1. Introduction
Here a method that involves the information communication theory and the relation of the DNA and protein molecules’ lengths is presented. It is done by equalization of the information contents of these two macromolecules and is based on the assumption that the information amount that is encrypted in a protein cannot be solely determined by residue variation along the length of a protein. Therefore, it is claimed here that the length (
or the size,
which is used here interchangeably) variations in the proteins could be implemented for a better comprehension. Accordingly, the method to be presented here is related to the evolution of the protein translation machinery and the other evolutionary processes since the protein sizes were not constant over time. So, protein-length is an evolutionary concern [
1,
2,
3] and its distribution was previously investigated for extracting the information on the evolutionary forces that are acting on the proteins [
2]. For instance, length distributions of the proteins evolving under weaker functional constraints were studied through different organisms [
2]. On the other hand, length is a concept that fundamentally retains dimensionality, or simply the size, as a feature in a material world. Therefore, the length of proteins can be analyzed through these relevant terms.
Shannon’s communication theory [
4,
5,
6] is the basis in calculating the information amount of a message that can be carried through molecules and hence it is utilized for the biological molecules [
7,
8,
9,
10]. The information contents of DNA and protein molecules can be calculated through the information entropies (Shannon’s entropy) of the DNA- and the protein-residues. However, this approach apparently results in a lowered information amount in the protein when it is performed separately for a protein of a specific size and a DNA that would be encoding a protein of the same length. The information that resides in a certain length of DNA is a potent criterion for defining the upper limit of the information amount of a certain length of protein, assuming no other relevant information communication is present. Accordingly, a protein-length-derived parameter is aimed to be introduced here as a new variable in the calculation of the information amount of the protein and for the equalization of the Shannon’s information amount of a protein and the DNA molecule that would be coding for the same length of a protein. This attitude is potentially of biological relevance.
3. Results and Discussion
Based on the inherent relation between the DNA and proteins, information amounts of the DNA and the proteins are aimed to be equalized. It is possible that there could be better approximations for the information amount equalization attempt that is described herein. However, the current one is useful in the sense that it seems to be approximating, to some extent, the increasing protein sizes with the genomes and the presence of introns in the eukaryotic genome.
