1. Introduction
Proteases represent a very wide and important group of enzymes for various biological systems. Their proteolytic activity determines final structure and utility of proteins [
1]. As a result of this, proteases also take part in different pathological processes, such as atherosclerosis, malabsorption syndrome, lymphocyte dysfunction, cancer, and serve an important role in human digestion [
2,
3]. Chymotrypsin, one of the proteases present in human digestion system, plays a significant role in protein cleavage in the intestines. Together with trypsin, in the zymogen form, chymotrypsinogen is ejected into the duodenum, where trypsin cleaves it into the active form [
4]. Chymotrypsin is a serine protease that cleaves peptide bonds by hydrolysis at the location of tryptophan, tyrosine, phenylalanine, or leucin [
5]. Its activity is closely tied to the activity of trypsin, which, along with plasmin, is an important enzyme in human milk. Activity of plasmin ties to the quality of milk. Higher protease activity cleaves the proteins, mainly caseins, in the micelles in milk and affects the milk shelf-life or cheese yield and flavor [
6]. Chymotrypsin and trypsin both cleave caseins and therefore biosensors made based on the cleavage of caseins are sensitive for both proteases and plasmin.
In pathology and medicine, chymotrypsin has also anti-inflammatory effects and has been successfully used to reduce post-operation complication after cataract surgery [
7]. Measuring chymotrypsin activity in medicine can help during differential diagnosis of acinar and endocrinal neoplasms [
8].
Thus, development of sensitive, inexpensive, fast, and easy to use methods for detection of chymotrypsin or other milk proteases would be beneficial for both medicine and dairy. The assay that fulfills these requirements is not yet available. Protease detection is currently based on the detection of α-amino groups cleaved from the protein substrate using optical or high-performance liquid chromatography (HPLC) methods. The method that can be used for fast analysis of the protease concentration is based on enzyme-linked immunosorbent assay (ELISA) with a limit of detection (LOD) of about 0.5 nM for chymotrypsin [
9,
10]. However, the above-mentioned method does not allow the study of the kinetics of substrate digestion.
