Functional Genomics Approaches to Studying Symbioses between Legumes and Nitrogen-Fixing Rhizobia



Abstract
:1. Introduction
2. Functional Genomics of Rhizobia-Legume Symbiosis
2.1. Transcriptomics
2.1.1. Microarrays versus RNA-Sequencing
2.1.2. Transcript Profiling of Alpha-Rhizobia
2.1.3. Transcript Profiling of Beta-Rhizobia
2.2. Proteomics
2.2.1. 2-Dimensional Gel Electrophoresis versus Liquid Chromatography Combined with Tandem Mass Spectrometry
2.2.2. Protein Profiling of Free-Living Alpha-Rhizobia
2.2.3. Protein Profiling of Alpha-Rhizobia Living Inside Nodules
2.3. Metabolomics
2.3.1. Nuclear Magnetic Resonance versus Mass Spectrometry
2.3.2. Metabolic Profiling of Nodules Induced by Alpha-Rhizobia
2.3.3. Metabolic Profiling of Nodules Induced by Beta-Rhizobia
3. Integration of Different Omics Technologies
4. Future Perspective
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Herridge, D.F.; Peoples, M.B.; Boddey, R.M. Global inputs of biological nitrogen fixation in agricultural systems. Plant Soil 2008, 311, 1–18. [Google Scholar] [CrossRef]
- Peix, A.; Ramírez-Bahena, M.H.; Velázquez, E.; Bedmar, E.J. Bacterial associations with legumes. Crit. Rev. Plant Sci. 2015, 34, 17–42. [Google Scholar] [CrossRef]
- Moulin, L.; Munive, A.; Dreyfus, B.; Boivin-Masson, C. Nodulation of legumes by members of the β-subclass of Proteobacteria. Nature 2001, 411, 948–950. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-M.; James, E.K.; Prescott, A.R.; Kierans, M.; Sprent, J.I. Nodulation of Mimosa spp. by the β-Proteobacterium Ralstonia taiwanensis. Mol. Plant. Microbe Interact. 2003, 16, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-M.; Moulin, L.; Bontemps, C.; Vandamme, P.; Bena, G.; Boivin-Masson, C. Legume symbiotic nitrogen fixation by β-proteobacteria is widespread in nature. J. Bacteriol. 2003, 185, 7266–7272. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.N.; Chen, W.-M.; Bontemps, C.; Chou, J.-H.; Young, J.P.W.; Sprent, J.I.; James, E.K. Nodulation of Cyclopia spp. (Leguminosae, Papilionoideae) by Burkholderia tuberum. Ann. Bot. 2007, 100, 1403–1411. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.N.; Chen, W.-M.; Chou, J.-H.; Wang, H.-C.; Sheu, S.-Y.; Perin, L.; Reis, V.M.; Moulin, L.; Simon, M.F.; Bontemps, C.; et al. Burkholderia phymatum is a highly effective nitrogen-fixing symbiont of Mimosa spp. and fixes nitrogen ex planta. New Phytol. 2007, 173, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Elliott, G.N.; Chou, J.-H.; Chen, W.-M.; Bloemberg, G.V.; Bontemps, C.; Martínez-Romero, E.; Velázquez, E.; Young, J.P.W.; Sprent, J.I.; James, E.K. Burkholderia spp. are the most competitive symbionts of Mimosa, particularly under N-limited conditions. Environ. Microbiol. 2009, 11, 762–778. [Google Scholar] [CrossRef] [PubMed]
- Angus, A.A.; Hirsch, A.M. Insights into the history of the legume-betaproteobacterial symbiosis. Mol. Ecol. 2010, 19, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, F.B., Jr.; Simon, M.F.; Gross, E.; Boddey, R.M.; Elliott, G.N.; Neto, N.E.; de Fatima Loureiro, M.; de Queiroz, L.P.; Scotti, M.R.; Chen, W.-M.; et al. Nodulation and nitrogen fixation by Mimosa spp. in the Cerrado and Caatinga biomes of Brazil. New Phytol. 2010, 186, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wei, S.; Wang, F.; James, E.K.; Guo, X.; Zagar, C.; Xia, L.G.; Dong, X.; Wang, Y.P. Burkholderia and Cupriavidus spp. are the preferred symbionts of Mimosa spp. in southern China. FEMS Microbiol. Ecol. 2012, 80, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.P.N.; Tisseyre, P.; Melkonian, R.; Chaintreuil, C.; Miché, L.; Klonowska, A.; Gonzalez, S.; Bena, G.; Laguerre, G.; Moulin, L. Genetic diversity of Mimosa pudica rhizobial symbionts in soils of French Guiana: Investigating the origin and diversity of Burkholderia phymatum and other beta-rhizobia. FEMS Microbiol. Ecol. 2012, 79, 487–503. [Google Scholar] [CrossRef] [PubMed]
- Gyaneshwar, P.; Hirsch, A.M.; Moulin, L.; Chen, W.-M.; Elliott, G.N.; Bontemps, C.; Estrada-de los Santos, P.; Gross, E.; dos Reis, F.B.; Sprent, J.I.; et al. Legume-nodulating betaproteobacteria: Diversity, host range, and future prospects. Mol. Plant. Microbe Interact. 2011, 24, 1276–1288. [Google Scholar] [CrossRef] [PubMed]
- Lemaire, B.; Chimphango, S.B.M.; Stirton, C.; Rafudeen, S.; Honnay, O.; Smets, E.; Chen, W.-M.; Sprent, J.; James, E.K.; Muasya, A.M. Biogeographical patterns of legume-nodulating Burkholderia spp.: From African fynbos to continental scales. Appl. Environ. Microbiol. 2016, 82, 5099–5115. [Google Scholar] [CrossRef] [PubMed]
- Sawana, A.; Adeolu, M.; Gupta, R.S. Molecular signatures and phylogenomic analysis of the genus Burkholderia: Proposal for division of this genus into the emended genus Burkholderia containing pathogenic organisms and a new genus Paraburkholderia gen. nov. harboring environmental species. Front. Genet. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.M. Environmental regulation of rhizobial symbiotic nitrogen fixation genes. Trends Microbiol. 1996, 4, 317–320. [Google Scholar] [CrossRef]
- Gage, D.J. Infection and invasion of roots by symbiotic, nitrogen-fixing rhizobia during nodulation of temperate legumes. Microbiol. Mol. Biol. Rev. 2004, 68, 280–300. [Google Scholar] [CrossRef] [PubMed]
