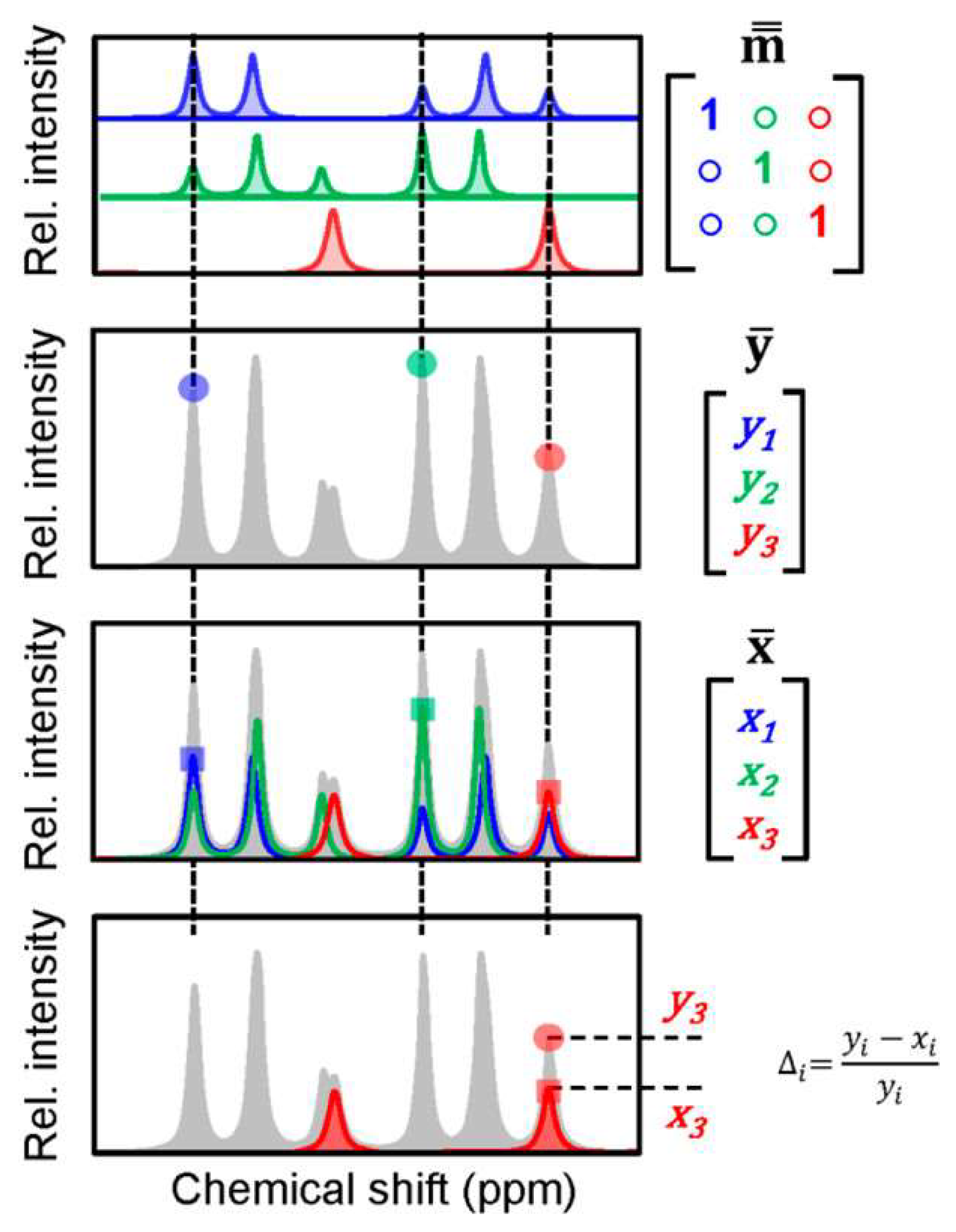
2.1. Quantitation of Metabolites
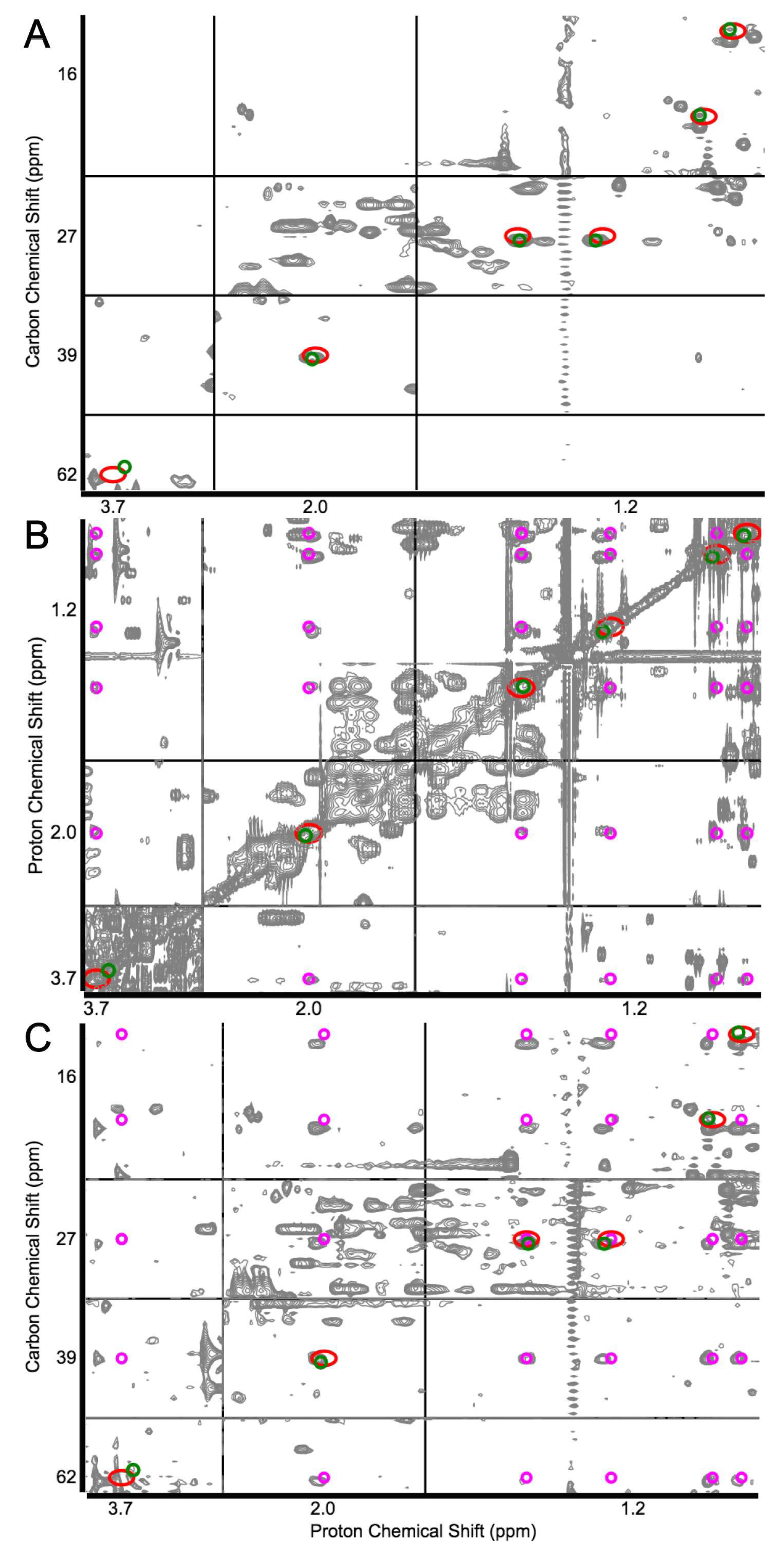
2.2. Identification of Known Metabolites with the Use of a Database for Matching
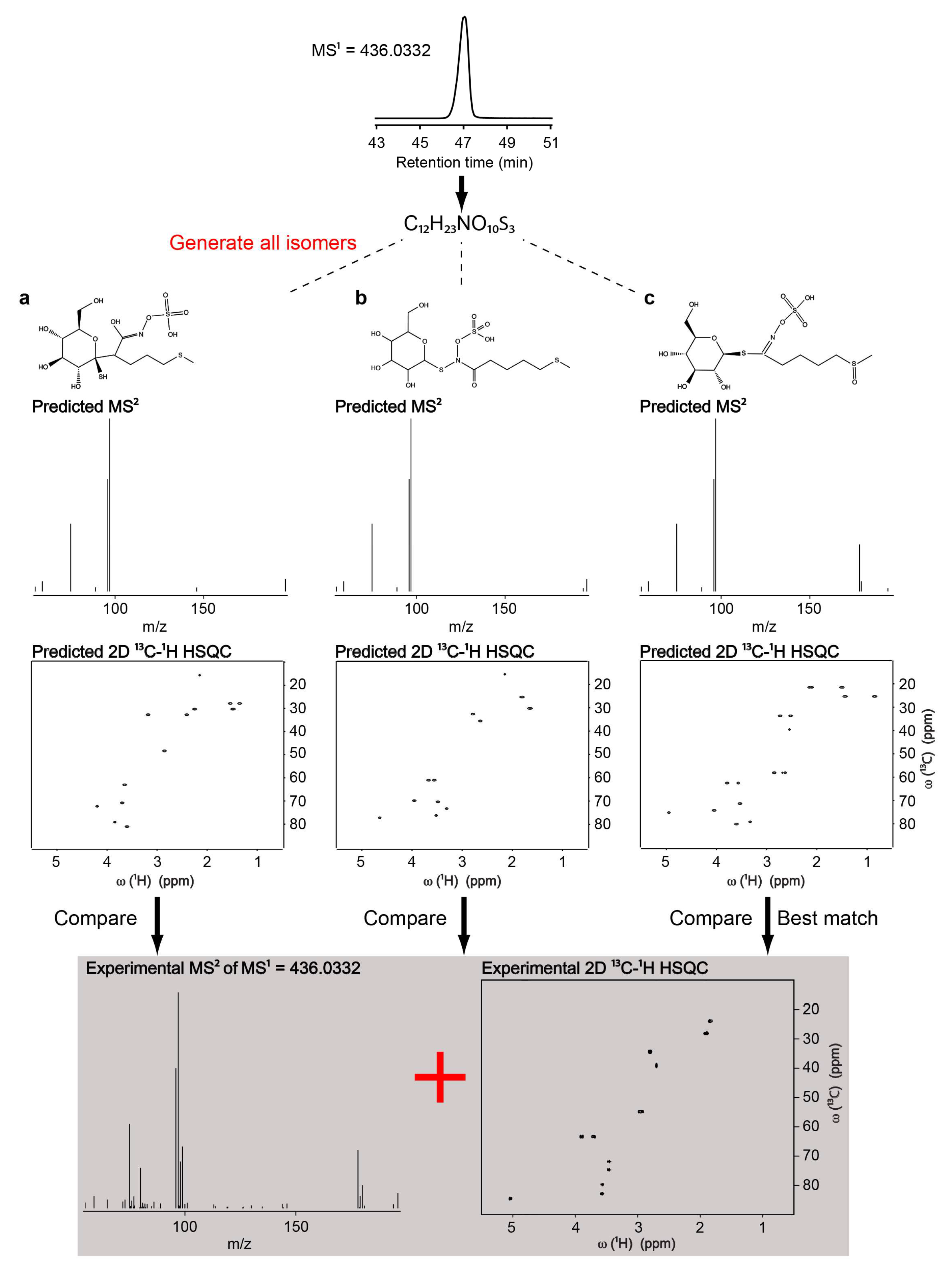
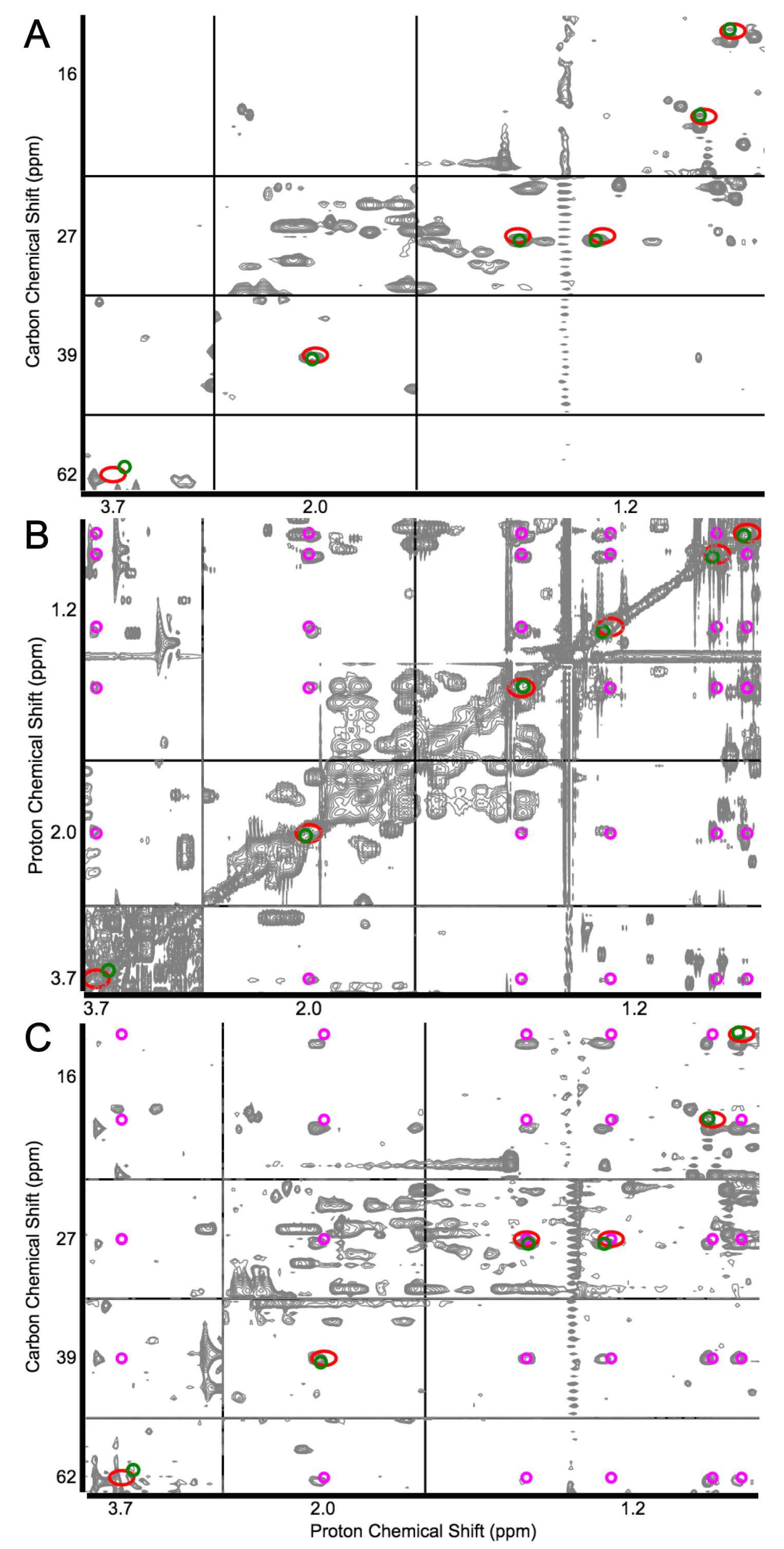
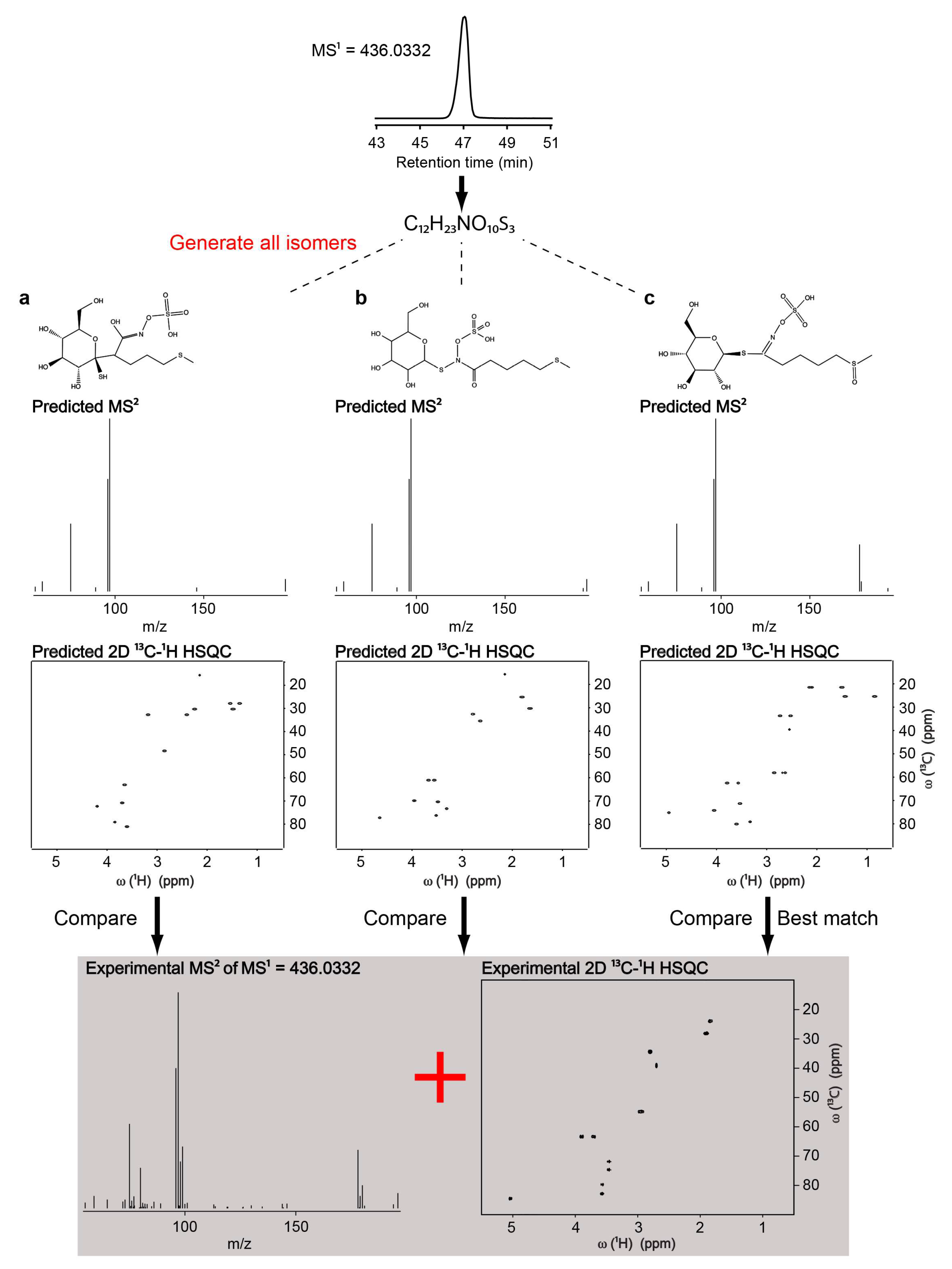
3. Untargeted Metabolomics
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Seshadri, R.; Varghese, N.J.; Eloe-Fadrosh, E.A.; Meier-Kolthoff, J.P.; Goker, M.; Coates, R.C.; Hadjithomas, M.; Pavlopoulos, G.A.; Paez-Espino, D.; et al. 1003 reference genomes of bacterial and archaeal isolates expand coverage of the tree of life. Nat. Biotechnol. 2017, 35, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Jansson, J.K.; Baker, E.S. A multi-omic future for microbiome studies. Nat. Microbiol. 2016, 1, 16049. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, M.; Sauer, U. Metabolomics-driven understanding of genotype-phenotype relations in model organisms. Curr. Opin. Syst. Biol. 2017, 6, 28–36. [Google Scholar] [CrossRef]
- White III, R.A.; Rivas-Ubach, A.; Borkum, M.I.; Köberl, M.; Bilbao, A.; Colby, S.M.; Hoyt, D.W.; Bingol, K.; Kim, Y.-M.; Wendler, J.P. The state of rhizospheric science in the era of multi-omics: A practical guide to omics technologies. Rhizosphere 2017, 3, 212–221. [Google Scholar] [CrossRef]
- Rueedi, R.; Mallol, R.; Raffler, J.; Lamparter, D.; Friedrich, N.; Vollenweider, P.; Waeber, G.; Kastenmuller, G.; Kutalik, Z.; Bergmann, S. Metabomatching: Using genetic association to identify metabolites in proton NMR spectroscopy. PLoS Comput. Biol. 2017, 13, e1005839. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Tohge, T.; Cuadros-Inostroza, Á.; Tong, H.; Tenenboim, H.; Kooke, R.; Méret, M.; Keurentjes, J.B.; Nikoloski, Z.; Fernie, A.R. Mapping the Arabidopsis metabolic landscape by untargeted metabolomics at different environmental conditions. Mol. Plant 2018, 11, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, O.; Urrutia, M.; Bernillon, S.; Giauffret, C.; Tardieu, F.; Le Gouis, J.; Langlade, N.; Charcosset, A.; Moing, A.; Gibon, Y. Fortune telling: Metabolic markers of plant performance. Metabolomics 2016, 12, 158. [Google Scholar] [CrossRef] [PubMed]