In this paper we propose two efficient methods for chymotrypsin detection: optical and gravimetric. Gravimetric method is based on measurement of the resonant frequency of shearing oscillations of AT-cut quartz crystal, as well as motional resistance, R
m, and is known as thickness shear mode method (TSM). The protease substrates, such as β-casein or short specific peptides, are immobilized in thin gold layers sputtered at a TSM transducer. High frequency voltage, typically in the range of 5–20 MHz, induces shearing oscillations of the crystal. The fundamental resonance frequency of the crystal, f
0, depends on the physical properties of the quartz (density, shearing modulus, and thickness), viscosity of the medium to which the crystal surface is exposed, as well as on the molecular interactions at the surface. The R
m value is sensitive to shearing viscosity, which is due to the molecular slip between the protein layer and surrounding water environment. Using Sauerbrey Equation (1) [
11], one can link the change in resonant frequency to the mass bound to the surface of the electrode.
where fo is the fundamental resonant frequency (Hz), A is the active crystal area (usually 0.2 cm2), ρq is quartz density (2.648 g·cm−3), Δm is the mass change (g), ρq is the shear modulus of the crystal (2.947 × 1011 g·cm−1s−2). This equation is valid only for rigid layer at vacuum. In a liquid environment and for relatively soft layers, the viscosity contribution can be estimated by measurements of Rm.
In this work, we modified the surface of the crystal with β-casein a natural substrate for chymotrypsin. The resulting mass added to the sensor caused the change of the frequency f and changes of motional resistance, R
m. Further changes of these values can be monitored following addition of chymotrypsin, which leads to cleavage of the β-casein. The mass-sensitive method has been already used for detection of other proteases. Gronewold [
12] monitored activity of bacterial proteases and esperase using proteins from bovine fetal serum. Poturnayova et al. used β-casein layers to detect activity of plasmin and trypsin with LOD around 0.65 nM [
13]. In another work of Tatarko et al., the multiharmonic quartz crystal microbalance (QCM) allowed detection of trypsin and plasmin with LOD of 0.2 nM and 0.5 nM, respectively. The machine learning algorithm applied in this work allowed us to distinguish these two proteases within 2 min [
14].
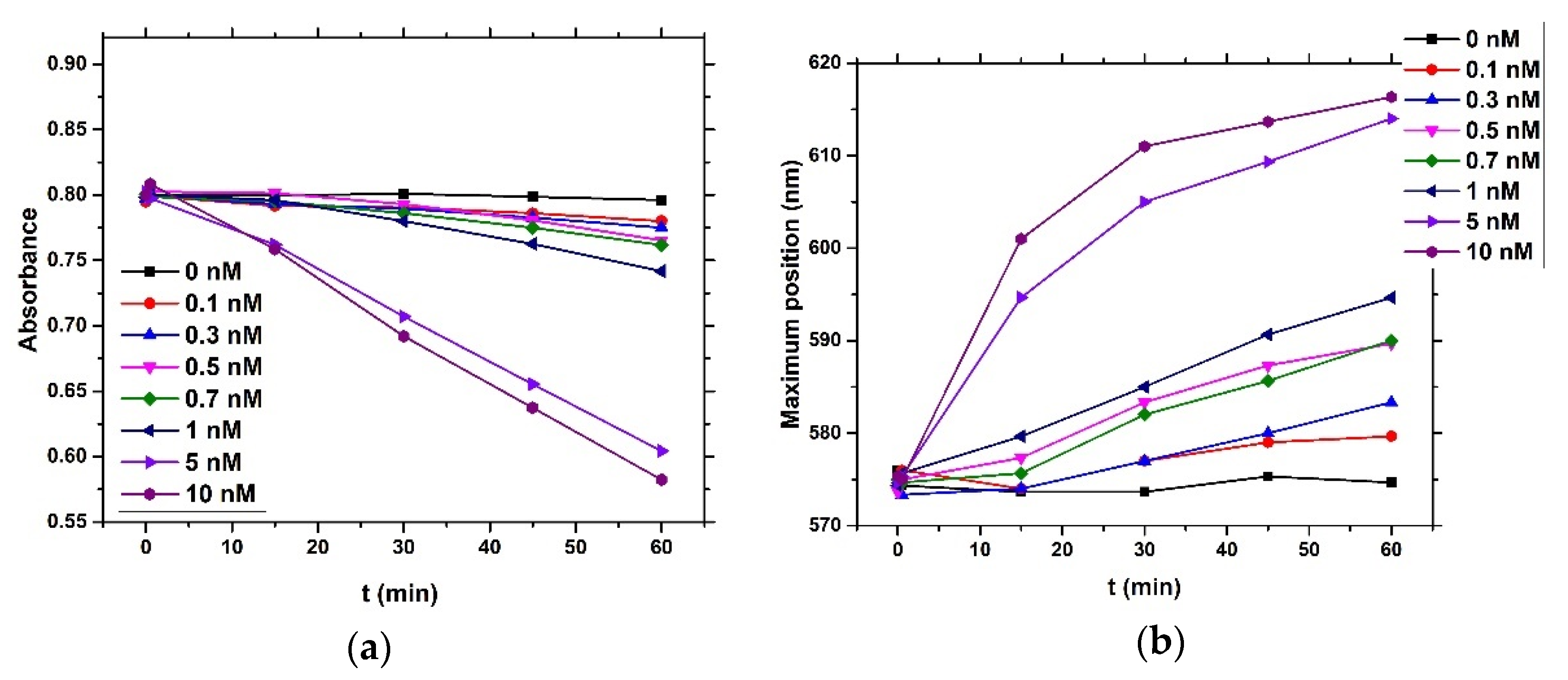
