
Application of High-Throughput Assays to Examine Phospho-Modulation of the Late Steps of Regulated Exocytosis



Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Isolation of Cortical Vesicles and Drug Treatments
2.3. High-Throughput Methodology
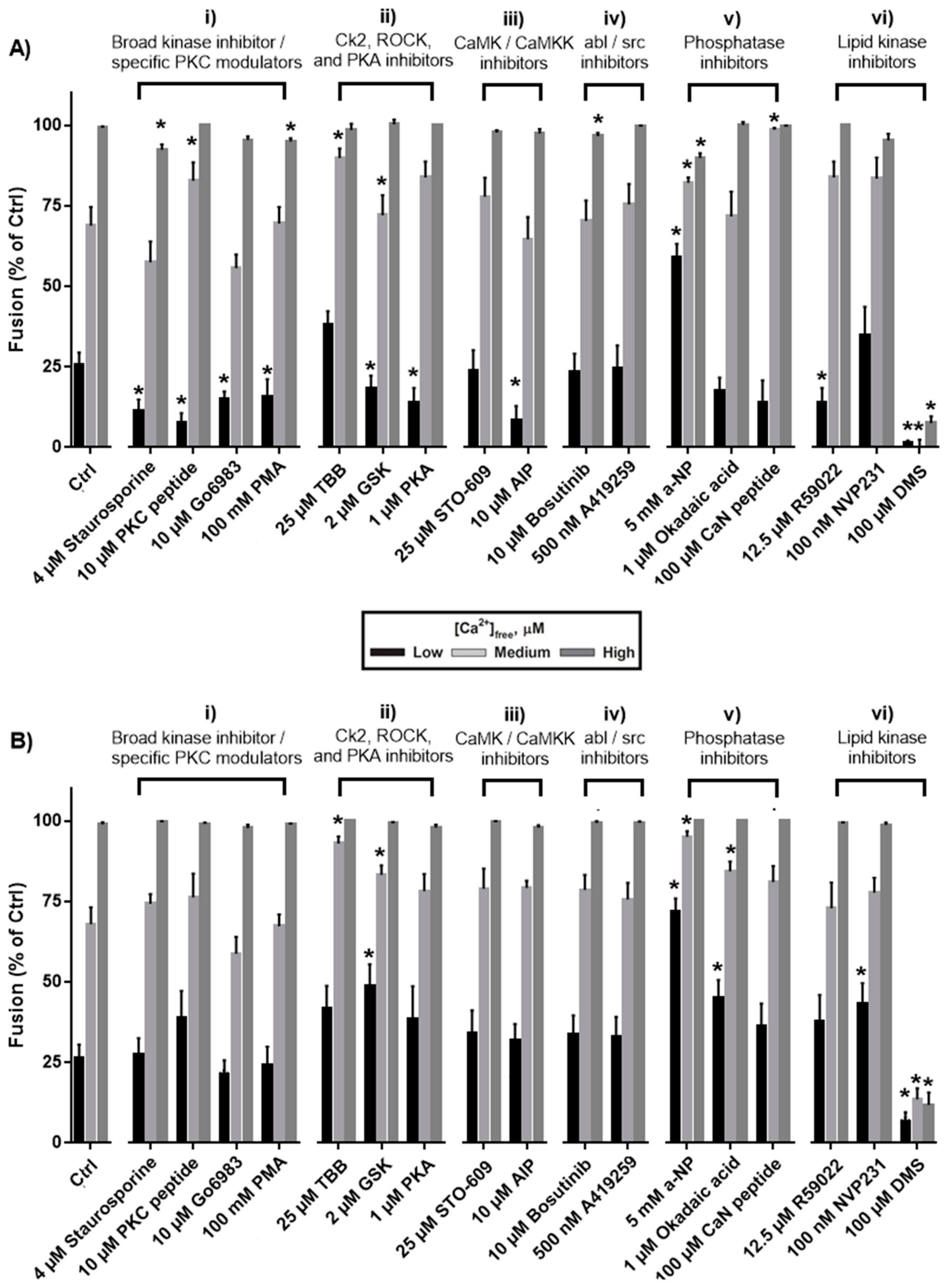
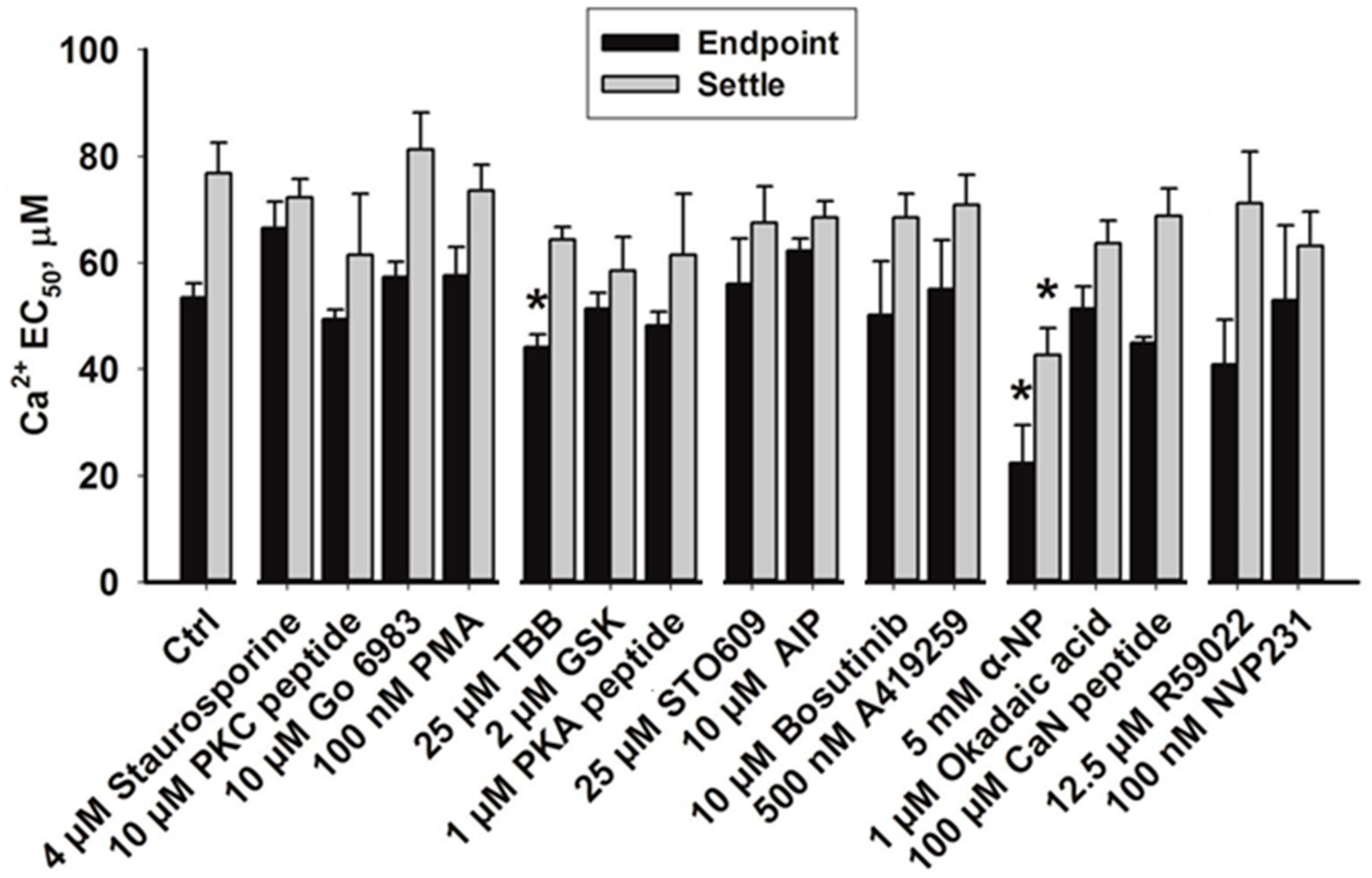
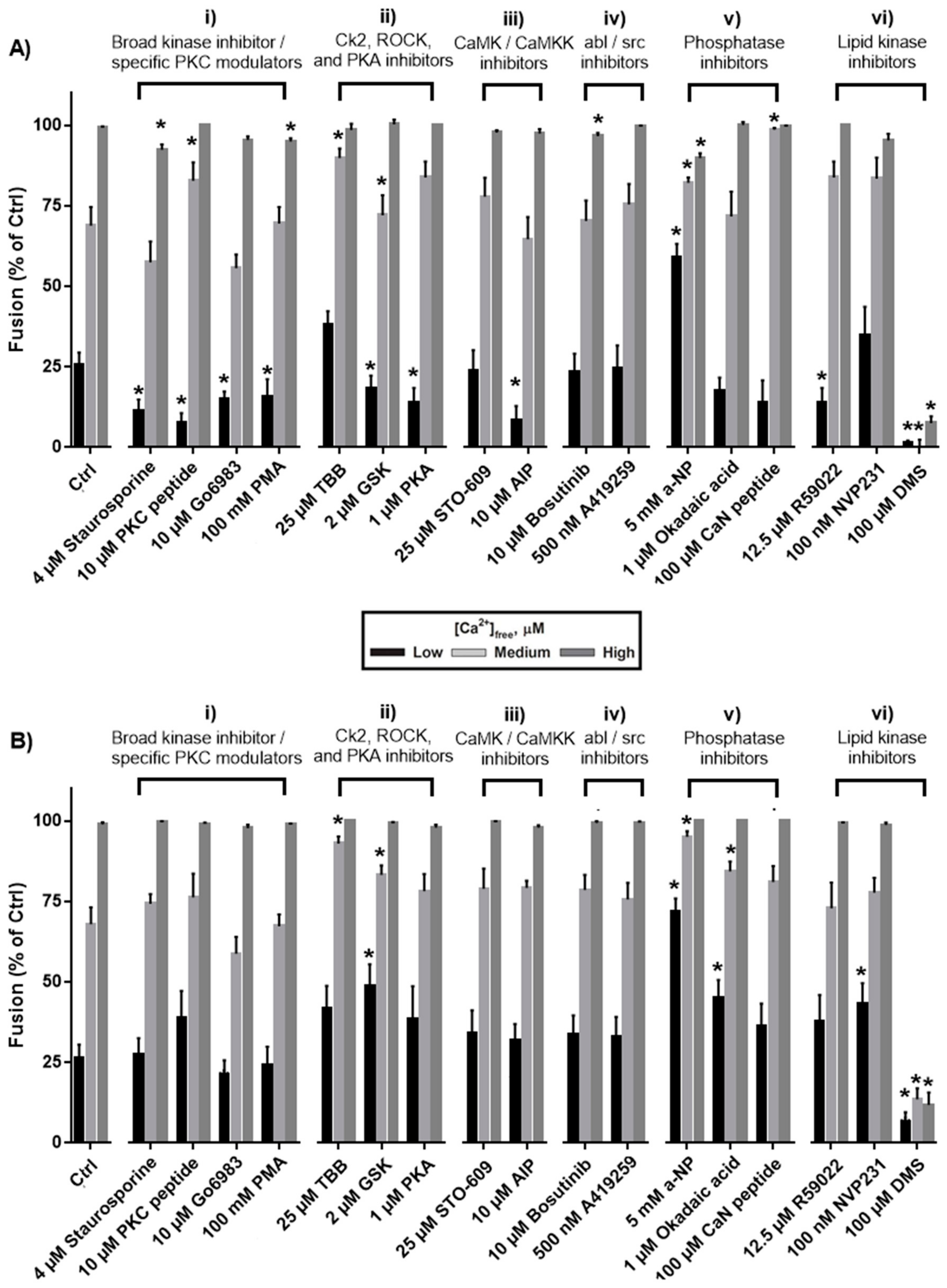
3. Results
3.1. Effects of Protein Kinase Inhibitors
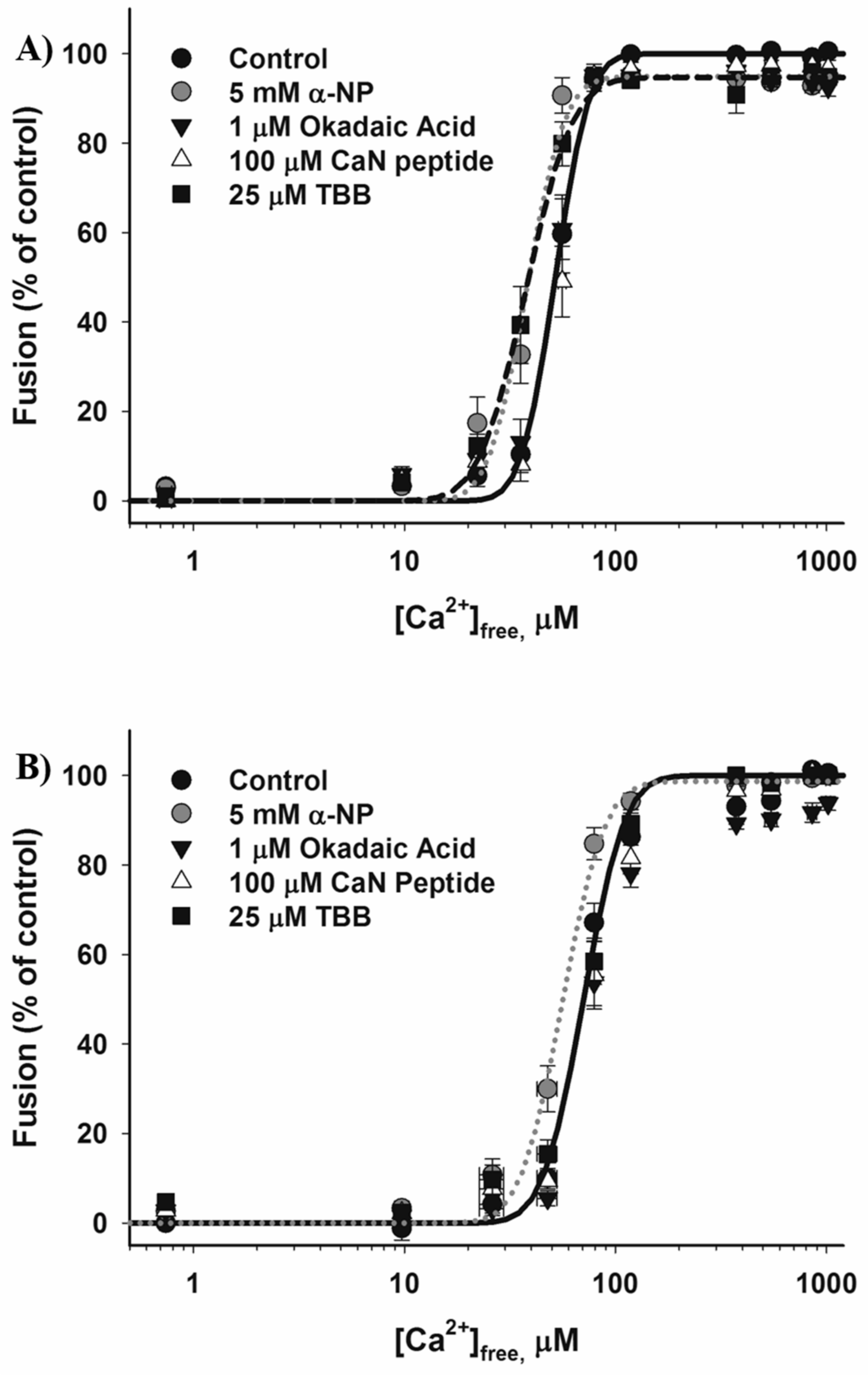
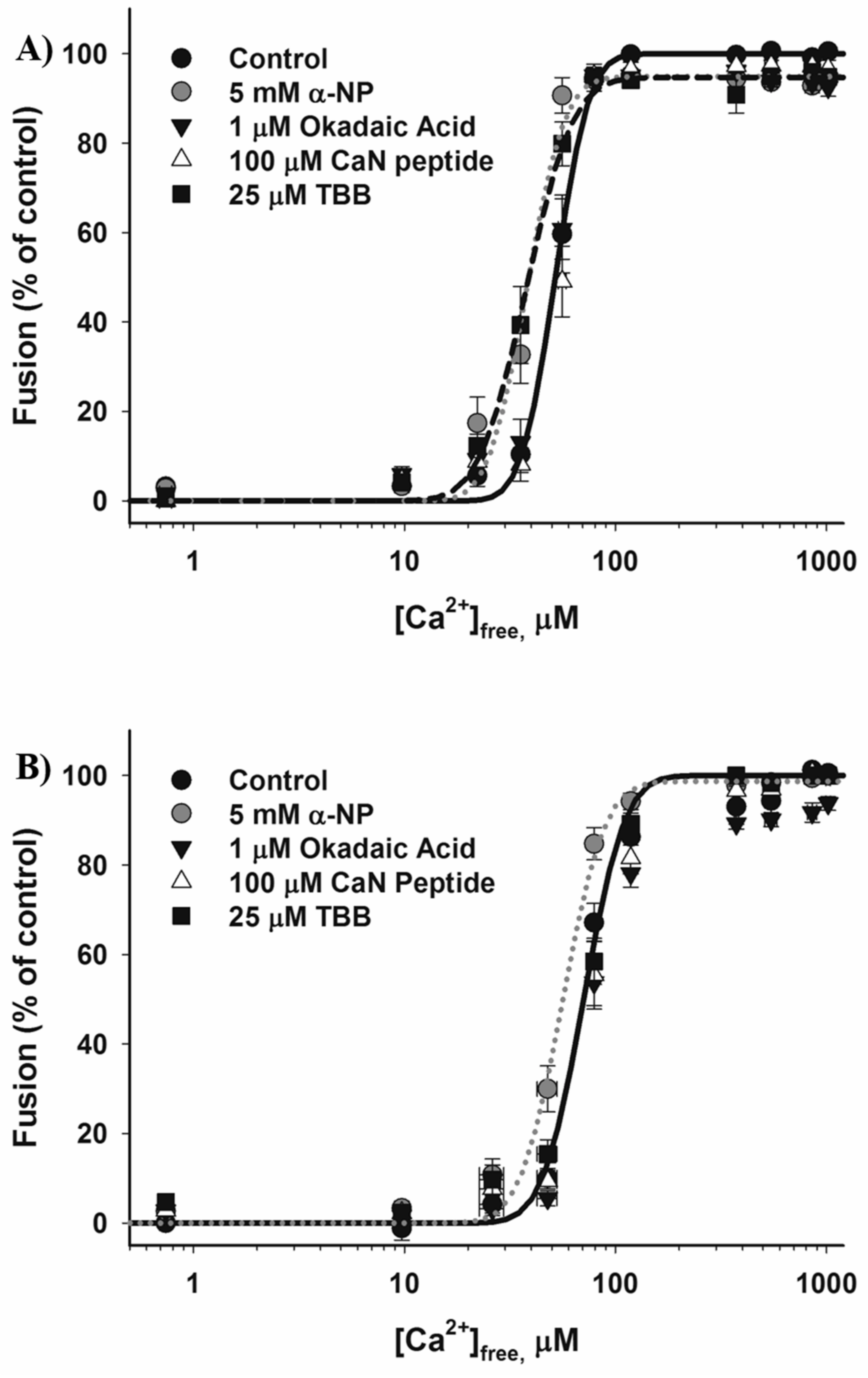
3.2. Effects of Phosphatase Inhibitors
3.3. Effects of Inhibiting Lipid Kinases
4. Discussion
4.1. Protein Kinase C, Protein Kinase A, Calmodulin-dependent Protein Kinase II
4.2. Rho Kinase
4.3. Casein Kinase 2
4.4. Dephosphorylation
4.5. Lipid Phosphorylation
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Churchward, M.A.; Coorssen, J.R. Cholesterol, regulated exocytosis and the physiological fusion machine. Biochem. J. 2009, 423, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Abbineni, P.S.; Hibbert, J.E.; Coorssen, J.R. Critical role of cortical vesicles in dissecting regulated exocytosis: Overview of insights into fundamental molecular mechanisms. Biol. Bull. 2013, 224, 200–217. [Google Scholar] [CrossRef] [PubMed]
- Furber, K.L.; Churchward, M.A.; Rogasevskaia, T.P.; Coorssen, J.R. Identifying critical components of native Ca2+-triggered membrane fusion. Integrating studies of proteins and lipids. Ann. N. Y. Acad. Sci. 2009, 1152, 121–134. [Google Scholar] [CrossRef] [PubMed]
- Becherer, U.; Rettig, J. Vesicle pools, docking, priming, and release. Cell Tissue Res. 2006, 326, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Abbineni, P.S.; Wright, E.P.; Rogasevskaia, T.P.; Killingsworth, M.C.; Malladi, C.S.; Coorssen, J.R. The sea urchin egg and cortical vesicles as model systems to dissect the fast, Ca2+-triggered steps of regulated exocytosis. In Exocytosis Methods; Thorn, P., Ed.; Humana Press: New York, NY, USA, 2013; pp. 221–241. [Google Scholar]
- Rizo, J.; Südhof, T.C. The membrane fusion enigma: SNAREs, Sec1/Munc18 proteins, and their accomplices—Guilty as charged? Ann. Rev. Cell. Dev. Biol. 2012, 28, 279–308. [Google Scholar] [CrossRef] [PubMed]