- Masson-Boivin, C.; Giraud, E.; Perret, X.; Batut, J. Establishing nitrogen-fixing symbiosis with legumes: How many rhizobium recipes? Trends Microbiol. 2009, 17, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.-M. Genetic regulation of nitrogen fixation in rhizobia. Microbiol. Rev. 1994, 58, 352–386. [Google Scholar] [PubMed]
- Spaink, H.P. Root nodulation and infection factors produced by rhizobial bacteria. Annu. Rev. Microbiol. 2000, 54, 257–288. [Google Scholar] [CrossRef] [PubMed]
- Long, S.R. Genes and signals in the Rhizobium-legume symbiosis. Plant Physiol. 2001, 125, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.; Kahn, D. Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2004, 2, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Hirsch, A.M. Signals and responses: Choreographing the complex interaction between legumes and α- and β-rhizobia. Plant Signal. Behav. 2006, 1, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Oldroyd, G.E.D.; Murray, J.D.; Poole, P.S.; Downie, J.A. The rules of engagement in the legume-rhizobial symbiosis. Annu. Rev. Genet. 2011, 45, 119–144. [Google Scholar] [CrossRef] [PubMed]
- Oldroyd, G.E.D. Speak, friend and enter: Signalling systems that promote beneficial symbiotic associations in plants. Nat. Rev. Microbiol. 2013, 11, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Udvardi, M.; Poole, P.S. Transport and metabolism in legume-rhizobia symbioses. Annu. Rev. Plant Biol. 2013, 64, 781–805. [Google Scholar] [CrossRef] [PubMed]
- Laranjo, M.; Alexandre, A.; Oliveira, S. Legume growth-promoting rhizobia: An overview on the Mesorhizobium genus. Microbiol. Res. 2014, 169, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Poole, P.; Ramachandran, V.; Terpolilli, J. Rhizobia: From saprophytes to endosymbionts. Nat. Rev. Microbiol. 2018, 16, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Minamisawa, K.; Uchiumi, T.; Sasamoto, S.; Watanabe, A.; Idesawa, K.; Iriguchi, M.; Kawashima, K.; et al. Complete genomic sequence of nitrogen-fixing symbiotic bacterium Bradyrhizobium japonicum USDA110 (supplement). DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2002, 9, 225–256. [Google Scholar] [CrossRef]
- Guo, X.; Castillo-Ramírez, S.; González, V.; Bustos, P.; Luís Fernández-Vázquez, J.; Santamaría, R.; Arellano, J.; Cevallos, M.A.; Dávila, G. Rapid evolutionary change of common bean (Phaseolus vulgaris L) plastome, and the genomic diversification of legume chloroplasts. BMC Genom. 2007, 8, 228. [Google Scholar] [CrossRef] [PubMed]
- Saski, C.; Lee, S.-B.; Daniell, H.; Wood, T.C.; Tomkins, J.; Kim, H.-G.; Jansen, R.K. Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol. Biol. 2005, 59, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Young, N.D.; Debellé, F.; Oldroyd, G.E.D.; Geurts, R.; Cannon, S.B.; Udvardi, M.K.; Benedito, V.A.; Mayer, K.F.X.; Gouzy, J.; Schoof, H.; et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 2011, 480, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Moulin, L.; Klonowska, A.; Caroline, B.; Booth, K.; Vriezen, J.A.C.; Melkonian, R.; James, E.K.; Young, J.P.W.; Bena, G.; Hauser, L.; et al. Complete Genome sequence of Burkholderia phymatum STM815T, a broad host range and efficient nitrogen-fixing symbiont of Mimosa species. Stand. Genomic Sci. 2014, 9, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Schena, M.; Shalon, D.; Davis, R.W.; Brown, P.O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 1995, 270, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Ekins, R.; Chu, F.W. Microarrays: Their origins and applications. Trends Biotechnol. 1999, 17, 217–218. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Sorek, R.; Cossart, P. Prokaryotic transcriptomics: A new view on regulation, physiology and pathogenicity. Nat. Rev. Genet. 2010, 11, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Mäder, U.; Nicolas, P.; Richard, H.; Bessières, P.; Aymerich, S. Comprehensive identification and quantification of microbial transcriptomes by genome-wide unbiased methods. Curr. Opin. Biotechnol. 2011, 22, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Westermann, A.J.; Gorski, S.A.; Vogel, J. Dual RNA-seq of pathogen and host. Nat. Rev. Microbiol. 2012, 10, 618–630. [Google Scholar] [CrossRef] [PubMed]
- Klonowska, A.; Melkonian, R.; Miché, L.; Tisseyre, P.; Moulin, L. Transcriptomic profiling of Burkholderia phymatum STM815, Cupriavidus taiwanensis LMG19424 and Rhizobium mesoamericanum STM3625 in response to Mimosa pudica root exudates illuminates the molecular basis of their nodulation competitiveness and symbiotic evolutionary history. BMC Genom. 2018, 19, 105. [Google Scholar] [CrossRef]
- Li, Y.; Tian, C.F.; Chen, W.F.; Wang, L.; Sui, X.H.; Chen, W.X. High-resolution transcriptomic analyses of Sinorhizobium sp. NGR234 bacteroids in determinate nodules of Vigna unguiculata and indeterminate nodules of Leucaena leucocephala. PLoS ONE 2013, 8, e70531. [Google Scholar] [CrossRef] [PubMed]
- Lardi, M.; Liu, Y.; Purtschert, G.; Bolzan de Campos, S.; Pessi, G. Transcriptome analysis of Paraburkholderia phymatum under nitrogen starvation and during symbiosis with Phaseolus vulgaris. Genes 2017, 8, 389. [Google Scholar] [CrossRef]