- Geng, S.; Misra, B.B.; Armas, E.; Huhman, D.V.; Alborn, H.T.; Sumner, L.W.; Chen, S. Jasmonate-mediated stomatal closure under elevated CO2 revealed by time-resolved metabolomics. Plant J. 2016, 88, 947–962. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Ito, K.; Date, Y. Environmental metabolomics with data science for investigating ecosystem homeostasis. Prog. Nucl. Magn. Reson. Spectrosc. 2018, 104, 56–88. [Google Scholar] [CrossRef] [PubMed]
- Ussher, J.R.; Elmariah, S.; Gerszten, R.E.; Dyck, J.R. The emerging role of metabolomics in the diagnosis and prognosis of cardiovascular disease. J. Am. Coll. Cardiol. 2016, 68, 2850–2870. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Perez, I.; Posma, J.M.; Gibson, R.; Chambers, E.S.; Hansen, T.H.; Vestergaard, H.; Hansen, T.; Beckmann, M.; Pedersen, O.; Elliott, P. Objective assessment of dietary patterns by use of metabolic phenotyping: A randomised, controlled, crossover trial. Lancet Diabetes Endocrinol. 2017, 5, 184–195. [Google Scholar] [CrossRef]
- Everett, J.R. NMR-based pharmacometabonomics: A new paradigm for personalised or precision medicine. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 102, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.N.; Fan, T.W. NMR-based stable isotope resolved metabolomics in systems biochemistry. Arch. Biochem. Biophys. 2017, 628, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Vinaixa, M.; Rodríguez, M.A.; Aivio, S.; Capellades, J.; Gómez, J.; Canyellas, N.; Stracker, T.H.; Yanes, O. Positional enrichment by proton analysis (PEPA): A one-dimensional 1H-NMR approach for 13C stable isotope tracer studies in metabolomics. Angew. Chem. Int. Ed. 2017, 56, 3531–3535. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Caflisch, L.; Lodi, A.; Brenner, A.J.; Tiziani, S. Metabolomic signature of brain cancer. Mol. Carcinog. 2017, 56, 2355–2371. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Brüschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of NMR-based metabolomics. Curr. Opin. Biotechnol. 