- Churchward, M.A.; Rogasevskaia, T.; Hofgen, J.; Bau, J.; Coorssen, J.R. Cholesterol facilitates the native mechanism of Ca2+-triggered membrane fusion. J. Cell Sci. 2005, 118, 4833–4848. [Google Scholar] [CrossRef] [PubMed]
- Rogasevskaia, T.P.; Coorssen, J.R. A new approach to the molecular analysis of docking, priming, and regulated membrane fusion. J. Chem. Biol. 2011, 4, 117–136. [Google Scholar] [CrossRef] [PubMed]
- Rogasevskaia, T.P.; Churchward, M.A.; Coorssen, J.R. Anionic lipids in Ca(2+)-triggered fusion. Cell Calcium 2012, 52, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Churchward, M.A.; Rogasevskaia, T.; Brandman, D.M.; Khosravani, H.; Nava, P.; Atkinson, J.K.; Coorssen, J.R. Specific lipids supply critical negative spontaneous curvature—An essential component of native Ca2+-triggered membrane fusion. Biophys. J. 2008, 94, 3976–3986. [Google Scholar] [CrossRef] [PubMed]
- Rogasevskaia, T.; Coorssen, J.R. Sphingomyelin-enriched microdomains define the efficiency of native Ca(2+)-triggered membrane fusion. J. Cell. Sci. 2006, 119, 2688–2694. [Google Scholar] [CrossRef] [PubMed]
- Ammala, C.; Eliasson, L.; Bokvist, K.; Berggren, P.O.; Honkanen, R.E.; Sjoholm, A.; Rorsman, P. Activation of protein kinases and inhibition of protein phosphatases play a central role in the regulation of exocytosis in mouse pancreatic β cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4343–4347. [Google Scholar] [CrossRef] [PubMed]
- Terbush, D.R.; Holz, R.W. Activation of protein kinase C is not required for exocytosis from bovine adrenal chromaffin cells. The effects of protein kinase C(19–31), Ca/CaM kinase II(291–317), and staurosporine. J. Biol. Chem. 1990, 265, 21179–21184. [Google Scholar] [PubMed]
- Scepek, S.; Coorssen, J.R.; Lindau, M. Fusion pore expansion in horse eosinophils is modulated by Ca2+ and protein kinase C via distinct mechanisms. EMBO J. 1998, 17, 4340–4345. [Google Scholar] [CrossRef] [PubMed]
- Shu, Y.; Liu, X.; Yang, Y.; Takahashi, M.; Gillis, K.D. Phosphorylation of SNAP-25 at Ser187 mediates enhancement of exocytosis by a phorbol ester in INS-1 cells. J. Neurosci. 2008, 28, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Kazanietz, M.G. Eyes wide shut: Protein kinase C isozymes are not the only receptors for the phorbol ester tumor promoters. Mol. Carcinog. 2000, 28, 5–11. [Google Scholar] [CrossRef]