We also applied an optical method based on measurement absorbance of the dispersion of gold nanoparticles (AuNPs) coated by β-casein and 1-mercaptohexanol (MCH). AuNPs reveal a surface plasmon resonance (SPR) effect, which can be measured using UV-Vis spectroscopy. The surface plasmon resonance (SPR) effect arises when the oscillating electromagnetic field of light rays gets into contact with the free electrons in metallic nanoparticles and induces their coherent oscillation. The maximum amplitude of this oscillation happens at a specific wavelength called SPR. The SPR position and intensity depends on various factors like metal type, particle size, shape, structure, and dielectric constant of surrounding medium [
15]. The absorbance of gold nanoparticles has a maximum in visible spectrum depending on the surrounding medium and on the distance between nanoparticles. AuNPs are rather popular in biosensing applications. In the work by Diouani, AuNPs modified with casein were used to detect Leishmania infantum using amperometric methods [
16]. Chen et al. showed the modified AuNPs with trypsin-specific peptide sequence [
17]. After the trypsin cleavage, the gold nanoparticles aggregated, which was visible by measuring UV-Vis spectrum. The detection limit of this method was 5 nM. Guaris et al. used dithiols to measure thrombin activity [
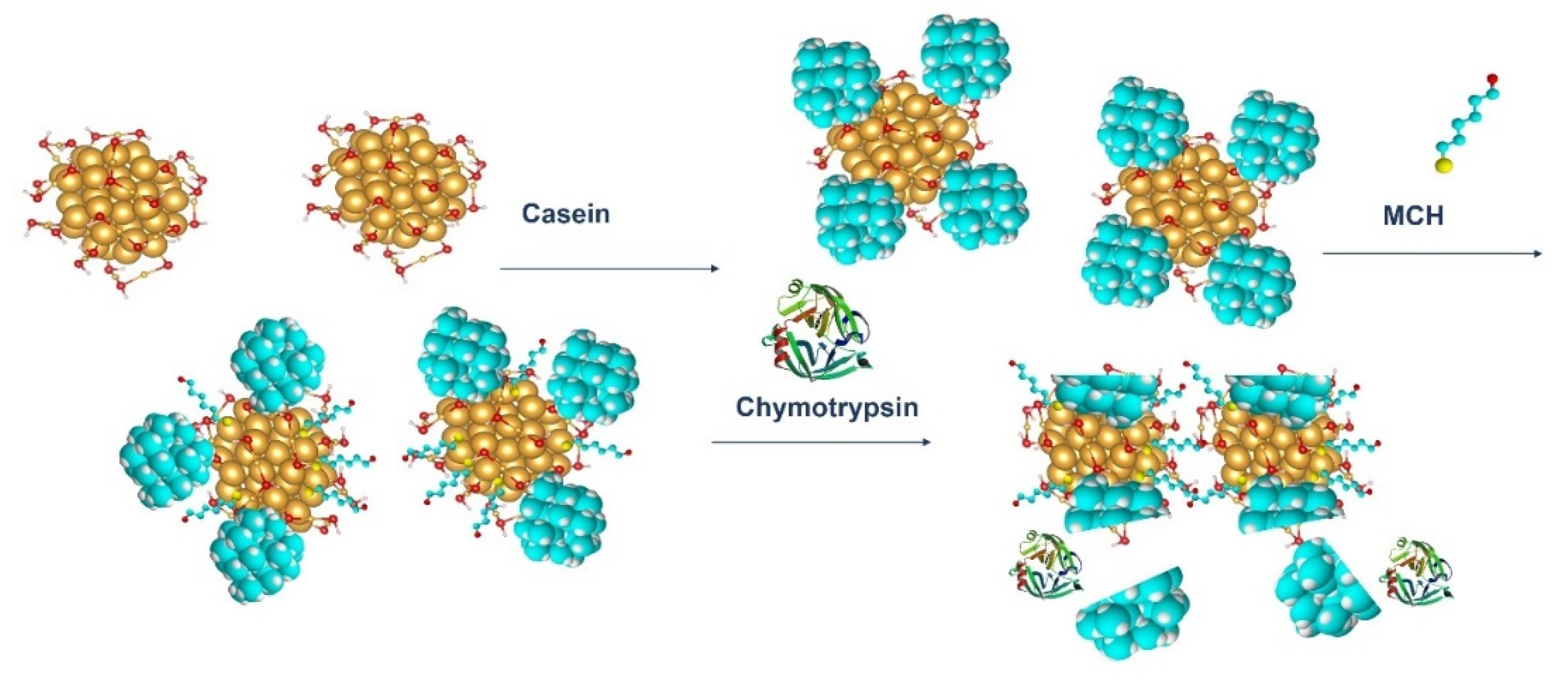
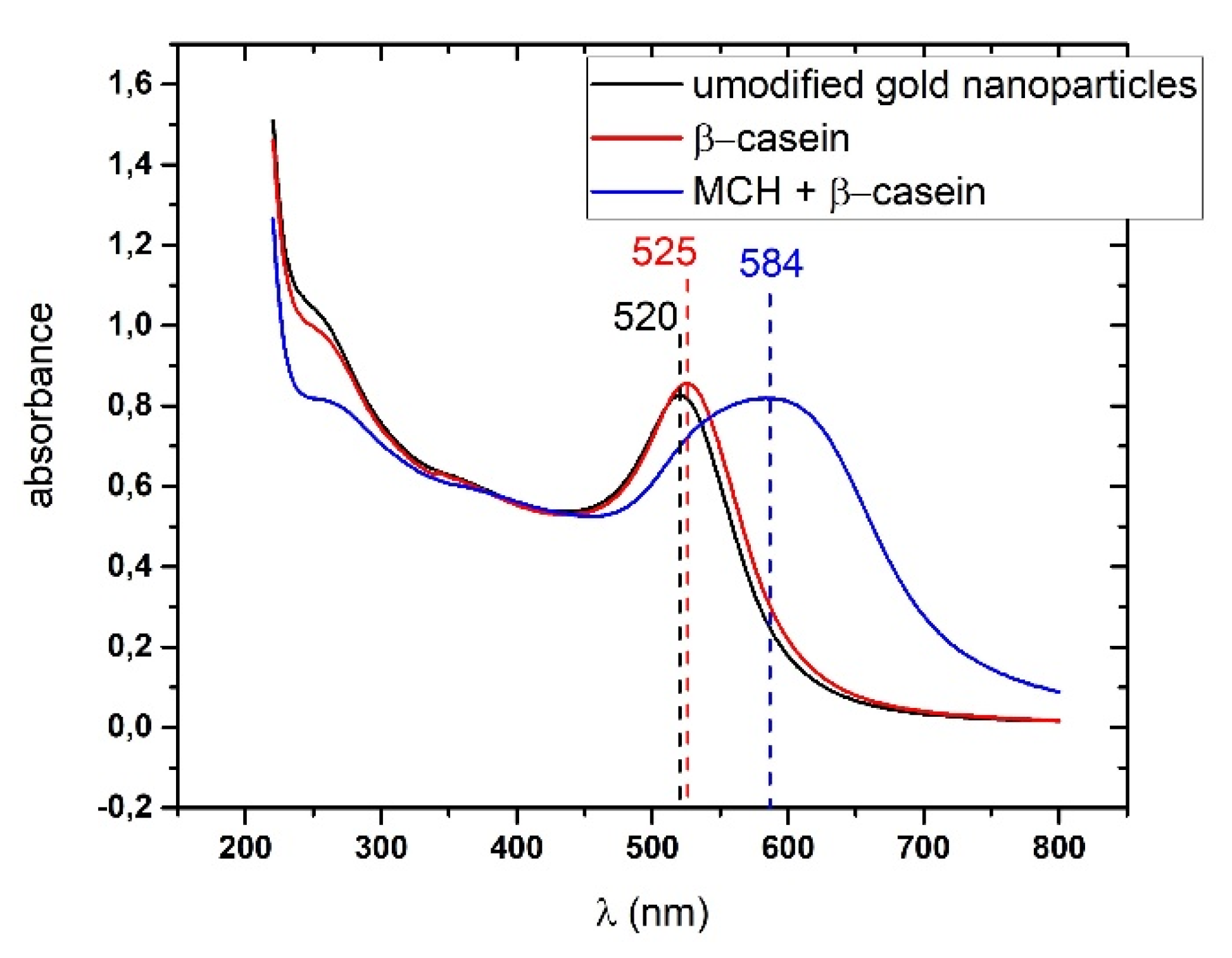
18]. In this work, a presence of substrate for thrombin resulted in aggregation of AuNPs, while addition of thrombin disaggregated the nanoparticles (LOD for thrombin detection was around 5 nM). Since AuNPs can be easily modified by thiol-group-containing peptides, this method makes it possible to prepare substrates with well-observable absorption properties. In our work, we modified the gold nanoparticles with β-casein and MCH. The β-casein protects the AuNPs from aggregation induced from the loss due to presence of MCH. Addition of the chymotrypsin and subsequent cleavage of the β-casein caused nanoparticles aggregation due to loss of the protective shell. This effect was possible to observe by measuring UV-Vis spectra of nanoparticle dispersion.
2. Materials and Methods
2.1. Chemicals