2017, 43, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Su, X.; Klein, M.S.; Lewis, I.A.; Fiehn, O.; Rabinowitz, J.D. Metabolite measurement: Pitfalls to avoid and practices to follow. Annu. Rev. Biochem. 2017, 86, 277–304. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Brüschweiler, R. Multidimensional approaches to NMR-based metabolomics. Anal. Chem. 2013, 86, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Nagana Gowda, G.; Raftery, D. Recent advances in NMR-based metabolomics. Anal. Chem. 2016, 89, 490–510. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Brüschweiler, R. Two elephants in the room: New hybrid nuclear magnetic resonance and mass spectrometry approaches for metabolomics. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Marshall, D.D.; Powers, R. Beyond the paradigm: Combining mass spectrometry and nuclear magnetic resonance for metabolomics. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 100, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Weljie, A.M.; Newton, J.; Mercier, P.; Carlson, E.; Slupsky, C.M. Targeted profiling: Quantitative analysis of 1H NMR metabolomics data. Anal. Chem. 2006, 78, 4430–4442. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wishart, D.S. Using metaboanalyst 3.0 for comprehensive metabolomics data analysis. Curr. Protoc. Bioinform. 2016, 55, 14.10.1–14.10.91. [Google Scholar] [CrossRef]
- Date, Y.; Kikuchi, J. Application of a deep neural network to metabolomics studies and its performance in determining important variables. Anal. Chem. 2018, 90, 1805–1810. [Google Scholar] [CrossRef] [PubMed]
- Takis, P.G.; Schäfer, H.; Spraul, M.; Luchinat, C. Deconvoluting interrelationships between concentrations and chemical shifts in urine provides a powerful analysis tool. Nat. Commun. 2017, 8, 1662. [Google Scholar] [CrossRef] [PubMed]
- Nagana Gowda, G.; Raftery, D. Whole blood metabolomics by 1H NMR spectroscopy provides a new opportunity to evaluate coenzymes and antioxidants. Anal. Chem. 2017, 89, 4620–4627. [Google Scholar] [CrossRef] [PubMed]