- Rhee, J.S.; Betz, A.; Pyott, S.; Reim, K.; Varoqueaux, F.; Augustin, I.; Hesse, D.; Sudhof, T.C.; Takahashi, M.; Rosenmund, C.; et al. β phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKCs. Cell 2002, 108, 121–133. [Google Scholar] [CrossRef]
- Oh, E.; Thurmond, D.C. The stimulus-induced tyrosine phosphorylation of Munc18c facilitates vesicle exocytosis. J. Biol. Chem. 2006, 281, 17624–17634. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, H.; Yamamori, S.; Ono, K.; Aoyagi, K.; Kondo, S.; Takahashi, M. A src family tyrosine kinase inhibits neurotransmitter release from neuronal cells. Proc. Natl. Acad. Sci. USA 2001, 98, 10930–10935. [Google Scholar] [CrossRef] [PubMed]
- Renstrom, E.; Ding, W.G.; Bokvist, K.; Rorsman, P. Neurotransmitter-induced inhibition of exocytosis in insulin-secreting β cells by activation of calcineurin. Neuron 1996, 17, 513–522. [Google Scholar] [CrossRef]
- Sihra, T.S.; Nairn, A.C.; Kloppenburg, P.; Lin, Z.; Pouzat, C. A role for calcineurin (protein phosphatase-2B) in the regulation of glutamate release. Biochem. Biophys. Res. Commun. 1995, 212, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Raufman, J.P.; Malhotra, R.; Raffaniello, R.D. Regulation of calcium-induced exocytosis from gastric chief cells by protein phosphatase-2B (calcineurin). Biochim. Biophys. Acta 1997, 1357, 73–80. [Google Scholar] [CrossRef]
- Kumashiro, S.; Lu, Y.F.; Tomizawa, K.; Matsushita, M.; Wei, F.Y.; Matsui, H. Regulation of synaptic vesicle recycling by calcineurin in different vesicle pools. Neurosci. Res. 2005, 51, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Coorssen, J.R.; Blank, P.S.; Tahara, M.; Zimmerberg, J. Biochemical and functional studies of cortical vesicle fusion: The snare complex and Ca2+ sensitivity. J. Cell Biol. 1998, 143, 1845–1857. [Google Scholar] [CrossRef] [PubMed]
- Tahara, M.; Coorssen, J.R.; Timmers, K.; Blank, P.S.; Whalley, T.; Scheller, R.; Zimmerberg, J. Calcium can disrupt the SNARE protein complex on sea urchin egg secretory vesicles without irreversibly blocking fusion. J. Biol. Chem. 1998, 273, 33667–33673. [Google Scholar] [CrossRef] [PubMed]