The chemicals needed to prepare the piezocrystal and gold nanoparticles were purchased from Sigma-Aldrich (Darmstadt, Germany), the chemicals needed to clean the crystals were purchased from Slavus (Bratislava, Slovakia). Deionized water was distilled directly at the flask using a Purelab Classic UV instrument (Elga, High Wycombe, UK). All standard chemicals, such as auric acid (HAuCl4), sodium citrate, β-casein, 1-mercaptohexanlo (MCH), phosphate buffered saline (PBS) tablets, 11-mercaptoundecanoic acid (MUA), N-(3-dimetylaminopropyl)-N′-etylcarbodiimid (EDC), N-hydroxisuccinimid (NHS), α-chymotrypsin, NaOH, HC NaOH, NH3, and H2O2 were of p.a. grade.
2.2. Optical Method
AuNPs were prepared by modified citrate method [
19]. In short, we prepared 100 mL of 0.01% chloroauric acid (HAuCl
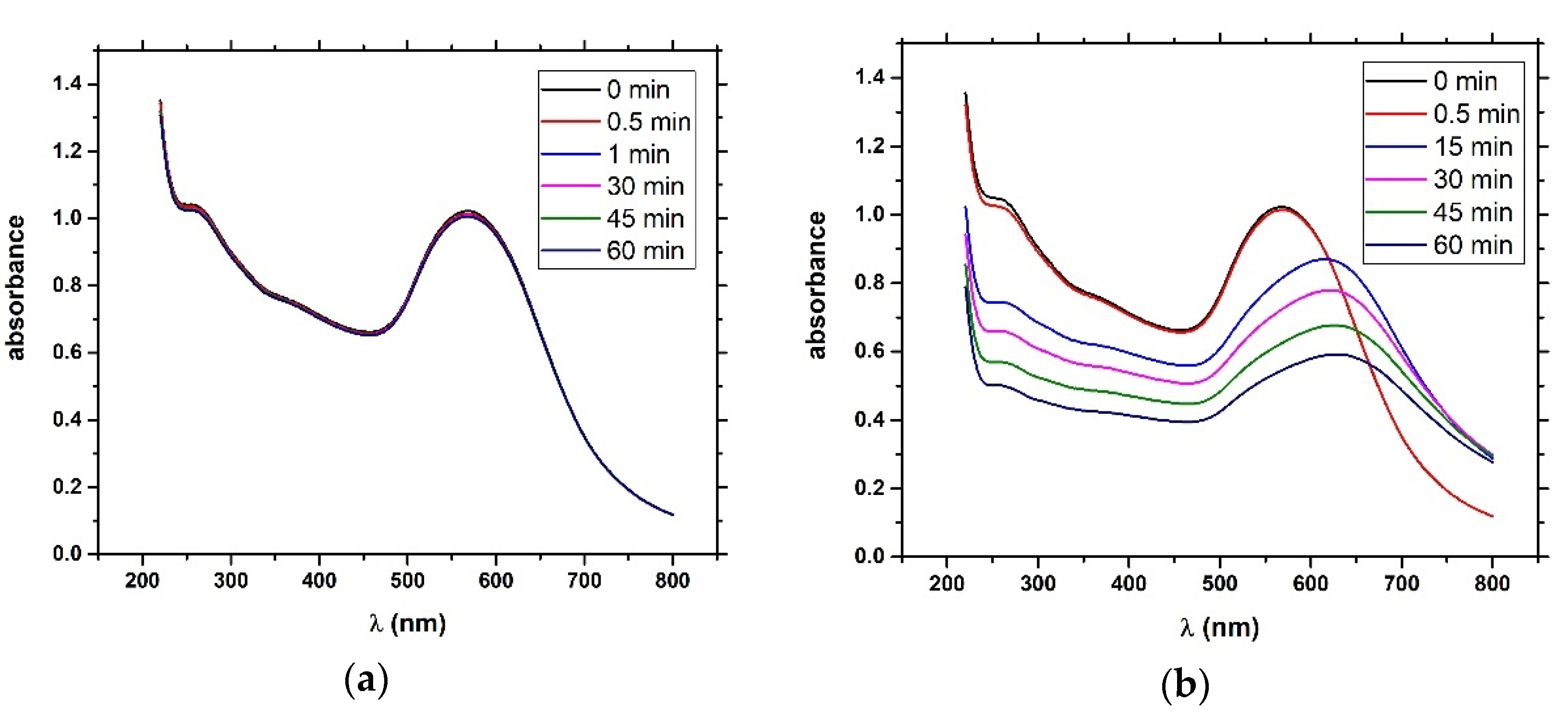
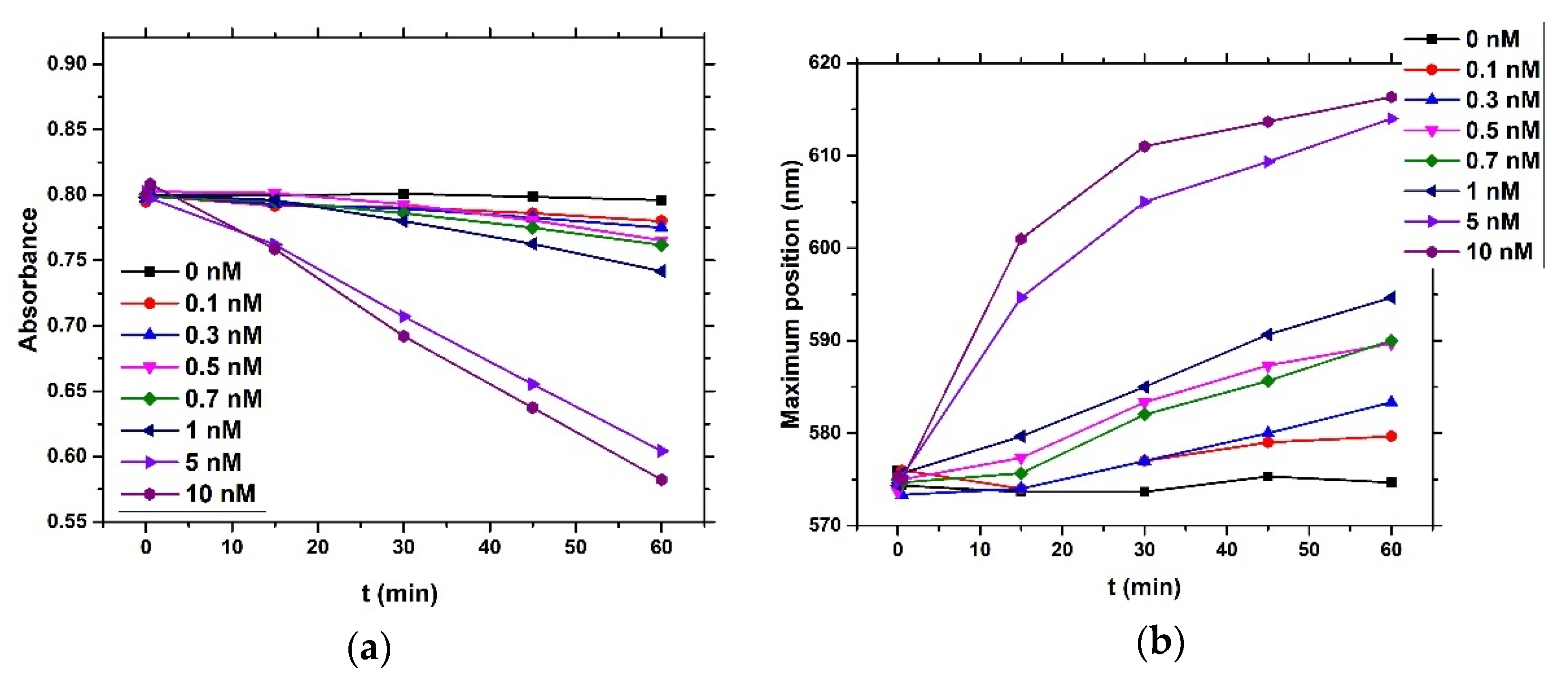
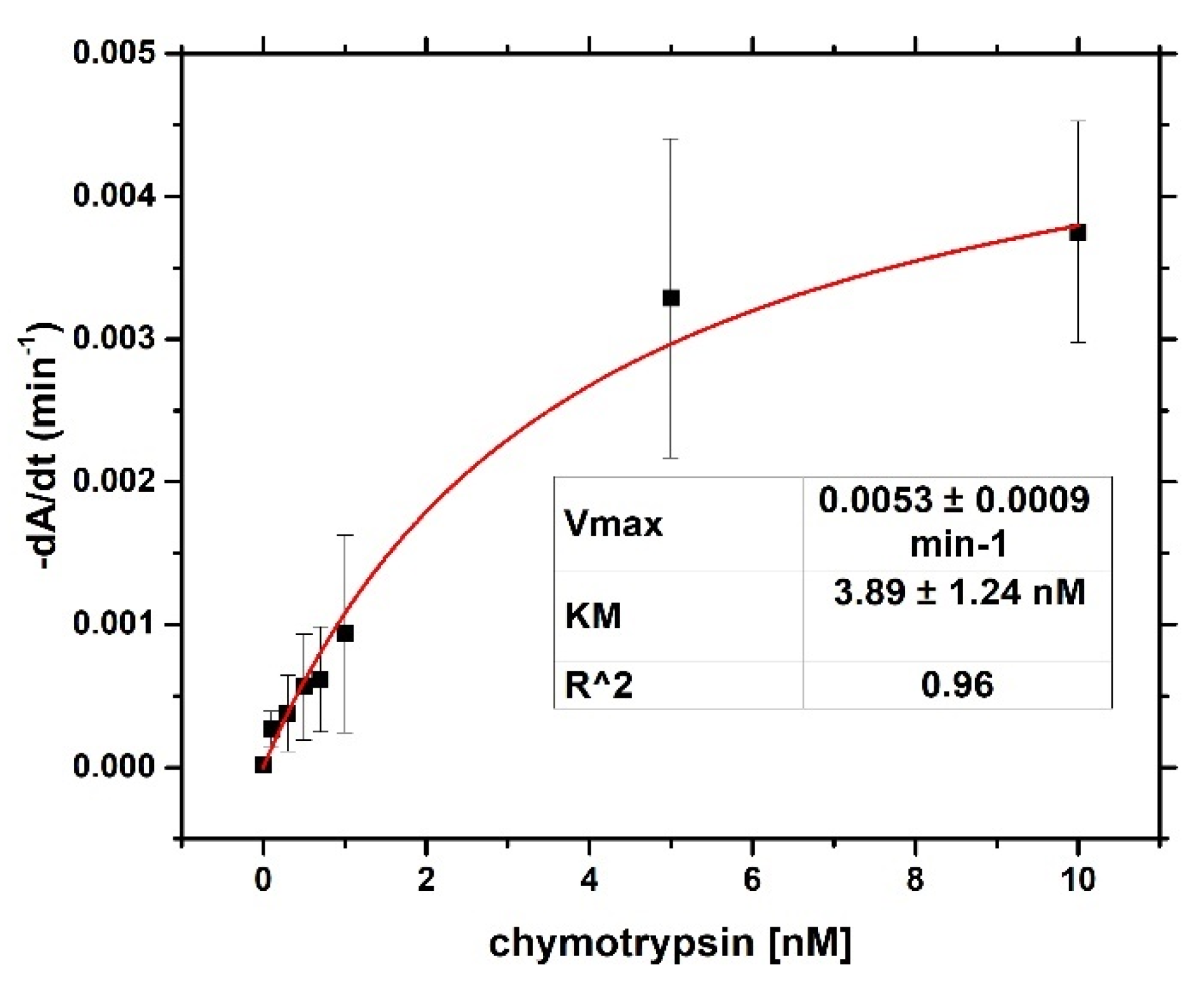
4) and brought it to a boil. We then added 5 mL of 1% sodium tris-citrate and left the solution boiling while stirring until it turned deep red. We then left the prepared AuNPs to cool down and stored them in the dark. To modify the gold nanoparticles with casein, we added 2 mL of 0.1 mg/mL β-casein into 18 mL of the AuNPs solution. After 2 h of incubation at room temperature without stirring, the gold nanoparticles were further incubated with 200 µL of 1 mM MCH overnight for approximately 18 h. MCH is important as it removes the surface charge of nanoparticles and thus protect them from aggregation. Addition of MCH leads to a color change to violet. However, nanoparticles (NPs) are protected from aggregation due to the presence of a β-casein layer. Addition of chymotrypsin caused cleavage of β-casein, and as a result, the NPs aggregated. This was reflected by changes of the color of the solution to blue and then it became colorless. For the experiments, we prepared 0.95 mL of NPs. Chymotrypsin was dissolved in deionized water and 0.05 mL of chymotrypsin from stock solution was added to each cuvette. The concentration of chymotrypsin in cuvettes was 0.1, 0.3, 0.5, 0.7, 1.5, and 10 at 1 mL total volume of solution. We also used a reference cuvette where only 0.05 mL of protease-free water was added to the AuNPs solution. We measured the spectra of the AuNPs before protease addition (0 min), just after protease addition (approximately 30 s) and then every 15 min up to 60 min. The measurement was repeated 3 times. We multiplied the value of absorbance at time t = 0 by the dilution factor to correct the changes in absorbance intensity caused by the initial protease addition. Absorbance was measured by UV-1700 spectrophotometer (Shimadzu, Kyoto, Japan). The scheme of AuNPs modification and chymotrypsin cleavage is presented in
Figure 1.