- Daly, R.A.; Borton, M.A.; Wilkins, M.J.; Hoyt, D.W.; Kountz, D.J.; Wolfe, R.A.; Welch, S.A.; Marcus, D.N.; Trexler, R.V.; MacRae, J.D. Microbial metabolisms in a 2.5-km-deep ecosystem created by hydraulic fracturing in shales. Nat. Microbiol. 2016, 1, 16146. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Liebeke, M.; Astle, W.; De Iorio, M.; Bundy, J.G.; Ebbels, T.M. Bayesian deconvolution and quantification of metabolites in complex 1D NMR spectra using batman. Nat. Protoc. 2014, 9, 1416–1427. [Google Scholar] [CrossRef] [PubMed]
- Ravanbakhsh, S.; Liu, P.; Bjordahl, T.C.; Mandal, R.; Grant, J.R.; Wilson, M.; Eisner, R.; Sinelnikov, I.; Hu, X.; Luchinat, C.; et al. Accurate, fully-automated NMR spectral profiling for metabolomics. PLoS ONE 2015, 10, e0124219. [Google Scholar] [CrossRef] [PubMed]
- Tardivel, P.J.; Canlet, C.; Lefort, G.; Tremblay-Franco, M.; Debrauwer, L.; Concordet, D.; Servien, R. Asics: An automatic method for identification and quantification of metabolites in complex 1D 1H NMR spectra. Metabolomics 2017, 13, 109. [Google Scholar] [CrossRef]
- Cañueto, D.; Gómez, J.; Salek, R.M.; Correig, X.; Cañellas, N. Rdolphin: A GUI R package for proficient automatic profiling of 1D 1H-NMR spectra of study datasets. Metabolomics 2018, 14, 24. [Google Scholar] [CrossRef]
- Röhnisch, H.E.; Eriksson, J.; Müllner, E.; Agback, P.; Sandström, C.; Moazzami, A.A. AQuA—An automated quantification algorithm for high-throughput NMR-based metabolomics and its application in human plasma. Anal. Chem. 2018, 90, 2095–2102. [Google Scholar] [CrossRef] [PubMed]
- Barrilero, R.; Gil, M.; Amigó, N.; Dias, C.B.; Wood, L.G.; Garg, M.L.; Ribalta, J.; Heras, M.; Vinaixa, M.; Correig, X. Lipspin: A new bioinformatics tool for quantitative 1H-NMR lipid profiling. Anal. Chem. 2018, 90, 2031–2040. [Google Scholar] [CrossRef] [PubMed]
- Mediani, A.; Khatib, A.; Ismail, A.; Hamid, M.; Lajis, N.H.; Shaari, K.; Abas, F. Application of BATMAN and BAYESIL for quantitative 1H-NMR based metabolomics of urine: Discriminant analysis of lean, obese and obese-diabetic rats. Metabolomics 2017, 13, 131. [Google Scholar] [CrossRef]