- Coorssen, J.R.; Blank, P.S.; Albertorio, F.; Bezrukov, L.; Kolosova, I.; Chen, X.; Backlund, P.S., Jr.; Zimmerberg, J. Regulated secretion: SNARE density, vesicle fusion and calcium dependence. J. Cell. Sci. 2003, 116, 2087–2097. [Google Scholar] [CrossRef] [PubMed]
- Rogasevskaia, T.P.; Coorssen, J.R. The role of phospholipase D in regulated exocytosis. J. Biol. Chem. 2015, 290, 28683–28696. [Google Scholar] [CrossRef] [PubMed]
- Whalley, T.; Crossley, I.; Whitaker, M. Phosphoprotein inhibition of calcium-stimulated exocytosis in sea urchin eggs. J. Cell Biol. 1991, 113, 769–778. [Google Scholar] [CrossRef] [PubMed]
- Sea Urchin Genome Sequencing Consortium; Sodergren, E.; Weinstock, G.M.; Davidson, E.H.; Cameron, R.A.; Gibbs, R.A.; Angerer, R.C.; Angerer, L.M.; Arnone, M.I.; Burgess, D.R.; et al. The genome of the sea urchin Strongylocentrotus purpuratus. Science 2006, 314, 941–952. [Google Scholar] [CrossRef] [PubMed]
- Bradham, C.A.; Foltz, K.R.; Beane, W.S.; Arnone, M.I.; Rizzo, F.; Coffman, J.A.; Mushegian, A.; Goel, M.; Morales, J.; Geneviere, A.M.; et al. The sea urchin kinome: A first look. Dev. Biol. 2006, 300, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Vogel, S.S.; Zimmerberg, J. Proteins on exocytic vesicles mediate calcium-triggered fusion. Proc. Natl. Acad. Sci. USA 1992, 89, 4749–4753. [Google Scholar] [CrossRef] [PubMed]
- Zimmerberg, J.; Blank, P.S.; Kolosova, I.; Cho, M.S.; Tahara, M.; Coorssen, J.R. A stage-specific preparation to study the Ca2+-triggered fusion steps of exocytosis: Rationale and perspectives. Biochimie 2000, 82, 303–314. [Google Scholar] [CrossRef]
- Vogel, S.S.; Beushausen, S.; Lester, D.S. Application of a membrane fusion assay for rapid drug screening. Pharm. Res. 1995, 12, 1417–1422. [Google Scholar] [CrossRef] [PubMed]
- Furber, K.L.; Brandman, D.M.; Coorssen, J.R. Enhancement of the Ca2+-triggering steps of native membrane fusion via thiol-reactivity. J. Chem. Biol. 2009, 2, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Abbineni, P.S.; Western Sydney University, Campbelltown, NSW, Australia; Coorssen, J.R.; Brock University, St. Catharines, ON, Canada. Unpublished work. 2017.