- Čuklina, J.; Hahn, J.; Imakaev, M.; Omasits, U.; Förstner, K.U.; Ljubimov, N.; Goebel, M.; Pessi, G.; Fischer, H.-M.; Ahrens, C.H.; et al. Genome-wide transcription start site mapping of Bradyrhizobium japonicum grown free-living or in symbiosis—A rich resource to identify new transcripts, proteins and to study gene regulation. BMC Genom. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Sharma, C.M.; Vogel, J. Differential RNA-seq: The approach behind and the biological insight gained. Curr. Opin. Microbiol. 2014, 19, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Pessi, G.; Ahrens, C.H.; Rehrauer, H.; Lindemann, A.; Hauser, F.; Fischer, H.-M.; Hennecke, H. Genome-wide transcript analysis of Bradyrhizobium japonicum bacteroids in soybean root nodules. Mol. Plant. Microbe Interact. 2007, 20, 1353–1363. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-S.; Franck, W.L.; Cytryn, E.; Jeong, S.; Joshi, T.; Emerich, D.W.; Sadowsky, M.J.; Xu, D.; Stacey, G. An oligonucleotide microarray resource for transcriptional profiling of Bradyrhizobium japonicum. Mol. Plant. Microbe Interact. 2007, 20, 1298–1307. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, A.; Moser, A.; Pessi, G.; Hauser, F.; Friberg, M.; Hennecke, H.; Fischer, H.-M. New target genes controlled by the Bradyrhizobium japonicum two-component regulatory system RegSR. J. Bacteriol. 2007, 189, 8928–8943. [Google Scholar] [CrossRef] [PubMed]
- Lang, K.; Lindemann, A.; Hauser, F.; Göttfert, M. The genistein stimulon of Bradyrhizobium japonicum. Mol. Genet. Genom. 2008, 279, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Mesa, S.; Reutimann, L.; Fischer, H.-M.; Hennecke, H. Posttranslational control of transcription factor FixK2, a key regulator for the Bradyrhizobium japonicum-soybean symbiosis. Proc. Natl. Acad. Sci. USA 2009, 106, 21860–21865. [Google Scholar] [CrossRef] [PubMed]
- Lardi, M.; Murset, V.; Fischer, H.-M.; Mesa, S.; Ahrens, C.H.; Zamboni, N.; Pessi, G. Metabolomic profiling of Bradyrhizobium diazoefficiens-induced root nodules reveals both host plant-specific and developmental signatures. Int. J. Mol. Sci. 2016, 17, 815. [Google Scholar] [CrossRef] [PubMed]
- Koch, M.; Delmotte, N.; Rehrauer, H.; Vorholt, J.A.; Pessi, G.; Hennecke, H. Rhizobial adaptation to hosts, a new facet in the legume root-nodule symbiosis. Mol. Plant. Microbe Interact. 2010, 23, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Jiang, X.; Guan, D.; Zhou, W.; Ma, M.; Zhao, B.; Cao, F.; Li, L.; Li, J. Transcriptional analysis of genes involved in competitive nodulation in Bradyrhizobium diazoefficiens at the presence of soybean root exudates. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Hao, B.; Liu, L.; Wang, S.; Ma, B.; Yang, Y.; Xie, F.; Li, Y. RNA-Seq and microarrays analyses reveal global differential transcriptomes of Mesorhizobium huakuii 7653R between bacteroids and free-living cells. PLoS ONE 2014, 9, e93626. [Google Scholar] [CrossRef] [PubMed]
- Uchiumi, T.; Ohwada, T.; Itakura, M.; Mitsui, H.; Nukui, N.; Dawadi, P.; Kaneko, T.; Tabata, S.; Yokoyama, T.; Tejima, K.; et al. Expression islands clustered on the symbiosis island of the Mesorhizobium loti genome. J. Bacteriol. 2004, 186, 2439–2448. [Google Scholar] [CrossRef] [PubMed]
- Salazar, E.; Diaz-Mejia, J.J.; Moreno-Hagelsieb, G.; Martinez-Batallar, G.; Mora, Y.; Mora, J.; Encarnacion, S. Characterization of the NifA-RpoN regulon in Rhizobium etli in free life and in symbiosis with Phaseolus vulgaris. Appl. Environ. Microbiol. 2010, 76, 4510–4520. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, R.; Ramachandran, V.K.; Seaman, J.C.; East, A.K.; Mouhsine, B.; Mauchline, T.H.; Prell, J.; Skeffington, A.; Poole, P.S. Transcriptomic analysis of Rhizobium leguminosarum biovar viciae in symbiosis with host plants Pisum sativum and Vicia cracca. J. Bacteriol. 2009, 191, 4002–4014. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, V.K.; East, A.K.; Karunakaran, R.; Downie, J.A.; Poole, P.S. Adaptation of Rhizobium leguminosarum to pea, alfalfa and sugar beet rhizospheres investigated by comparative transcriptomics. Genome Biol. 2011, 12, R106. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Montaño, F.; del Cerro, P.; Jiménez-Guerrero, I.; López-Baena, F.J.; Cubo, M.T.; Hungria, M.; Megías, M.; Ollero, F.J. RNA-seq analysis of the Rhizobium tropici CIAT 899 transcriptome shows similarities in the activation patterns of symbiotic genes in the presence of apigenin and salt. BMC Genom. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Del Cerro, P.; Pérez-Montaño, F.; Gil-Serrano, A.; López-Baena, F.J.; Megías, M.; Hungria, M.; Ollero, F.J. The Rhizobium tropici CIAT 899 NodD2 protein regulates the production of Nod factors under salt stress in a flavonoid-independent manner. Sci. Rep. 2017, 7, 46712. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Montaño, F.; Jiménez-Guerrero, I.; Acosta-Jurado, S.; Navarro-Gómez, P.; Ollero, F.J.; Ruiz-Sainz, J.E.; López-Baena, F.J.; Vinardell, J.M. A transcriptomic analysis of the effect of genistein on Sinorhizobium fredii HH103 reveals novel rhizobial genes putatively involved in symbiosis. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Ampe, F.; Kiss, E.; Sabourdy, F.; Batut, J. Transcriptome analysis of Sinorhizobium meliloti during symbiosis. Genome Biol. 2003, 4, R15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A.; Bergès, H.; Krol, E.; Bruand, C.; Rüberg, S.; Capela, D.; Lauber, E.; Meilhoc, E.; Ampe, F.; de Bruijn, F.J.; et al. Global changes in gene expression in Sinorhizobium meliloti 1021 under microoxic and symbiotic conditions. Mol. Plant. Microbe Interact. 2004, 17, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Capela, D.; Filipe, C.; Bobik, C.; Batut, J.; Bruand, C. Sinorhizobium meliloti differentiation during symbiosis with alfalfa: A transcriptomic dissection. Mol. Plant. Microbe Interact. 2006, 19, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Capela, D.; Carrere, S.; Batut, J. Transcriptome-based identification of the Sinorhizobium meliloti NodD1 regulon. Appl. Environ. Microbiol. 2005, 71, 4910–4913. [Google Scholar] [CrossRef] [PubMed]
- Barnett, M.J.; Toman, C.J.; Fisher, R.F.; Long, S.R. A dual-genome symbiosis chip for coordinate study of signal exchange and development in a prokaryote-host interaction. Proc. Natl. Acad. Sci. USA 2004, 101, 16636–16641. [Google Scholar] [CrossRef] [PubMed]
- Bobik, C.; Meilhoc, E.; Batut, J. FixJ: A major regulator of the oxygen limitation response and late symbiotic functions of Sinorhizobium meliloti. J. Bacteriol. 2006, 188, 4890–4902. [Google Scholar] [CrossRef] [PubMed]
- Sallet, E.; Roux, B.; Sauviac, L.; Jardinaud, M.-F.; Carrere, S.; Faraut, T.; de Carvalho-Niebel, F.; Gouzy, J.; Gamas, P.; Capela, D.; et al. Next-generation annotation of prokaryotic genomes with EuGene-P: Application to Sinorhizobium meliloti 2011. DNA Res. 2013, 20, 339–354. [Google Scholar] [CrossRef] [PubMed]
- Roux, B.; Rodde, N.; Jardinaud, M.-F.; Timmers, T.; Sauviac, L.; Cottret, L.; Carrère, S.; Sallet, E.; Courcelle, E.; Moreau, S.; et al. An integrated analysis of plant and bacterial gene expression in symbiotic root nodules using laser-capture microdissection coupled to RNA sequencing. Plant J. 2014, 77, 817–837. [Google Scholar] [CrossRef] [PubMed]
- Lardi, M.; Liu, Y.; Giudice, G.; Ahrens, C.; Zamboni, N.; Pessi, G. Metabolomics and transcriptomics identify multiple downstream targets of Paraburkholderia phymatum σ54 during symbiosis with Phaseolus vulgaris. Int. J. Mol. Sci. 2018, 19, 1049. [Google Scholar] [CrossRef] [PubMed]
- Sarma, A.D.; Emerich, D.W. Global protein expression pattern of Bradyrhizobium japonicum bacteroids: A prelude to functional proteomics. Proteomics 2005, 5, 4170–4184. [Google Scholar] [CrossRef] [PubMed]
- Sarma, A.D.; Emerich, D.W. A comparative proteomic evaluation of culture grown vs. nodule isolated Bradyrhizobium japonicum. Proteomics 2006, 6, 3008–3028. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, N.; Ahrens, C.H.; Knief, C.; Qeli, E.; Koch, M.; Fischer, H.-M.; Vorholt, J.A.; Hennecke, H.; Pessi, G. An integrated proteomics and transcriptomics reference data set provides new insights into the Bradyrhizobium japonicum bacteroid metabolism in soybean root nodules. Proteomics 2010, 10, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Süss, C.; Hempel, J.; Zehner, S.; Krause, A.; Patschkowski, T.; Göttfert, M. Identification of genistein-inducible and type III-secreted proteins of Bradyrhizobium japonicum. J. Biotechnol. 2006, 126, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Hempel, J.; Zehner, S.; Göttfert, M.; Patschkowski, T. Analysis of the secretome of the soybean symbiont Bradyrhizobium japonicum. J. Biotechnol. 2009, 140, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Dainese-Hatt, P.; Fischer, H.-M.; Hennecke, H.; James, P. Classifying symbiotic proteins from Bradyrhizobium japonicum into functional groups by proteome analysis of altered gene expression levels. Electrophoresis 1999, 20, 3514–3520. [Google Scholar] [CrossRef]
- Liu, Y.; Guan, D.; Jiang, X.; Ma, M.; Li, L.; Cao, F.; Chen, H.; Shen, D.; Li, J. Proteins involved in nodulation competitiveness of two Bradyrhizobium diazoefficiens strains induced by soybean root exudates. Biol. Fertil. Soils 2015, 51, 251–260. [Google Scholar] [CrossRef]
- Da Silva Batista, J.S.; Hungria, M. Proteomics reveals differential expression of proteins related to a variety of metabolic pathways by genistein-induced Bradyrhizobium japonicum strains. J. Proteom. 2012, 75, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Delmotte, N.; Mondy, S.; Alunni, B.; Fardoux, J.; Chaintreuil, C.; Vorholt, J.; Giraud, E.; Gourion, B. A proteomic approach of Bradyrhizobium/Aeschynomene root and stem symbioses reveals the importance of the fixA locus for symbiosis. Int. J. Mol. Sci. 2014, 15, 3660–3670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatsukami, Y.; Nambu, M.; Morisaka, H.; Kuroda, K.; Ueda, M. Disclosure of the differences of Mesorhizobium loti under the free-living and symbiotic conditions by comparative proteome analysis without bacteroid isolation. BMC Microbiol. 2013, 13, 180. [Google Scholar] [CrossRef] [PubMed]