- Zangger, K. Pure shift NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2015, 86, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Moutzouri, P.; Chen, Y.; Foroozandeh, M.; Kiraly, P.; Phillips, A.R.; Coombes, S.R.; Nilsson, M.; Morris, G.A. Ultraclean pure shift NMR. Chem. Commun. 2017, 53, 10188–10191. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Brüschweiler, R. Deconvolution of chemical mixtures with high complexity by NMR consensus trace clustering. Anal. Chem. 2011, 83, 7412–7417. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Zhang, F.; Bruschweiler-Li, L.; Brüschweiler, R. Quantitative analysis of metabolic mixtures by two-dimensional 13C constant-time TOCSY NMR spectroscopy. Anal. Chem. 2013, 85, 6414–6420. [Google Scholar] [CrossRef] [PubMed]
- Marchand, J.; Martineau, E.; Guitton, Y.; Dervilly-Pinel, G.; Giraudeau, P. Multidimensional NMR approaches towards highly resolved, sensitive and high-throughput quantitative metabolomics. Curr. Opin. Biotechnol. 2017, 43, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Bornet, A.L.; Maucourt, M.L.; Deborde, C.; Jacob, D.; Milani, J.; Vuichoud, B.; Ji, X.; Dumez, J.-N.; Moing, A.; Bodenhausen, G. Highly repeatable dissolution dynamic nuclear polarization for heteronuclear NMR metabolomics. Anal. Chem. 2016, 88, 6179–6183. [Google Scholar] [CrossRef] [PubMed]
- Lerche, M.H.; Yigit, D.; Frahm, A.B.; Ardenkjær-Larsen, J.H.; Malinowski, R.M.; Jensen, P.R. Stable isotope-resolved analysis with quantitative dissolution dynamic nuclear polarization. Anal. Chem. 2017, 90, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Kostidis, S.; Choi, Y.H. NMR analysis of fecal samples. Clin. Metab. 2018, 317–328. [Google Scholar] [CrossRef]
- Johns, C.W.; Lee, A.B.; Springer, T.I.; Rosskopf, E.N.; Hong, J.C.; Turechek, W.; Kokalis-Burelle, N.; Finley, N.L. Using NMR-based metabolomics to monitor the biochemical composition of agricultural soils: A pilot study. Eur. J. Soil Biol. 2017, 83, 98–105. [Google Scholar] [CrossRef]