- Blank, P.S.; Cho, M.S.; Vogel, S.S.; Kaplan, D.; Kang, A.; Malley, J.; Zimmerberg, J. Submaximal responses in calcium-triggered exocytosis are explained by differences in the calcium sensitivity of individual secretory vesicles. J. Gen. Physiol. 1998, 112, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Pocotte, S.L.; Frye, R.A.; Senter, R.A.; TerBush, D.R.; Lee, S.A.; Holz, R.W. Effects of phorbol ester on catecholamine secretion and protein phosphorylation in adrenal medullary cell cultures. Proc. Natl. Acad. Sci. USA 1985, 82, 930–934. [Google Scholar] [CrossRef] [PubMed]
- Gillis, K.D.; Mossner, R.; Neher, E. Protein kinase c enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron 1996, 16, 1209–1220. [Google Scholar] [CrossRef]
- Coorssen, J.R.; Haslam, R.J. GTP gamma S and phorbol ester act synergistically to stimulate both Ca2+-independent secretion and phospholipase d activity in permeabilized human platelets. Inhibition by BAPTA and analogues. FEBS Lett. 1993, 316, 170–174. [Google Scholar] [CrossRef]
- Shen, S.S.; Burgart, L.J. 1,2-diacylglycerols mimic phorbol 12-myristate 13-acetate activation of the sea urchin egg. J. Cell. Physiol. 1986, 127, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Wert, M.M.; Palfrey, H.C. Divergence in the anti-apoptotic signalling pathways used by nerve growth factor and basic fibroblast growth factor (bFGF) in PC12 cells: Rescue by bFGF involves protein kinase C delta. Biochem. J. 2000, 352, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Masuda, K.; Toguchi, M.; Ohoka, Y.; Sakai, T.; Furuyama, T.; Inagaki, S. Neurotrophic effect of semaphorin 4D in PC12 cells. Biochem. Biophys. Res. Commun. 2003, 301, 304–310. [Google Scholar] [CrossRef]
- Young, L.H.; Balin, B.J.; Weis, M.T. Go 6983: A fast acting protein kinase C inhibitor that attenuates myocardial ischemia/reperfusion injury. Cardiovasc. Drug Rev. 2005, 23, 255–272. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.C.; Alexander, J.S. Endothelial PKCδ activation attenuates neutrophil transendothelial migration. Inflammat. Res. 2008, 57, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Piyanuch, R.; Sukhthankar, M.; Wandee, G.; Baek, S.J. Berberine, a natural isoquinoline alkaloid, induces NAG-1 and ATF3 expression in human colorectal cancer cells. Cancer Lett. 2007, 258, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Bridle, K.R.; Ramm, G.A.; O'Neill, R.; Britton, R.S.; Bacon, B.R. Effect of phorbol ester and platelet-derived growth factor on protein kinase C in rat hepatic stellate cells. Liver Int. 2007, 27, 1066–1075. [Google Scholar] [CrossRef] [PubMed]
- Slish, D.F.; Welsh, D.G.; Brayden, J.E. Diacylglycerol and protein kinase C activate cation channels involved in myogenic tone. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H2196–H2201. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Matti, U.; Nehring, R.B.; Binz, T.; Rettig, J.; Neher, E.; Sorensen, J.B. Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J. NeuroSci. 2002, 22, 9278–9286. [Google Scholar] [PubMed]
- Koh, J.Y.; Wie, M.B.; Gwag, B.J.; Sensi, S.L.; Canzoniero, L.M.; Demaro, J.; Csernansky, C.; Choi, D.W. Staurosporine-induced neuronal apoptosis. Exp. Neurol. 1995, 135, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Ling, D.S.F.; Benardo, L.S.; Serrano, P.A.; Blace, N.; Kelly, M.T.; Crary, J.F.; Sacktor, T.C. Protein kinase M zeta is necessary and sufficient for LTP maintenance. Nat. Neurosci. 2002, 5, 295–296. [Google Scholar] [CrossRef] [PubMed]
- Toullec, D.; Pianetti, P.; Coste, H.; Bellevergue, P.; Grandperret, T.; Ajakane, M.; Baudet, V.; Boissin, P.; Boursier, E.; Loriolle, F.; et al. The bisindolylmaleimide GF 109203x is a potent and selective inhibitor of protein kinase C. J. Biol. Chem. 1991, 266, 15771–15781. [Google Scholar] [PubMed]
- Barber, L.A.; Vasko, M.R. Activation of protein kinase C augments peptide release from rat sensory neurons. J. Neurochem. 1996, 67, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Baggiolini, M. The protein-kinase inhibitor staurosporine, like phorbol esters, induces the association of protein kinase C with membranes. Biochem. Biophys. Res. Commun. 1988, 154, 1273–1279. [Google Scholar] [CrossRef]
- Ohmichi, M.; Decker, S.J.; Pang, L.; Saltiel, A.R. Inhibition of the cellular actions of nerve growth factor by staurosporine and K252A results from the attenuation of the activity of the trk tyrosine kinase. Biochemistry 1992, 31, 4034–4039. [Google Scholar] [CrossRef] [PubMed]
- Dewald, B.; Thelen, M.; Wymann, M.P.; Baggiolini, M. Staurosporine inhibits the respiratory burst and induces exocytosis in human neutrophils. BioChem. J. 1989, 264, 879–884. [Google Scholar] [CrossRef] [PubMed]
- Sako, T.; Tauber, A.I.; Jeng, A.Y.; Yuspa, S.H.; Blumberg, P.M. Contrasting actions of staurosporine, a protein kinase C inhibitor, on human neutrophils and primary mouse epidermal cells. Cancer Res. 1988, 48, 4646–4650. [Google Scholar] [PubMed]
- Watson, S.P.; McNally, J.; Shipman, L.J.; Godfrey, P.P. The action of the protein kinase C inhibitor, staurosporine, on human platelets. Evidence against a regulatory role for protein kinase C in the formation of inositol trisphosphate by thrombin. BioChem. J. 1988, 249, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Covian-Nares, J.F.; Smith, R.M.; Vogel, S.S. Two independent forms of endocytosis maintain embryonic cell surface homeostasis during early development. Dev. Biol. 2008, 316, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Kuo, A.L.; Cappelluti, S.; Cervantes-Cervantes, M.; Rodriguez, M.; Bush, D.S. Okadaic acid, a protein phosphatase inhibitor, blocks calcium changes, gene expression, and cell death induced by gibberellin in wheat aleurone cells. Plant Cell 1996, 8, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Dobrowsky, R.T.; Hannun, Y.A. Ceramide stimulates a cytosolic protein phosphatase. J. Biol. Chem. 1992, 267, 5048–5051. [Google Scholar] [PubMed]
- Yatsunami, J.; Komori, A.; Ohta, T.; Suganuma, M.; Fujiki, H. Hyperphosphorylation of retinoblastoma protein and p53 by okadaic acid, a tumor promoter. Cancer Res. 1993, 53, 239–241. [Google Scholar] [PubMed]