- Nambu, M.; Tatsukami, Y.; Morisaka, H.; Kuroda, K.; Ueda, M. Quantitative time-course proteome analysis of Mesorhizobium loti during nodule maturation. J. Proteom. 2015, 125, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Meneses, N.; Taboada, H.; Dunn, M.F.; del Carmen Vargas, M.; Buchs, N.; Heller, M.; Encarnación, S. The naringenin-induced exoproteome of Rhizobium etli CE3. Arch. Microbiol. 2017, 199, 737–755. [Google Scholar] [CrossRef] [PubMed]
- Tolin, S.; Arrigoni, G.; Moscatiello, R.; Masi, A.; Navazio, L.; Sablok, G.; Squartini, A. Quantitative analysis of the naringenin-inducible proteome in Rhizobium leguminosarum by isobaric tagging and mass spectrometry. Proteomics 2013, 13, 1961–1972. [Google Scholar] [CrossRef] [PubMed]
- Arrigoni, G.; Tolin, S.; Moscatiello, R.; Masi, A.; Navazio, L.; Squartini, A. Calcium-dependent regulation of genes for plant nodulation in Rhizobium leguminosarum detected by iTRAQ quantitative proteomic analysis. J. Proteome Res. 2013, 12, 5323–5330. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, N.; Redmond, J.W.; Rolfe, B.G.; Djordjevic, M.A. New Rhizobium leguminosarum flavonoid-induced proteins revealed by proteome analysis of differentially displayed proteins. Mol. Plant. Microbe Interact. 1997, 10, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Natera, S.H.A.; Guerreiro, N.; Djordjevic, M.A. Proteome analysis of differentially displayed proteins as a tool for the investigation of symbiosis. Mol. Plant. Microbe Interact. 2000, 13, 995–1009. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, M.A.; Chen, H.C.; Natera, S.; Van Noorden, G.; Menzel, C.; Taylor, S.; Renard, C.; Geiger, O.; Weiller, G.F. A global analysis of protein expression profiles in Sinorhizobium meliloti: Discovery of new genes for nodule occupancy and stress adaptation. Mol. Plant. Microbe Interact. 2003, 16, 508–524. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, M.A. Sinorhizobium meliloti metabolism in the root nodule: A proteomic perspective. Proteomics 2004, 4, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Larrainzar, E.; Wienkoop, S.; Weckwerth, W.; Ladrera, R.; Arrese-Igor, C.; Gonzalez, E.M. Medicago truncatula root nodule proteome analysis reveals differential plant and bacteroid responses to drought stress. Plant Physiol. 2007, 144, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Marx, H.; Minogue, C.E.; Jayaraman, D.; Richards, A.L.; Kwiecien, N.W.; Siahpirani, A.F.; Rajasekar, S.; Maeda, J.; Garcia, K.; Del Valle-Echevarria, A.R.; et al. A proteomic atlas of the legume Medicago truncatula and its nitrogen-fixing endosymbiont Sinorhizobium meliloti. Nat. Biotechnol. 2016, 34, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Higgins, J.; Oresnik, I.J.; Hynes, M.F.; Natera, S.; Djordjevic, M.A.; Weinman, J.J.; Rolfe, B.G. Proteome analysis demonstrates complex replicon and luteolin interactions in pSyma-cured derivatives of Sinorhizobium meliloti strain 2011. Electrophoresis 2000, 21, 3833–3842. [Google Scholar] [CrossRef]
- Brechenmacher, L.; Lei, Z.; Libault, M.; Findley, S.; Sugawara, M.; Sadowsky, M.J.; Sumner, L.W.; Stacey, G. Soybean metabolites regulated in root hairs in response to the symbiotic bacterium Bradyrhizobium japonicum. Plant Physiol. 2010, 153, 1808–1822. [Google Scholar] [CrossRef] [PubMed]
- Vauclare, P.; Bligny, R.; Gout, E.; Widmer, F. An overview of the metabolic differences between Bradyrhizobium japonicum 110 bacteria and differentiated bacteroids from soybean (Glycine max) root nodules: An in vitro 13C- and 31P-nuclear magnetic resonance spectroscopy study. FEMS Microbiol. Lett. 2013, 343, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Colebatch, G.; Desbrosses, G.; Ott, T.; Krusell, L.; Montanari, O.; Kloska, S.; Kopka, J.; Udvardi, M.K. Global changes in transcription orchestrate metabolic differentiation during symbiotic nitrogen fixation in Lotus japonicus. Plant J. 2004, 39, 487–512. [Google Scholar] [CrossRef] [PubMed]
- Desbrosses, G.G.; Kopka, J.; Udvardi, M.K. Lotus japonicus metabolic profiling. Development of gas chromatography-mass spectrometry resources for the study of plant-microbe interactions. Plant Physiol. 2005, 137, 1302–1318. [Google Scholar] [CrossRef] [PubMed]
- Barsch, A.; Tellström, V.; Patschkowski, T.; Küster, H.; Niehaus, K. Metabolite profiles of nodulated alfalfa plants indicate that distinct stages of nodule organogenesis are accompanied by global physiological adaptations. Mol. Plant. Microbe Interact. 2006, 19, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Gemperline, E.; Jayaraman, D.; Maeda, J.; Ané, J.-M.; Li, L. Multifaceted investigation of metabolites during nitrogen fixation in Medicago via high resolution MALDI-MS imaging and ESI-MS. J. Am. Soc. Mass Spectrom. 2015, 26, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Guerrero, I.; Acosta-Jurado, S.; del Cerro, P.; Navarro-Gómez, P.; López-Baena, F.; Ollero, F.; Vinardell, J.; Pérez-Montaño, F. Transcriptomic studies of the effect of nod gene-inducing molecules in rhizobia: Different weapons, one purpose. Genes 2017, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, J.-P.; Reinkensmeier, J.; Daschkey, S.; Evguenieva-Hackenberg, E.; Janssen, S.; Jänicke, S.; Becker, J.D.; Giegerich, R.; Becker, A. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genom. 2010, 11, 245. [Google Scholar] [CrossRef] [PubMed]