- Rådjursöga, M.; Karlsson, G.B.; Lindqvist, H.M.; Pedersen, A.; Persson, C.; Pinto, R.C.; Ellegård, L.; Winkvist, A. Metabolic profiles from two different breakfast meals characterized by 1H NMR-based metabolomics. Food Chem. 2017, 231, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Li, D.W.; Bruschweiler-Li, L.; Cabrera, O.A.; Megraw, T.; Zhang, F.; Brüschweiler, R. Unified and isomer-specific NMR metabolomics database for the accurate analysis of 13C–1H HSQC spectra. ACS Chem. Biol. 2014, 10, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Zhang, F.; Bruschweiler-Li, L.; Brüschweiler, R. TOCCATA: A customized carbon total correlation spectroscopy NMR metabolomics database. Anal. Chem. 2012, 84, 9395–9401. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Bruschweiler-Li, L.; Li, D.-W.; Brüschweiler, R. Customized metabolomics database for the analysis of NMR 1H–1H TOCSY and 13C–1H HSQC-TOCSY spectra of complex mixtures. Anal. Chem. 2014, 86, 5494–5501. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Li, D.-W.; Zhang, B.; Brüschweiler, R. Comprehensive metabolite identification strategy using multiple two-dimensional NMR spectra of a complex mixture implemented in the COLMARm web server. Anal. Chem. 2016, 88, 12411–12418. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Brüschweiler, R. NMR/MS translator for the enhanced simultaneous analysis of metabolomics mixtures by NMR spectroscopy and mass spectrometry: Application to human urine. J. Proteome Res. 2015, 14, 2642–2648. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.R.; Hoyt, D.W.; Walker, S.M.; Ward, J.K.; Nicora, C.D.; Bingol, K. Unambiguous metabolite identification in high-throughput metabolomics by hybrid 1D 1H NMR/ESI MS1 approach. Magn. Reson. Chem. 2016, 54, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Molinski, T.F. Microscale methodology for structure elucidation of natural products. Curr. Opin. Biotechnol. 2010, 21, 819–826. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Bruschweiler-Li, L.; Li, D.; Zhang, B.; Xie, M.; Brüschweiler, R. Emerging new strategies for successful metabolite identification in metabolomics. Bioanalysis 2016, 8, 557–573. [Google Scholar] [CrossRef] [PubMed]