- Felix, M.A.; Cohen, P.; Karsenti, E. Cdc2 H1 kinase is negatively regulated by a type 2A phosphatase in the Xenopus early embryonic cell cycle: evidence from the effects of okadaic acid. EMBO J. 1990, 9, 675–683. [Google Scholar] [PubMed]
- Gromada, J.; Hoy, M.; Buschard, K.; Salehi, A.; Rorsman, P. Somatostatin inhibits exocytosis in rat pancreatic alpha-cells by Gi2-dependent activation of calcineurin and depriming of secretory granules. J. Physiol. 2001, 535, 519–532. [Google Scholar] [CrossRef] [PubMed]
- Nagy, G.; Reim, K.; Matti, U.; Brose, N.; Binz, T.; Rettig, J.; Neher, E.; Sorensen, J.B. Regulation of releasable vesicle pool sizes by protein kinase A-dependent phosphorylation of SNAP-25. Neuron 2004, 41, 417–429. [Google Scholar] [CrossRef]
- Baba, T.; Sakisaka, T.; Mochida, S.; Takai, Y. PKA-catalyzed phosphorylation of tomosyn and its implication in Ca2+-dependent exocytosis of neurotransmitter. J. Cell Biol. 2005, 170, 1113–1125. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, K.; Nomura, K.; Suzuki, N. Cyclic-amp-dependent activation of an inter-phylum hybrid histone-kinase complex reconstituted from sea urchin sperm-regulatory subunits and bovine heart catalytic subunits. Eur. J. Biochem. FEBS 1997, 243, 612–623. [Google Scholar] [CrossRef]
- Sarno, S.; Reddy, H.; Meggio, F.; Ruzzene, M.; Davies, S.P.; Donella-Deana, A.; Shugar, D.; Pinna, L.A. Selectivity of 4,5,6,7-tetrabromobenzotriazole, an ATP site-directed inhibitor of protein kinase CK2 (‘casein kinase-2’). FEBS Lett. 2001, 496, 44–48. [Google Scholar] [CrossRef]
- Sanz-Clemente, A.; Matta, J.A.; Isaac, J.T.; Roche, K.W. Casein kinase 2 regulates the NR2 subunit composition of synaptic NMDA receptors. Neuron 2010, 67, 984–996. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, T.; Kitazawa, K. Size-dependent heterogeneity of contractile Ca2+ sensitization in rat arterial smooth muscle. J. Physiol. 2012, 590, 5401–5423. [Google Scholar] [CrossRef] [PubMed]
- Tersteeg, C.; Heijnen, H.F.; Eckly, A.; Pasterkamp, G.; Urbanus, R.T.; Maas, C.; Hoefer, I.E.; Nieuwland, R.; Farndale, R.W.; Gachet, C.; et al. Flow-induced protrusions (FLIPRs): A platelet-derived platform for the retrieval of microparticles by monocytes and neutrophils. Circ. Res. 2014, 114, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Pedraza, C.E.; Taylor, C.; Pereira, A.; Seng, M.; Tham, C.S.; Izrael, M.; Webb, M. Induction of oligodendrocyte differentiation and in vitro myelination by inhibition of Rho-associated kinase. ASN Neuro 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Kitazawa, T. Contractile signaling pathways in mouse prostate smooth muscle. Prostate 2013, 73, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Golas, J.M.; Arndt, K.; Etienne, C.; Lucas, J.; Nardin, D.; Gibbons, J.; Frost, P.; Ye, F.; Boschelli, D.H.; Boschelli, F. SKI-606, a 4-anilino-3-quinolinecarbonitrile dual inhibitor of Src and Abl kinases, is a potent antiproliferative agent against chronic myelogenous leukemia cells in culture and causes regression of K562 xenografts in nude mice. Cancer Res. 2003, 63, 375–381. [Google Scholar] [PubMed]
- Vultur, A.; Buettner, R.; Kowolik, C.; Liang, W.; Smith, D.; Boschelli, F.; Jove, R. SKI-606 (bosutinib), a novel Src kinase inhibitor, suppresses migration and invasion of human breast cancer cells. Mol. Cancer Ther. 2008, 7, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Borriello, A.; Caldarelli, I.; Basile, M.A.; Bencivenga, D.; Tramontano, A.; Perrotta, S.; Della Ragione, F.; Oliva, A. The tyrosine kinase inhibitor dasatinib induces a marked adipogenic differentiation of human multipotent mesenchymal stromal cells. PLoS ONE 2011, 6, e28555. [Google Scholar] [CrossRef] [PubMed]
- Giansanti, P.; Preisinger, C.; Huber, K.V.M.; Gridling, M.; Superti-Furga, G.; Bennett, K.L.; Heck, A.J.R. Evaluating the promiscuous nature of tyrosine kinase inhibitors assessed in A431 epidermoid carcinoma cells by both chemical- and phosphoproteomics. ACS Chem. Biol. 2014, 9, 1490–1498. [Google Scholar] [CrossRef] [PubMed]
- Xi, S.C.; Zhang, Q.; Dyer, K.F.; Lerner, E.C.; Smithgall, T.E.; Gooding, W.E.; Kamens, J.; Grandis, J.R. Src kinases mediate STAT growth pathways in squamous cell carcinoma of the head and neck. J. Biol. Chem. 2003, 278, 31574–31583. [Google Scholar] [CrossRef] [PubMed]
- Hossaina, M.I.; Hoquel, A.; Lessene, G.; Kamaruddin, M.A.; Chu, P.W.Y.; Ng, I.H.W.; Irtegun, S.; Ng, D.C.H.; Bogoyevitch, M.A.; Burgess, A.W.; et al. Dual role of Src kinase in governing neuronal survival. Brain Res. 2015, 1594, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Pene-Dumitrescu, T.; Peterson, L.F.; Donato, N.J.; Smithgall, T.E. An inhibitor-resistant mutant of Hck protects CML cells against the antiproliferative and apoptotic effects of the broad-spectrum Src family kinase inhibitor A-419259. Oncogene 2008, 27, 7055–7069. [Google Scholar] [CrossRef] [PubMed]
- Meyn, M.A.; Schreiner, S.J.; Dumitrescu, T.P.; Nau, G.J.; Smithgall, T.E. Src family kinase activity is required for murine embryonic stem cell growth and differentiation. Mol. Pharmacol. 2005, 68, 1320–1330. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.W.; Lee, P.L.; Chen, C.T.; Mi, F.L.; Juang, J.H.; Hwang, S.M.; Ho, Y.C.; Sung, H.W. Elucidating the signaling mechanism of an epithelial tight-junction opening induced by chitosan. Biomaterials 2012, 33, 6254–6263. [Google Scholar] [CrossRef] [PubMed]
- Shakiryanova, D.; Klose, M.K.; Zhou, Y.; Gu, T.; Deitcher, D.L.; Atwood, H.L.; Hewes, R.S.; Levitan, E.S. Presynaptic ryanodine receptor-activated calmodulin kinase II increases vesicle mobility and potentiates neuropeptide release. J. NeuroSci. 2007, 27, 7799–7806. [Google Scholar] [CrossRef] [PubMed]
- Gardner, A.J.; Knott, J.G.; Jones, K.T.; Evans, J.P. CAMKII can participate in but is not sufficient for the establishment of the membrane block to polyspermy in mouse eggs. J. Cell. Physiol. 2007, 212, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Yurimoto, S.; Fujimoto, T.; Magari, M.; Kanayama, N.; Kobayashi, R.; Tokumitsu, H. In vitro substrate phosphorylation by Ca2+/calmodulin-dependent protein kinase kinase using guanosine-5′-triphosphate as a phosphate donor. BMC Biochem. 2012, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Inuzuka, H.; Ishikawa, Y.; Kobayashi, R. A single amino acid difference between α and β Ca2+/calmodulin-dependent protein kinase kinase dictates sensitivity to the specific inhibitor, STO-609. J. Biol. Chem. 2003, 278, 10908–10913. [Google Scholar] [CrossRef] [PubMed]