- Madhugiri, R.; Pessi, G.; Voss, B.; Hahn, J.; Sharma, C.M.; Reinhardt, R.; Vogel, J.; Hess, W.R.; Fischer, H.-M.; Evguenieva-Hackenberg, E. Small RNAs of the Bradyrhizobium/Rhodopseudomonas lineage and their analysis. RNA Biol. 2012, 9, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Vercruysse, M.; Fauvart, M.; Cloots, L.; Engelen, K.; Thijs, I.M.; Marchal, K.; Michiels, J. Genome-wide detection of predicted non-coding RNAs in Rhizobium etli expressed during free-living and host-associated growth using a high-resolution tiling array. BMC Genom. 2010, 11, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Quesada, O.; Reinkensmeier, J.; Schlüter, J.-P.; Robledo, M.; Peregrina, A.; Giegerich, R.; Toro, N.; Becker, A.; Jiménez-Zurdo, J.I. Genome-wide profiling of Hfq-binding RNAs uncovers extensive post-transcriptional rewiring of major stress response and symbiotic regulons in Sinorhizobium meliloti. RNA Biol. 2014, 11, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Schlüter, J.-P.; Reinkensmeier, J.; Barnett, M.J.; Lang, C.; Krol, E.; Giegerich, R.; Long, S.R.; Becker, A. Global mapping of transcription start sites and promoter motifs in the symbiotic α-proteobacterium Sinorhizobium meliloti 1021. BMC Genom. 2013, 14, 156. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Overlöper, A.; Schlüter, J.-P.; Reinkensmeier, J.; Robledo, M.; Giegerich, R.; Narberhaus, F.; Evguenieva-Hackenberg, E. Riboregulation in plant-associated α-proteobacteria. RNA Biol. 2014, 11, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Melkonian, R.; Moulin, L.; Béna, G.; Tisseyre, P.; Chaintreuil, C.; Heulin, K.; Rezkallah, N.; Klonowska, A.; Gonzalez, S.; Simon, M.; et al. The geographical patterns of symbiont diversity in the invasive legume Mimosa pudica can be explained by the competitiveness of its symbionts and by the host genotype: Competition for nodulation in α- and β-rhizobia. Environ. Microbiol. 2014, 16, 2099–2111. [Google Scholar] [CrossRef] [PubMed]
- Ahrens, C.H.; Brunner, E.; Qeli, E.; Basler, K.; Aebersold, R. Generating and navigating proteome maps using mass spectrometry. Nat. Rev. Mol. Cell Biol. 2010, 11, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Qeli, E.; Ahrens, C.H. Peptide Classifier for protein inference and targeted quantitative proteomics. Nat. Biotechnol. 2010, 28, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Stekhoven, D.J.; Omasits, U.; Quebatte, M.; Dehio, C.; Ahrens, C.H. Proteome-wide identification of predominant subcellular protein localizations in a bacterial model organism. J. Proteom. 2014, 99, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Nesvizhskii, A.I. Proteogenomics: Concepts, applications and computational strategies. Nat. Methods 2014, 11, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Omasits, U.; Varadarajan, A.R.; Schmid, M.; Goetze, S.; Melidis, D.; Bourqui, M.; Nikolayeva, O.; Québatte, M.; Patrignani, A.; Dehio, C.; et al. An integrative strategy to identify the entire protein coding potential of prokaryotic genomes by proteogenomics. Genome Res. 2017, 27, 2083–2095. [Google Scholar] [CrossRef] [PubMed]
- Omasits, U.; Quebatte, M.; Stekhoven, D.J.; Fortes, C.; Roschitzki, B.; Robinson, M.D.; Dehio, C.; Ahrens, C.H. Directed shotgun proteomics guided by saturated RNA-seq identifies a complete expressed prokaryotic proteome. Genome Res. 2013, 23, 1916–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Pessi, G.; Riedel, K.; Eberl, L. Identification of AHL- and BDSF-controlled proteins in Burkholderia cenocepacia by proteomics. Methods Mol. Biol. 2018, 1673, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Rogowska-Wrzesinska, A.; Le Bihan, M.-C.; Thaysen-Andersen, M.; Roepstorff, P. 2D gels still have a niche in proteomics. J. Proteom. 2013, 88, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Washburn, M.P.; Wolters, D.; Yates, J.R. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 2001, 19, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Larrainzar, E.; Wienkoop, S. A proteomic view on the role of legume symbiotic interactions. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Fry, J.; Wood, M.; Poole, P.S. Investigation of myo-inositol catabolism in Rhizobium leguminosarum bv. viciae and its effect on nodulation competitiveness. Mol. Plant. Microbe Interact. 2001, 14, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Geddes, B.A.; González, J.E.; Oresnik, I.J. Exopolysaccharide production in response to medium acidification is correlated with an increase in competition for nodule occupancy. Mol. Plant. Microbe Interact. 2014, 27, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Winzer, T.; Bairl, A.; Linder, M.; Linder, D.; Werner, D.; Müller, P. A novel 53-kDa nodulin of the symbiosome membrane of soybean nodules, controlled by Bradyrhizobium japonicum. Mol. Plant. Microbe Interact. 1999, 12, 218–226. [Google Scholar] [CrossRef] [PubMed]
- Panter, S.; Thomson, R.; de Bruxelles, G.; Laver, D.; Trevaskis, B.; Udvardi, M. Identification with proteomics of novel proteins associated with the peribacteroid membrane of soybean root nodules. Mol. Plant. Microbe Interact. 2000, 13, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Nomura, M.; Arunothayanan, H.; Van dao, T.; Le, H.T.-P.; Kaneko, T.; Sato, S.; Tabata, S.; Tajima, S. Differential protein profiles of Bradyrhizobium japonicum USDA110 bacteroid during soybean nodule development. Soil Sci. Plant Nutr. 2010, 56, 579–590. [Google Scholar] [CrossRef]