- Allard, P.-M.; Genta-Jouve, G.; Wolfender, J.-L. Deep metabolome annotation in natural products research: Towards a virtuous cycle in metabolite identification. Curr. Opin. Chem. Biol. 2017, 36, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8, 3. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Ruttkies, C.; Krauss, M.; Brouard, C.; Kind, T.; Dührkop, K.; Allen, F.; Vaniya, A.; Verdegem, D.; Böcker, S. Critical assessment of small molecule identification 2016: Automated methods. J. Cheminform. 2017, 9, 22. [Google Scholar] [CrossRef] [PubMed]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A technology platform for identifying knowns and unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [PubMed]
- Lai, Z.; Tsugawa, H.; Wohlgemuth, G.; Mehta, S.; Mueller, M.; Zheng, Y.; Ogiwara, A.; Meissen, J.; Showalter, M.; Takeuchi, K.; et al. Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat. Methods 2018, 15, 53. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Zhang, F.; Bruschweiler-Li, L.; Brüschweiler, R. Carbon backbone topology of the metabolome of a cell. J. Am. Chem. Soc. 2012, 134, 9006–9011. [Google Scholar] [CrossRef] [PubMed]
- Clendinen, C.S.; Pasquel, C.; Ajredini, R.; Edison, A.S. 13C NMR metabolomics: Inadequate network analysis. Anal. Chem. 2015, 87, 5698–5706. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Ohishi, R.; Shino, A.; Kikuchi, J. Structure and metabolic-flow analysis of molecular complexity in a 13C-labeled tree by 2D and 3D NMR. Angew. Chem. 2016, 128, 6104–6107. [Google Scholar] [CrossRef]
- Bingol, K.; Bruschweiler-Li, L.; Yu, C.; Somogyi, A.; Zhang, F.; Brüschweiler, R. Metabolomics beyond spectroscopic databases: A combined MS/NMR strategy for the rapid identification of new metabolites in complex mixtures. Anal. Chem. 2015, 87, 3864–3870. [Google Scholar] [CrossRef] [PubMed]
- Clendinen, C.S.; Stupp, G.S.; Wang, B.; Garrett, T.J.; Edison, A.S. 13C metabolomics: NMR and IROA for unknown identification. Curr. Metab. 2016, 4, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; He, L.; Li, D.-W.; Bruschweiler-Li, L.; Marshall, A.G.; Brüschweiler, R. Accurate identification of unknown and known metabolic mixture components by combining 3D NMR with fourier transform ion cyclotron resonance tandem mass spectrometry. J. Proteome Res. 2017, 16, 3774–3786. [Google Scholar] [CrossRef] [PubMed]