- Tokumitsu, H.; Inuzuka, H.; Ishikawa, Y.; Ikeda, M.; Saji, I.; Kobayashi, R. STO-609, a specific inhibitor of the Ca2+/calmodulin-dependent protein kinase kinase. J. Biol. Chem. 2002, 277, 15813–15818. [Google Scholar] [CrossRef] [PubMed]
- Holden, N.J.; Savage, C.O.S.; Young, S.P.; Wakelam, M.J.; Harper, L.; Williams, J.M. A dual role for diacylglycerol kinase generated phosphatidic acid in autoantibody-induced neutrophil exocytosis. Mol. Med. 2011, 17, 1242–1252. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, T.; Hiura, M.; Yoshida, K.; Okamura, N.; Ishibashi, S. A diacylglycerol kinase inhibitor, R 59 022, potentiates superoxide anion production and 46-kDa protein phosphorylation in guinea pig polymorphonuclear leukocytes. J. Biol. Chem. 1990, 265, 15418–15423. [Google Scholar] [PubMed]
- Niwa, S.; Graf, C.; Bornancin, F. Ceramide kinase deficiency impairs microendothelial cell angiogenesis in vitro. Microvasc. Res. 2009, 77, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Bornancin, F. Ceramide kinase: The first decade. Cell. Signal. 2011, 23, 999–1008. [Google Scholar] [CrossRef] [PubMed]
- Graf, C.; Rovina, P.; Bornancin, F. A secondary assay for ceramide kinase inhibitors based on cell growth inhibition by short-chain ceramides. Anal. Biochem. 2009, 384, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Graf, C.; Klumpp, M.; Habig, M.; Rovina, P.; Billich, A.; Baumruker, T.; Oberhauser, B.; Bornancin, F. Targeting ceramide metabolism with a potent and specific ceramide kinase inhibitor. Mol. Pharmacol. 2008, 74, 925–932. [Google Scholar] [CrossRef] [PubMed]
- McDonald, R.A.; Pyne, S.; Pyne, N.J.; Grant, A.; Wainwright, C.L.; Wadsworth, R.M. The sphingosine kinase inhibitor N,N-dimethylsphingosine inhibits neointimal hyperplasia. Br. J. Pharmacol. 2010, 159, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Ruegg, U.T.; Burgess, G.M. Staurosporine, K-252 and UCN-0: potent but nonspecific inhibitors of protein kinases. Trends Pharmacol. Sci. 1989, 10, 218–220. [Google Scholar] [CrossRef]
- Meggio, F.; Deana, A.D.; Ruzzene, M.; Brunati, A.M.; Cesaro, L.; Guerra, B.; Meyer, T.; Mett, H.; Fabbro, D.; Furet, P.; et al. Different susceptibility of protein kinases to staurosporine inhibition. Kinetic-studies and molecular-bases for the resistance of protein-kinase CK2. Eur. J. Biochem. 1995, 234, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Kishida, M.; Watanabe, S.; Hashimoto, Y.; Fukamizu, K.; Tomonaga, T. Proteome-wide discovery of unknown ATP-binding proteins and kinase inhibitor target proteins using an ATP probe. J. Proteom. Res. 2014, 13, 5461–5470. [Google Scholar] [CrossRef] [PubMed]
- Messa, M.; Congia, S.; Defranchi, E.; Valtorta, F.; Fassio, A.; Onofri, F.; Benfenati, F. Tyrosine phosphorylation of synapsin I by Src regulates synaptic-vesicle trafficking. J. Cell Sci. 2010, 123, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Gromada, J.; Hoy, M.; Renstrom, E.; Bokvist, K.; Eliasson, L.; Gopel, S.; Rorsman, P. CaM kinase II-dependent mobilization of secretory granules underlies acetylcholine-induced stimulation of exocytosis in mouse pancreatic B-cells. J. Physiol. 1999, 518, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.S.; Wu, L.G. Protein kinase C increases the apparent affinity of the release machinery to Ca2+ by enhancing the release machinery downstream of the Ca2+ sensor. J. Neurosci. 2001, 21, 7928–7936. [Google Scholar] [PubMed]
- Yawo, H. Protein kinase C potentiates transmitter release from the chick ciliary presynaptic terminal by increasing the exocytotic fusion probability. J. Physiol. 1999, 515, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Gillis, K.D. A highly Ca2+-sensitive pool of granules is regulated by glucose and protein kinases in insulin-secreting INS-1 cells. J. Gen. Physiol. 2004, 124, 641–651. [Google Scholar] [CrossRef] [PubMed]
- Coorssen, J.R.; Davidson, M.M.; Haslam, R.J. Factors affecting dense and α-granule secretion from electropermeabilized human platelets: Ca2+-independent actions of phorbol ester and GTPγ S. Cell Regul. 1990, 1, 1027–1041. [Google Scholar] [CrossRef] [PubMed]
- Berg, L.K.; Wessel, G.M. Cortical granules of the sea urchin translocate early in oocyte maturation. Development 1997, 124, 1845–1850. [Google Scholar] [PubMed]
- Sakisaka, T.; Baba, T.; Tanaka, S.; Izumi, G.; Yasumi, M.; Takai, Y. Regulation of SNAREs by tomosyn and ROCK: Implication in extension and retraction of neurites. J. Cell Biol. 2004, 166, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Hammar, E.; Tomas, A.; Bosco, D.; Halban, P.A. Role of the Rho-ROCK (Rho-associated kinase) signaling pathway in the regulation of pancreatic β-cell function. Endocrinology 2009, 150, 2072–2079. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Coppola, T.; Regazzi, R. Involvement of Rho GTPases and their effectors in the secretory process of PC12 cells. Exp. Cell Res. 2002, 273, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Rickman, C.; Duncan, R.R. Munc18/syntaxin interaction kinetics control secretory vesicle dynamics. J. Biol. Chem. 2010, 285, 3965–3972. [Google Scholar] [CrossRef] [PubMed]
- Nielander, H.B.; Onofri, F.; Valtorta, F.; Schiavo, G.; Montecucco, C.; Greengard, P.; Benfenati, F. Phosphorylation of VAMP/synaptobrevin in synaptic vesicles by endogenous protein kinases. J. Neurochem. 1995, 65, 1712–1720. [Google Scholar] [CrossRef] [PubMed]
- Shata, A.; Saisu, H.; Odani, S.; Abe, T. Phosphorylated synaphin/complexin found in the brain exhibits enhanced SNARE complex binding. Biochem. Biophys. Res. Commun. 2007, 354, 808–813. [Google Scholar] [CrossRef] [PubMed]
- Gil, C.; Falques, A.; Sarro, E.; Cubi, R.; Blasi, J.; Aguilera, J.; Itarte, E. Protein kinase CK2 associates to lipid rafts and its pharmacological inhibition enhances neurotransmitter release. FEBS Lett. 2011, 585, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Snyder, D.A.; Kelly, M.L.; Woodbury, D.J. SNARE complex regulation by phosphorylation. Cell BioChem. Biophys 2006, 45, 111–123. [Google Scholar] [CrossRef]