- Farkas, A.; Maroti, G.; Durg, H.; Gyorgypal, Z.; Lima, R.M.; Medzihradszky, K.F.; Kereszt, A.; Mergaert, P.; Kondorosi, E. Medicago truncatula symbiotic peptide NCR247 contributes to bacteroid differentiation through multiple mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 5183–5188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penterman, J.; Abo, R.P.; De Nisco, N.J.; Arnold, M.F.F.; Longhi, R.; Zanda, M.; Walker, G.C. Host plant peptides elicit a transcriptional response to control the Sinorhizobium meliloti cell cycle during symbiosis. Proc. Natl. Acad. Sci. USA 2014, 111, 3561–3566. [Google Scholar] [CrossRef] [PubMed]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Metabolomics: The apogee of the omics trilogy: Innovation. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Fuhrer, T.; Zamboni, N. High-throughput discovery metabolomics. Curr. Opin. Biotechnol. 2015, 31, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Brüschweiler, R.; Edison, A.S.; Eghbalnia, H.R.; Powers, R.; Raftery, D.; Wishart, D.S. The future of NMR-based metabolomics. Curr. Opin. Biotechnol. 2017, 43, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Veenstra, T.D. Metabolomics: The final frontier? Genome Med. 2012, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Chuang, W.-G.; Cui, X.; Cheng, R.-H.; Chiu, K.; Chen, Z.; Ding, S. High resolution 31P NMR spectroscopy generates a quantitative evolution profile of phosphorous translocation in germinating sesame seed. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Want, E.J.; Nordström, A.; Morita, H.; Siuzdak, G. From exogenous to endogenous: The inevitable imprint of mass spectrometry in metabolomics. J. Proteome Res. 2007, 6, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Yin, Y. Strategies for large-scale targeted metabolomics quantification by liquid chromatography-mass spectrometry. Analyst 2016, 141, 6362–6373. [Google Scholar] [CrossRef] [PubMed]
- Scalbert, A.; Brennan, L.; Fiehn, O.; Hankemeier, T.; Kristal, B.S.; van Ommen, B.; Pujos-Guillot, E.; Verheij, E.; Wishart, D.; Wopereis, S. Mass-spectrometry-based metabolomics: Limitations and recommendations for future progress with particular focus on nutrition research. Metabolomics 2009, 5, 435–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, F.; Lindemann, A.; Vuilleumier, S.; Patrignani, A.; Schlapbach, R.; Fischer, H.M.; Hennecke, H. Design and validation of a partial-genome microarray for transcriptional profiling of the Bradyrhizobium japonicum symbiotic gene region. Mol. Genet. Genom. 2006, 275, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Caspi, R.; Foerster, H.; Fulcher, C.A.; Kaipa, P.; Krummenacker, M.; Latendresse, M.; Paley, S.; Rhee, S.Y.; Shearer, A.G.; Tissier, C.; et al. The MetaCyc Database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2008, 36, D623–D631. [Google Scholar] [CrossRef] [PubMed]
- Van Opijnen, T.; Bodi, K.L.; Camilli, A. Tn-seq: High-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat. Methods 2009, 6, 767–772. [Google Scholar] [CrossRef] [PubMed]
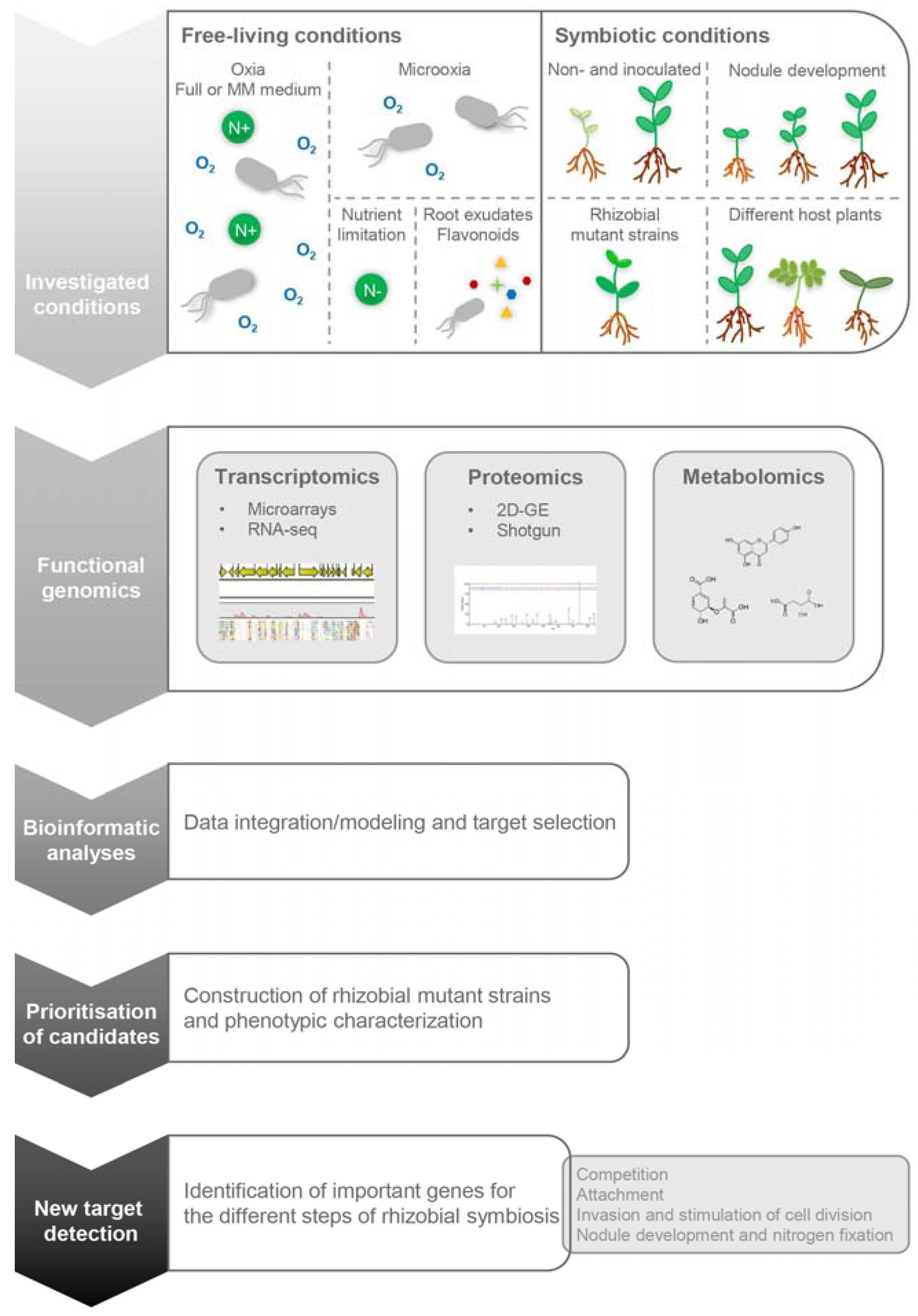

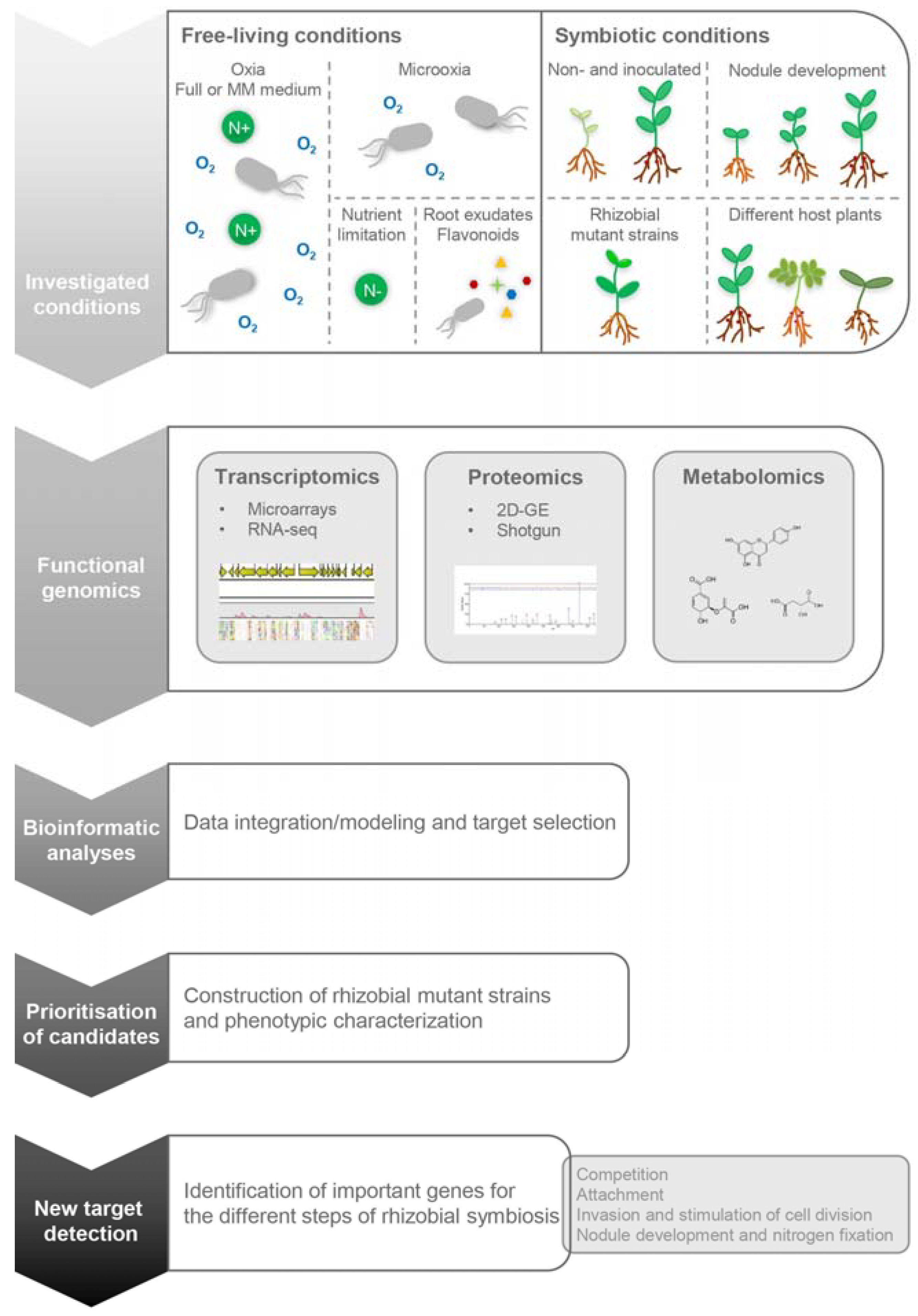{kind=link}
| Bacteria | Plant Host | Strain | Conditions | Reference |
|---|---|---|---|---|
| Transcriptomics (microarrays and RNA-seq *) | ||||
| Alpha-rhizobia | ||||
| Bradyrhizobium diazoefficiens USDA110 | Glycine max | wt, rpoN double mt | microoxia (0.5% O2), nodule development (10, 13, 21 and 31 dpi) | [46] |
| Bradyrhizobium diazoefficiens USDA110 | G. max | wt | bacteroids (28 dpi), salt stress | [47] |
| Bradyrhizobium diazoefficiens USDA110 | G. max | wt | nodules (21 dpi) | [44] * |
| Bradyrhizobium diazoefficiens USDA110 | G. max | wt, regR mt | microoxia (0.5% O2), nodule development (13 and 21 dpi) | [48] |
| Bradyrhizobium diazoefficiens USDA110 | wt, nodW mt, nodW-nswA double mt with over-expression of nwsB | application of genistein | [49] | |
| Bradyrhizobium diazoefficiens USDA110 | G. max | wt, fixk2 mt, fixJ mt | nodules (21 dpi) | [50] |
| Bradyrhizobium diazoefficiens USDA110 | G. max | wt, nifH mt, nifA mt | nodules (21 dpi) | [51] |
| Bradyrhizobium diazoefficiens USDA110 | G. max, Macroptilium atropurpureum, Vigna unguiculata | wt | nodules (21 or 31 dpi [M. atropurpureum]) | [52] |
| Bradyrhizobium diazoefficiens 4534, 4222 | wt | application of root exudates | [53] * | |
| Mesorhizobium huakuii 7653R | Astragalus sinicus | wt | bacteroids (32 dpi) | [54] * |
| Mesorhizobium loti MAFF303099 | Lotus japonicus | wt | microoxia (1.5% O2), bacteroids (42 dpi) | [55] |
| Rhizobium etli CFN42 | Phaseolus vulgaris | wt, nifA mt | microoxia (1% O2), nodules (11 dpi) | [56] |
| Rhizobium leguminosarum biovar viciae 3841 | Pisum sativum, Vicia cracca | wt | bacteroids (28 dpi), bacteroid development (7, 15, and 21 dpi) | [57] |
| Rhizobium leguminosarum biovar viciae 3841 | P. sativum, Medicago sativa, Beta vulgaris | wt | application of root exudates, rhizosphere | [58] |
| Rhizobium mesoamericanum STM3625 | Mimosa pudica | wt | application of root exudates | [41] * |