- Bingol, K.; Brüschweiler, R. Knowns and unknowns in metabolomics identified by multidimensional NMR and hybrid MS/NMR methods. Curr. Opin. Biotechnol. 2017, 43, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Boiteau, R.M.; Hoyt, D.W.; Nicora, C.D.; Kinmonth-Schultz, H.A.; Ward, J.K.; Bingol, K. Structure elucidation of unknown metabolites in metabolomics by combined NMR and MS/MS prediction. Metabolites 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Liebeke, M.; Sommer, U.; Viant, M.R.; Bundy, J.G.; Ebbels, T.M. Statistical correlations between NMR spectroscopy and direct infusion FT-ICR mass spectrometry aid annotation of unknowns in metabolomics. Anal. Chem. 2016, 88, 2583–2589. [Google Scholar] [CrossRef] [PubMed]
- Gowda, G.N.; Djukovic, D.; Bettcher, L.F.; Gu, H.; Raftery, D. NMR-guided mass spectrometry for absolute quantitation of human blood metabolites. Anal. Chem. 2018, 90, 2001–2009. [Google Scholar] [CrossRef] [PubMed]
- Ardenkjaer-Larsen, J.H.; Boebinger, G.S.; Comment, A.; Duckett, S.; Edison, A.S.; Engelke, F.; Griesinger, C.; Griffin, R.G.; Hilty, C.; Maeda, H. Facing and overcoming sensitivity challenges in biomolecular NMR spectroscopy. Angew. Chem. Int. Ed. 2015, 54, 9162–9185. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bingol, K. Recent Advances in Targeted and Untargeted Metabolomics by NMR and MS/NMR Methods. High-Throughput 2018, 7, 9. https://doi.org/10.3390/ht7020009
Bingol K. Recent Advances in Targeted and Untargeted Metabolomics by NMR and MS/NMR Methods. High-Throughput. 2018; 7(2):9. https://doi.org/10.3390/ht7020009
Chicago/Turabian StyleBingol, Kerem. 2018. "Recent Advances in Targeted and Untargeted Metabolomics by NMR and MS/NMR Methods" High-Throughput 7, no. 2: 9. https://doi.org/10.3390/ht7020009
APA StyleBingol, K. (2018). Recent Advances in Targeted and Untargeted Metabolomics by NMR and MS/NMR Methods. High-Throughput, 7(2), 9. https://doi.org/10.3390/ht7020009