2.3. Thickness Shear Mode (TSM) Method
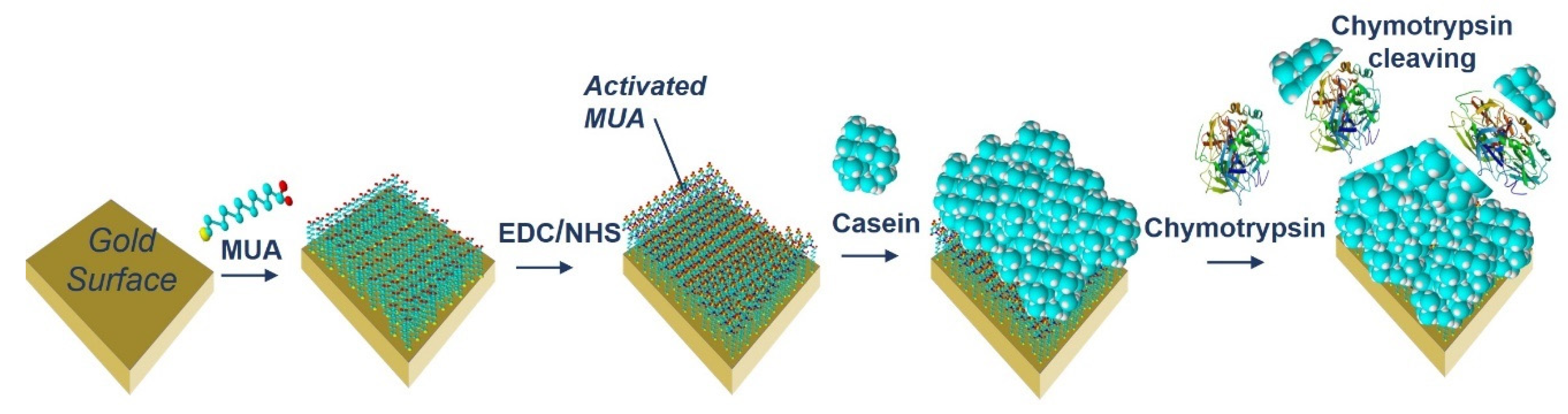
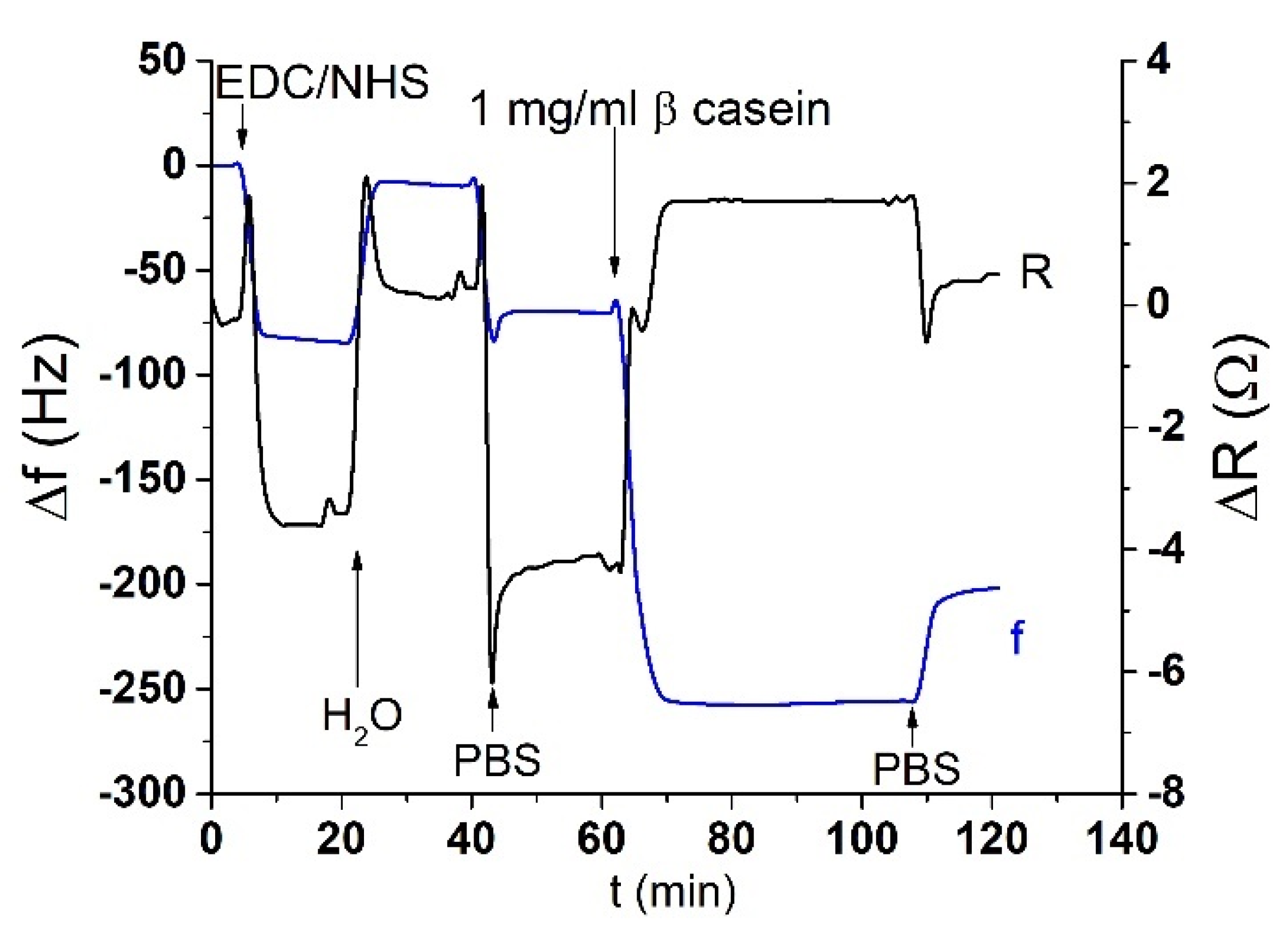
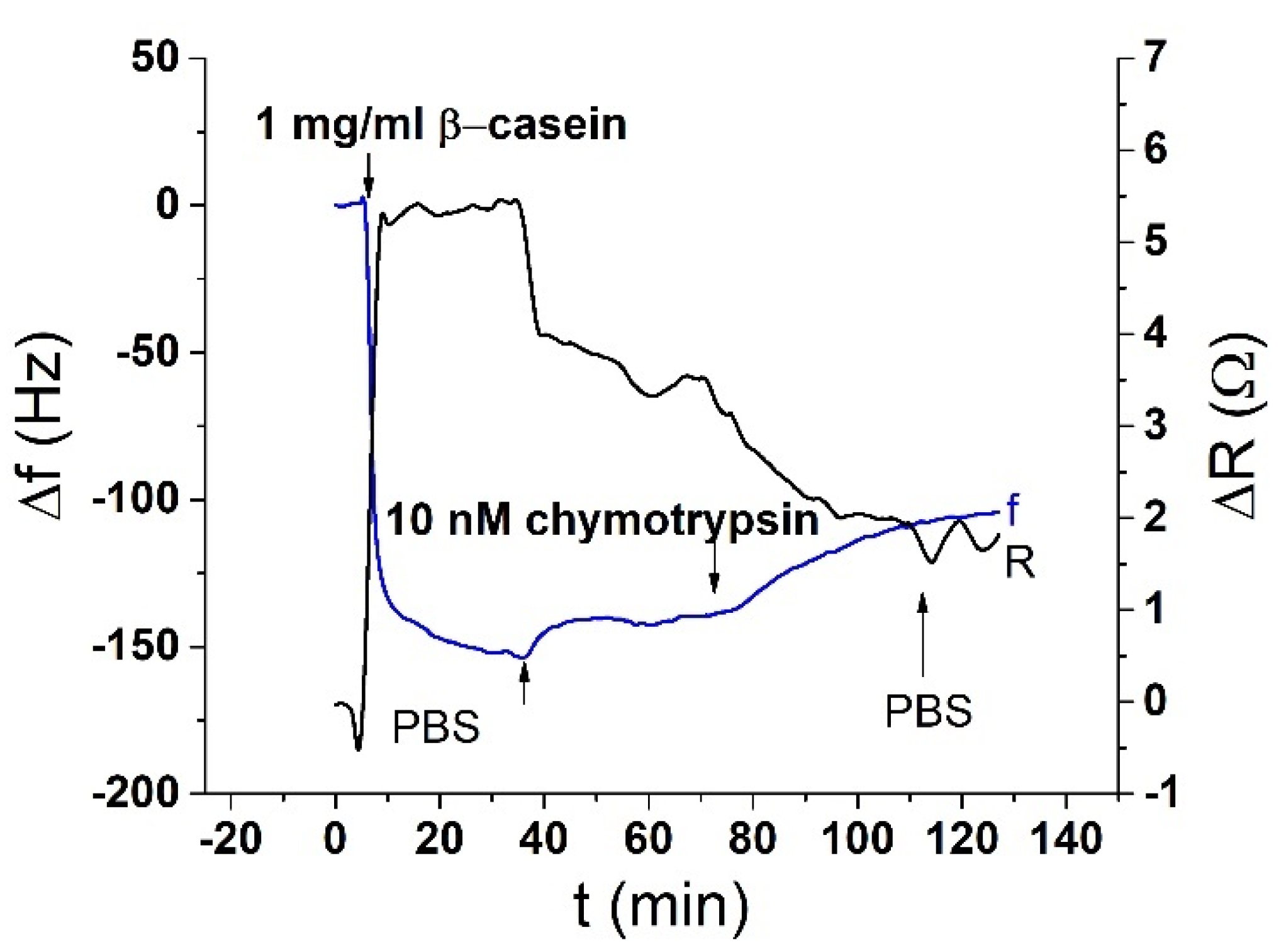
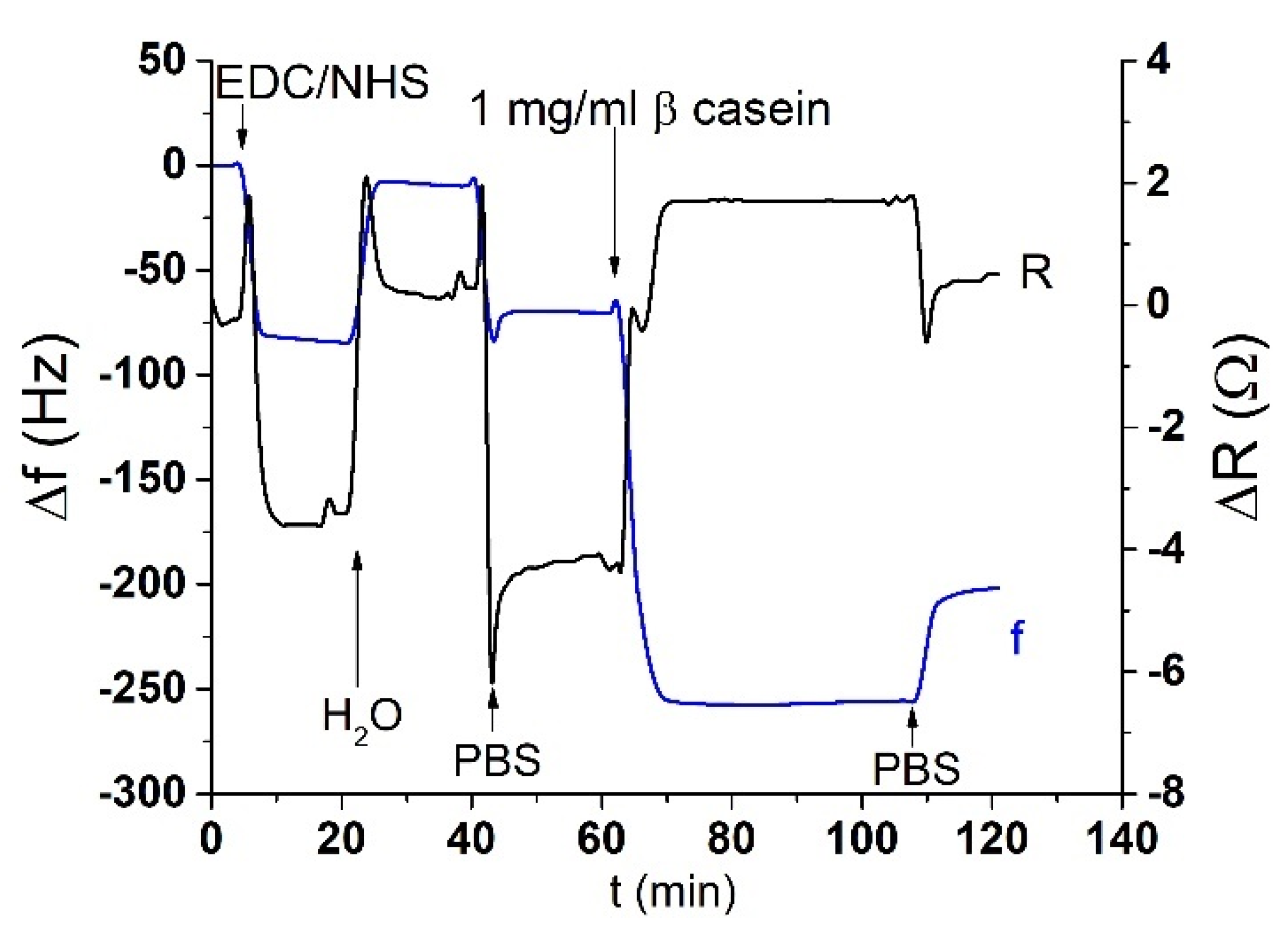
The acoustic TSM sensor was prepared on a gold quartz crystal layer for QCM (8 MHz, ICM, Oklahoma City, OK, USA, electrode area: A = 0.2 cm2). First, the crystal was carefully cleaned as follows. It has been exposed to a basic piranha solution (H2O2:NH3:H2O = 1:1:5 mL). The crystals were immersed for 25 min in this solution, in beakers in a water bath set at approximately 75 °C. Subsequently, the crystals were withdrawn, rinsed with distilled water, and returned to the beaker with a new dose of piranha solution on the reverse side of the crystal. This was repeated three times. On the last extraction, the crystals were washed three times with distilled water and then washed with ethanol and placed in a bottle with ethanol for storage at room temperature. We applied this procedure for each