| Rhizobium tropici CIAT 899 | wt, nodD1 mt, nodD2 mt | application of apigenin, salt stress | [59,60] * | |
| Sinorhizobium fredii HH103 | wt, nodD1 mt, ttsI mt | application of genistein | [61] * | |
| Sinorhizobium meliloti 1021 | Medicago truncatula, M. sativa | wt, bacA mt | application of luteolin, microoxia (<1 µM O2), nodule development (8 and 18 dpi for Fix+ nodules and 11 dpi for Fix−) | [62] |
| Sinorhizobium meliloti 1021 | M. sativa | wt | microoxia (<1 µM O2), bacteroid (18–22 dpi) | [63] |
| Sinorhizobium meliloti 1021 | M. sativa | wt, bacA mt | nodule development (5, 8, 11, 14, or 18 dpi) | [64] |
| Sinorhizobium meliloti 1021 | wt, wt with over-expression of nodD1 | application of luteolin | [65] | |
| Sinorhizobium meliloti 1021 | M. truncatula | wt, nodD123 triple mt, nodD123 triple mt over-expressing nodD1 or nodD3, rpoN mt, fixJ mt | application of luteolin, nodules (33–35 dpi) | [66] |
| Sinorhizobium meliloti 1021 | M. sativa | wt, fixJ mt, nifA mt, fixK mt, nifH mt | microoxia (2% O2), nodules (14 dpi) | [67] |
| Sinorhizobium meliloti 2011 | M. truncatula | wt | nodules (10 dpi) | [68] * |
| Sinorhizobium meliloti 2011 | M. truncatula | wt | nodule development (10 or 15 dpi, laser dissection) | [69] * |
| Sinorhizobium sp. NGR234 | V. unguiculata, Leucaena leucocephala | wt | bacteroids (21 dpi or 31 dpi for L. leucocephala) | [42] * |
| Beta-rhizobia | ||||
| Cupriavidus taiwanensis LMG19424 | M. pudica | wt | application of root exudates | [41] * |
| Paraburkholderia phymatum STM815 | P. vulgaris | wt, rpoN mt | nodules (21 dpi) | [43,70] * |
| Paraburkholderia phymatum STM815 | M. pudica | wt | application of root exudates | [41] * |
| Paraburkholderia phymatum STM815 | wt | nitrogen starvation | [43] * | |
| Proteomics (2-D GE and LC-MS/MS *) | ||||
| Alpha-rhizobia | ||||
| Bradyrhizobium diazoefficiens USDA110 | G. max | wt | bacteroids (28 dpi or 21 dpi) | [71,72] [46,73] * |
| Bradyrhizobium diazoefficiens USDA110 | G. max, M. atropurpureum, V. unguiculata | wt | bacteroids (21 or 31 dpi [M. atropurpureum]) | [52] * |
| Bradyrhizobium diazoefficiens USDA110 | wt, flagellin-ttsI double mt, flagellin-T3SS double mt | application of genistein | [74] | |
| Bradyrhizobium diazoefficiens USDA110 | wt, flagellin mt | application of genistein | [75] * | |
| Bradyrhizobium diazoefficiens USDA110 | wt | microoxia (2% O2), anoxia | [76] | |
| Bradyrhizobium diazoefficiens 4534, 4222 | wt | application of root exudates | [77] | |
| Bradyrhizobium diazoefficiens CPAC 15 | wt | application of genistein | [78] | |
| Bradyrhizobium sp. ORS278 | Aeschynomene indica | wt | bacteroid development (14 and 21 dpi) | [79] * |
| Mesorhizobium loti MAFF303099 | L. japonicus | wt | nodules (49 dpi) | [80] * |
| Mesorhizobium loti MAFF303099 | L. japonicus | wt | bacteroid development (14, 21 and 28 dpi) | [81] * |
| Rhizobium etli CFN42 | P. vulgaris | wt, nifA mt | bacteroids (11 dpi) | [56] |
| Rhizobium etli CE3 | wt | application of naringenin | [82] * | |
| Rhizobium leguminosarum bv viciae 3841 | wt | application of naringenin | [83,84] * | |
| Rhizobium leguminosarum bv trifolii ANU843 | wt | application of 7,4′-dihydroxyflavone (DHF) | [85] | |
| Sinorhizobium meliloti 1021 | M. truncatula, Melilotus alba | wt | nodules, bacteroids (12 dpi) | [86,87,88] |
| Sinorhizobium meliloti 2011 | M. truncatula | wt | bacteroids, nodule development (+ or − drought stress) (3 and 6 dpi) | [89] * |
| Sinorhizobium meliloti | M. truncatula | wt | bacteroids, nodule development (10, 14 and 28 dpi) | [90] * |
| Sinorhizobium meliloti 2011 | wt, pRm211aΔ14-16, pRm2011a cured | application of luteolin | [91] | |
| Metabolomics | ||||
| Alpha-rhizobia | ||||
| Bradyrhizobium diazoefficiens USDA110 | G. max, M. atropurpureum, V. unguiculata, Vigna radiata | wt, nifA mt, nifH mt | nodule development (13, 21 and 31 dpi) | [51] |
| Bradyrhizobium diazoefficiens USDA110 | G. max | wt | root hairs | [92] |
| Bradyrhizobium diazoefficiens USDA110 | G. max | wt | bacteroids (28–32 dpi) | [93] |
| Mesorhizobium loti R7A | L. japonicus | wt | nodules (84 dpi) | [94,95] |
| Sinorhizobium meliloti 1021 | M. sativa | wt, exoY mt, nifH mt | nodules (21 dpi) | [96] |
| Sinorhizobium meliloti 1021 | M. truncatula | wt, fixJ mt | nodules (14 dpi) | [97] |
| Beta-rhizobia | ||||
| Paraburkholderia phymatum STM815 | P. vulgaris | wt, rpoN mt | nodules (21 dpi) | [70] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lardi, M.; Pessi, G. Functional Genomics Approaches to Studying Symbioses between Legumes and Nitrogen-Fixing Rhizobia. High-Throughput 2018, 7, 15. https://doi.org/10.3390/ht7020015
Lardi M, Pessi G. Functional Genomics Approaches to Studying Symbioses between Legumes and Nitrogen-Fixing Rhizobia. High-Throughput. 2018; 7(2):15. https://doi.org/10.3390/ht7020015
Chicago/Turabian StyleLardi, Martina, and Gabriella Pessi. 2018. "Functional Genomics Approaches to Studying Symbioses between Legumes and Nitrogen-Fixing Rhizobia" High-Throughput 7, no. 2: 15. https://doi.org/10.3390/ht7020015
APA StyleLardi, M., & Pessi, G. (2018). Functional Genomics Approaches to Studying Symbioses between Legumes and Nitrogen-Fixing Rhizobia. High-Throughput, 7(2), 15. https://doi.org/10.3390/ht7020015