- Morgan, A.; Burgoyne, R.D.; Barclay, J.W.; Craig, T.J.; Prescott, G.R.; Ciufo, L.F.; Evans, G.J.; Graham, M.E. Regulation of exocytosis by protein kinase C. BioChem. Soc. Trans. 2005, 33, 1341–1344. [Google Scholar] [CrossRef] [PubMed]
- Domart, M.C.; Hobday, T.M.; Peddie, C.J.; Chung, G.H.; Wang, A.; Yeh, K.; Jethwa, N.; Zhang, Q.; Wakelam, M.J.; Woscholski, R.; et al. Acute manipulation of diacylglycerol reveals roles in nuclear envelope assembly & endoplasmic reticulum morphology. PLoS ONE 2012, 7, e51150. [Google Scholar]
- Darios, F.; Wasser, C.; Shakirzyanova, A.; Giniatullin, A.; Goodman, K.; Munoz-Bravo, J.L.; Raingo, J.; Jorgacevski, J.; Kreft, M.; Zorec, R.; et al. Sphingosine facilitates snare complex assembly and activates synaptic vesicle exocytosis. Neuron 2009, 62, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Kajimoto, T.; Okada, T.; Yu, H.; Goparaju, S.K.; Jahangeer, S.; Nakamura, S. Involvement of sphingosine-1-phosphate in glutamate secretion in hippocampal neurons. Mol. Cell. Biol. 2007, 27, 3429–3440. [Google Scholar] [CrossRef] [PubMed]
- Flasker, A.; Jorgacevski, J.; Calejo, A.I.; Kreft, M.; Zorec, R. Vesicle size determines unitary exocytic properties and their sensitivity to sphingosine. Mol. Cell. Endocrinol. 2013, 376, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Morrell, C.N.; Lowenstein, C.J. Sphingosine 1-phosphate activates weibel-palade body exocytosis. Proc. Natl. Acad. Sci. USA 2004, 101, 11483–11487. [Google Scholar] [CrossRef] [PubMed]
- Suhaiman, L.; De Blas, G.A.; Obeid, L.M.; Darszon, A.; Mayorga, L.S.; Belmonte, S.A. Sphingosine 1-phosphate and sphingosine kinase are involved in a novel signaling pathway leading to acrosomal exocytosis. J. Biol. Chem. 2010, 285, 16302–16314. [Google Scholar] [CrossRef] [PubMed]
- Trkov, S.; Stenovec, M.; Kreft, M.; Potokar, M.; Parpura, V.; Davletov, B.; Zorec, R. Fingolimod—A sphingosine-like molecule inhibits vesicle mobility and secretion in astrocytes. Glia 2012, 60, 1406–1416. [Google Scholar] [CrossRef] [PubMed]
- Camoletto, P.G.; Vara, H.; Morando, L.; Connell, E.; Marletto, F.P.; Giustetto, M.; Sassoe-Pognetto, M.; Van Veldhoven, P.P.; Ledesma, M.D. Synaptic vesicle docking: Sphingosine regulates syntaxin1 interaction with Munc18. PLoS ONE 2009, 4, e5310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goni, F.M.; Alonso, A. Biophysical properties and membrane organization of ceramides, ceramide-1-phosphate and other simple sphingolipids. Chem. Phys. Lip. 2009, 160, S2. [Google Scholar] [CrossRef]
- Kohansal-Nodehi, M.; Chua, J.J.; Urlaub, H.; Jahn, R.; Czernik, D. Analysis of protein phosphorylation in nerve terminal reveals extensive changes in active zone proteins upon exocytosis. eLife 2016, 5, e14530. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Compound | Molecular Target | Dose Used | Refs |
|---|---|---|---|
| Phorbol 12-myristate 13-acetate (PMA) | PKC activator | 100 nM | [12,14,37,38,39,40] |
| Gö 6983 | PKC inhibitor | 10 µM | [41,42,43,44,45,46] |
| PKC inhibitory peptide (19–36; RFARKGALRQKNVHEVKN) | PKC inhibitor | 10 µM | [13,47,48] |
| Staurosporine | Broad spectrum kinase inhibitor | 4 µM | [49,50,51,52,53,54,55,56,57,58] |
| α-naphthyl phosphate (α-NP) | Broad spectrum phosphatase inhibitor | 5 mM | [14] |
| Okadaic acid | Dual protein phosphatase 1/2A inhibitor | 1 µM | [12,20,28,59,60,61,62] |
| Calcineurin (CaN) inhibitory peptide (457–482; ITSFEEAKGLDRINERMPPRRDAMP) | CaN inhibitor | 100 µM | [20,63] |
| PKA inhibitory peptide (6–22; TYADFIASGRTGRRNAI) | Protein kinase A inhibitor | 1 µM | [64,65,66] |
| 4,5,6,7-tetrabromobenzotriazole (TBB) | Casein kinase 2 (Ck2) inhibitor | 25 µM | [67,68] |
| GSK 429286 | Rho kinase (ROCK) inhibitor | 2 µM | [69,70,71,72] |
| Bosutinib | Dual abl/src kinase inhibitor | 10 µM | [73,74,75,76] |
| A419259 | Src kinase inhibitor | 500 nM | [77,78,79,80,81] |
| Autocamtide-2-related inhibitory peptide (AIP) (KKALRRQEAVDAL) | Calmodulin-dependent protein kinase II (CaMK II) inhibitor | 10 µM | [82,83] |
| STO609 | Ca2+/calmodulin-dependent protein kinase kinase (CaMK kinase) inhibitor | 25 µM | [84,85,86,87] |
| R59022 | Diacylglycerol kinase inhibitor | 12.5 µM | [88,89] |
| NVP231 | Ceramide kinase inhibitor | 100 nM | [90,91,92,93] |
| Dimethylsphingosine (DMS) | Sphingosine kinase inhibitor | 100 µM | [94] |
| Enzyme Name | Percent Identity | Percent Similarity |
|---|---|---|
| Protein kinase C | 68.4 | 80 |
| PKC conserved region 1 (phorbol ester binding site) | 90 | 92 |
| Serine/threonine catalytic domain | 81.1 | 92.7 |
| cAMP-dependent protein kinase catalytic subunit αisoform | 78.7 | 89.8 |
| Casein kinase 2 α subunit | 79.1 | 86.9 |
| Abl-related protein tyrosine kinase | 69.1 | 80.6 |
| Src-family protein tyrosine kinase | 54.4 | 67.9 |
| Rho kinase | 47.2 | 64.4 |
| Rho kinase associated coiled coil (containing PH domain) | 55.4 | 69.6 |
| Calmodulin-dependent protein kinase 1 | 57.6 | 76.6 |
| Calmodulin-dependent protein kinase kinase | 33.4 | 44.3 |
| Protein phosphatase 2A catalytic subunit | 94.4 | 97.4 |
| Calcineurin B homologous protein | 62.2 | 74.0 |
| Diacylglycerol kinase | 52.8 | 65.8 |
| Ceramide kinase | 44.8 | 66.5 |
| Sphingosine kinase | 43.3 | 61.0 |
| Sphingosine kinase conserved domains: | ||
| C 1 | 64.7 | 76.5 |
| C 2 | 40.0 | 72.0 |
| C 3 | 75.0 | 91.7 |
| C 4 | 57.1 | 82.1 |
| C 5 | 80.0 | 100.0 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abbineni, P.S.; Coorssen, J.R. Application of High-Throughput Assays to Examine Phospho-Modulation of the Late Steps of Regulated Exocytosis. High-Throughput 2017, 6, 17. https://doi.org/10.3390/ht6040017
Abbineni PS, Coorssen JR. Application of High-Throughput Assays to Examine Phospho-Modulation of the Late Steps of Regulated Exocytosis. High-Throughput. 2017; 6(4):17. https://doi.org/10.3390/ht6040017
Chicago/Turabian StyleAbbineni, Prabhodh S., and Jens R. Coorssen. 2017. "Application of High-Throughput Assays to Examine Phospho-Modulation of the Late Steps of Regulated Exocytosis" High-Throughput 6, no. 4: 17. https://doi.org/10.3390/ht6040017
APA StyleAbbineni, P. S., & Coorssen, J. R. (2017). Application of High-Throughput Assays to Examine Phospho-Modulation of the Late Steps of Regulated Exocytosis. High-Throughput, 6(4), 17. https://doi.org/10.3390/ht6040017