crystal and then used it repeatedly for measurement. The clean crystal was incubated overnight for 16–18 h at room temperature with 2 mM MUA dissolved in ethanol. MUA is a carboxylic acid with a sulfide group (SH). The sulfide moiety interacts with the gold on the crystal to form a self-assembled layer. After incubation, the crystal was washed with ethanol, distilled water, and 20 mM EDC and 50 mM NHS were applied for 25 min. These substances react with the carboxyl moiety of MUA and activate them to form a covalent bond with other amino acids. Subsequently, we cleaned the crystal again, dried it with nitrogen, and placed it in a cell. The cell was filled with PBS buffer using a Genie plus 2011 step pump (Kent Scientific, Torrington, CT, USA) at a flow rate of 200 μL/min. After filling the cell, we switched the flow to the rate of 50 μL/min. Then, 1 mg/mL of β-casein dissolved in PBS was then added to the MUA layer. After 35 min, only pure PBS was added to remove the unbound fragments. β-casein layer formation, and in some cases EDC/NHS layer activation, was recorded using an RQCM instrument (Maxtek, East Syracuse, NY, USA).
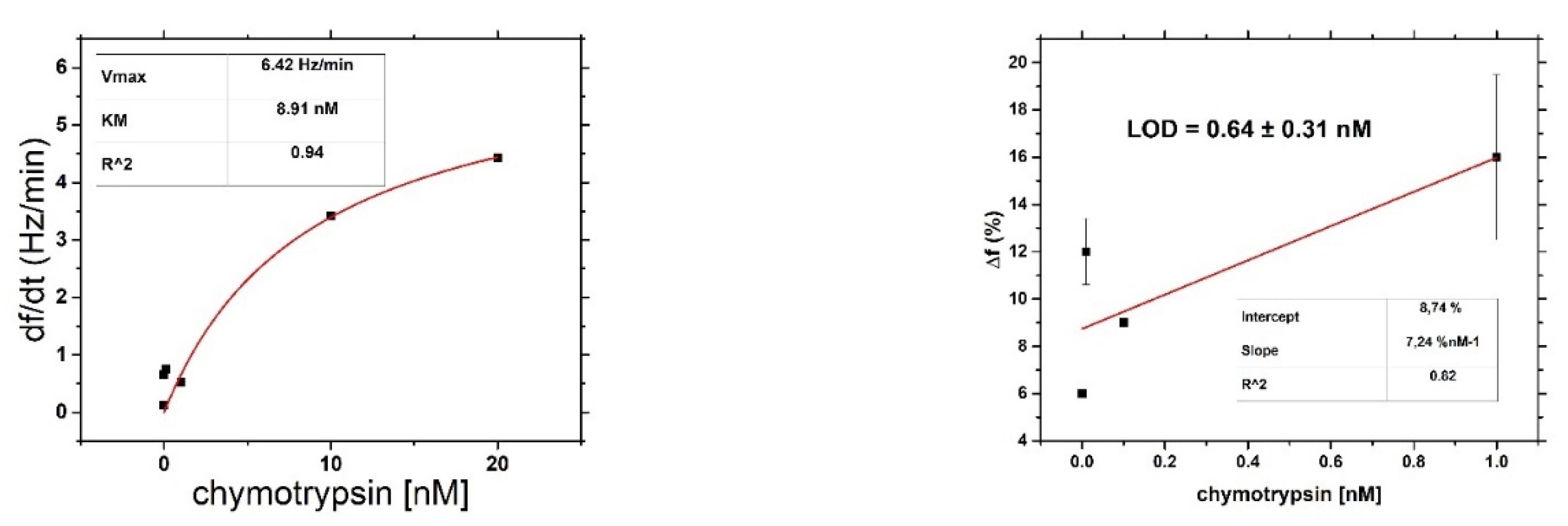
After binding of β-casein to the electrode surface and stabilizing the frequency (washing out all unbound residues), we applied chymotrypsin to the crystal at concentrations of 1 pM, 10 pM, 100 pM, 1 nM, 10 nM, and 20 nM. After 35 min of chymotrypsin application, the PBS was let into the cell until the resonant frequency stabilized. The change in resonant frequency from chymotrypsin application to stabilization in PBS was taken as the amount of casein cleaved from the layer. After lower concentrations (1 pM, 10 pM, 100 pM), we applied a higher concentration of chymotrypsin (at least at a concentration 2 orders of magnitude higher). In such measurements, we analyzed the degree of cleavage as the change in frequency from the initial state to a steady value. All measurements were performed at PBS, pH 7.4.
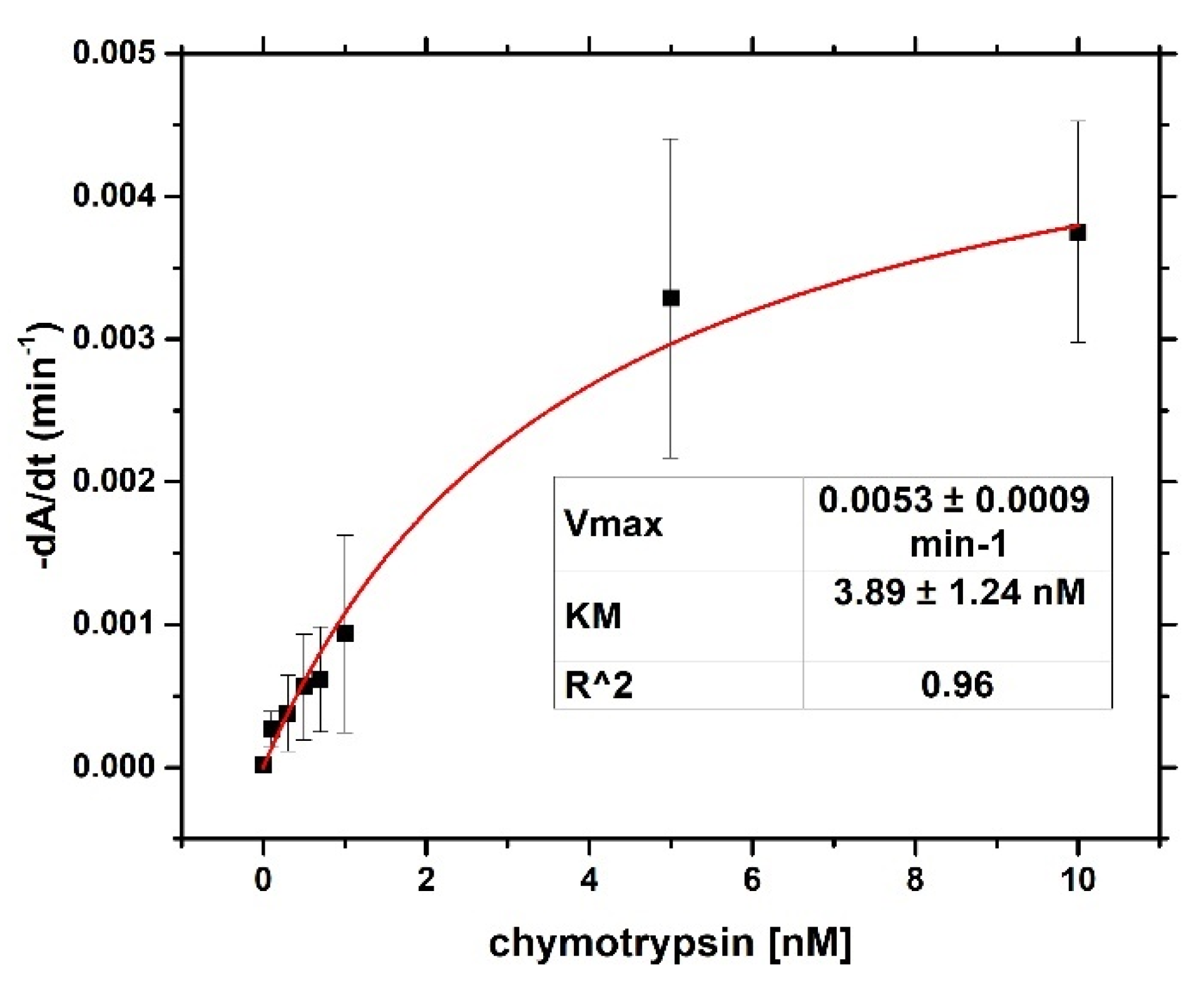
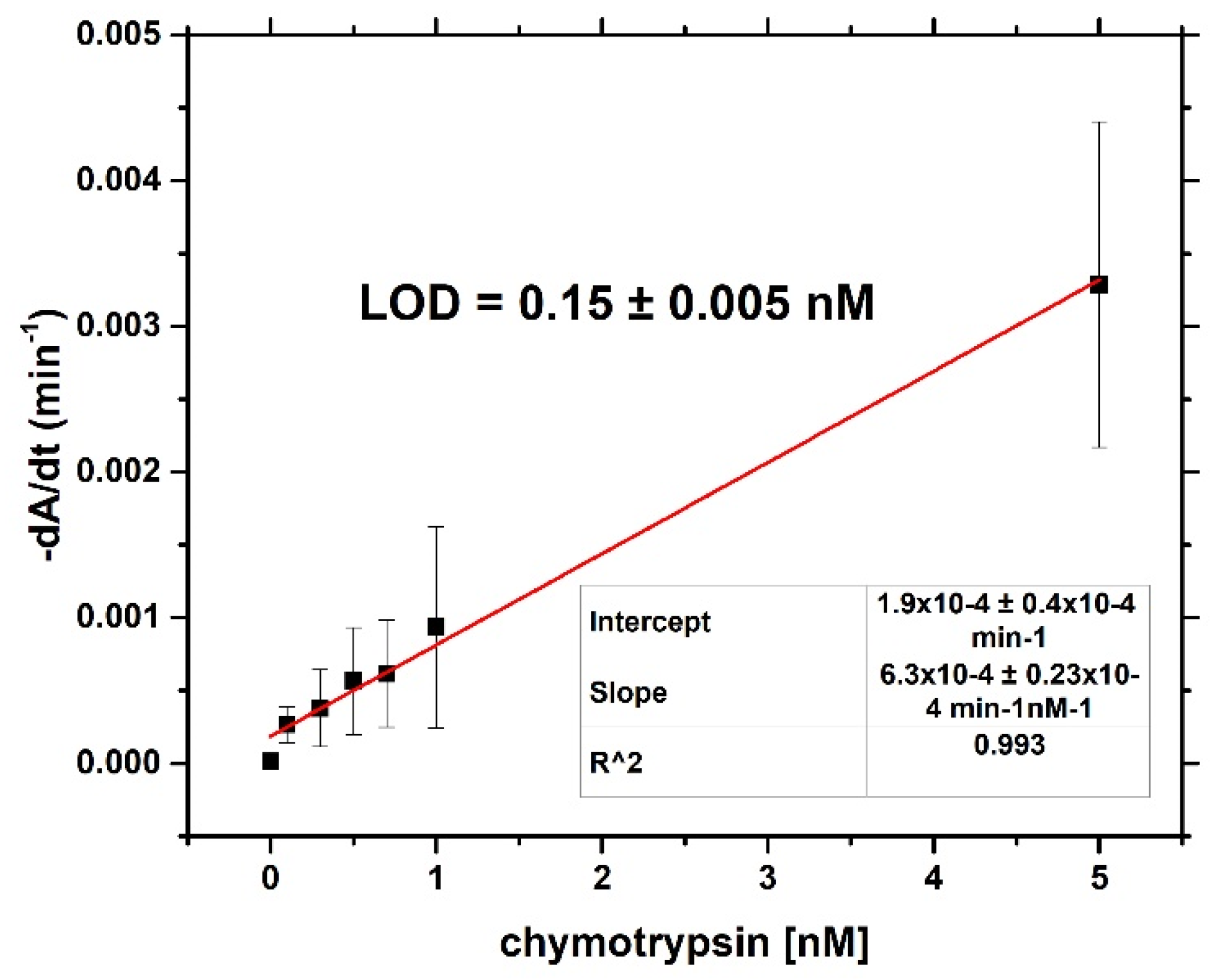
For both optical and gravimetric methods, the LOD was determined using equation: LOD = 3.3*(SD)/S, where SD is standard deviation of the sample with lowest concentration and S is slope calculated from fit of linear part of the calibration curve. Scheme of modification of TSM piezo and chymotrypsin cleavage is presented in
Figure 2.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}