When a length-wise parameter is included in the calculations as a multiplication factor (see in Equation (3)), information amount of the proteins comes to be rather comparable to that of DNA that would be coding the proteins with the same sizes as those of interest. This observation is significant since it can be biologically relevant to the observations on the organism- and species-specific distributions of the protein lengths, which are limited to certain ranges.
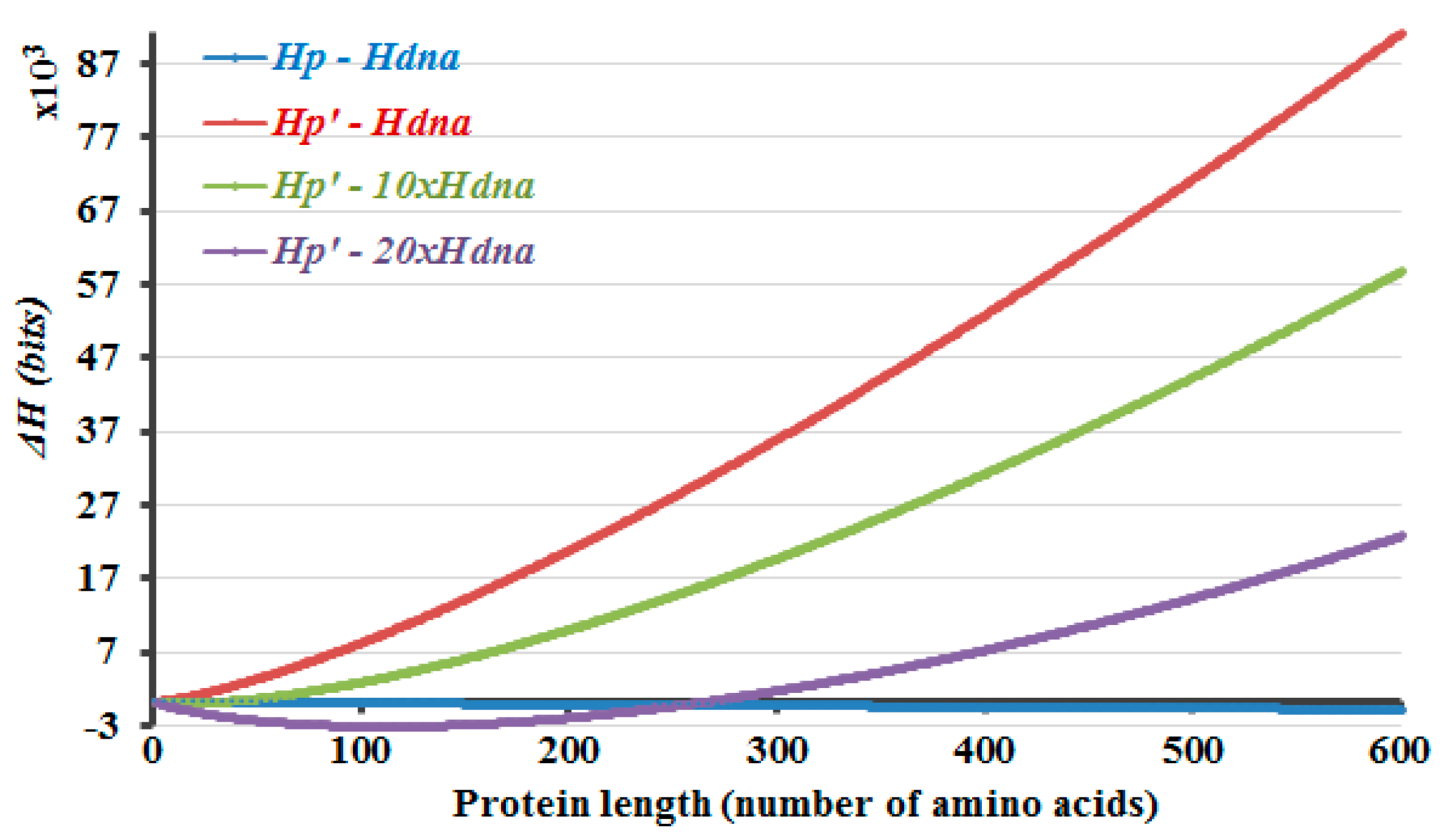
Protein-length-dependent variation of Δ
H values that are calculated differently but under the condition that the exponent
m is 1/3, are shown in
Figure 1, as an example, and also as the results that reveal the closest approximation to the literature values (
Figure 2). Actually, this work could be summarized along with the data that is shown in
Figure 1. It is observed that the Δ
H values that are calculated by taking the difference of
Hp and Hdna (
Figure 1, blue curve) decrease gradually and become more negative, as the protein length increases. Preventing this is the initial and equalization of the difference in the information amounts of the DNA and protein molecules is the latter aim of the current study. So, the mentioned decline in the information amount of proteins compared to the corresponding lengths of DNA is prevented (
Figure 1, red curve) by introducing a protein-length dependent parameter in the calculations, which is described in the
methods section. Although it may seem obvious to some, one may still ask why additional information amount is assumed to be present only in case of the protein molecules, in the form of information that is residing in the size, structure, and/or function features of the proteins. This is due to the fact that the direction of information flow in the process of protein translation is towards the protein, which is being translated by using the information that is encrypted in the DNA. Therefore the message-transfer event is prone to noise in that direction, from the DNA to the protein, and this can affect the success of the information transfer process. Here, regardless of such facts about the information transfer during protein translation in the biological realm, it is assumed that there is no loss (and actually also no gain) of information amount, during the translation of a protein from its encoding gene.
Turning back to
Figure 1, the Δ
H values that are calculated by taking the difference of
Hp′ and
Hdna (
Figure 1, red curve) prevents the decay in the information amount in the protein, compared to that of the DNA that would be encoding the same length of proteins. However, it is obvious that the equalization is observed only at around certain length ranges of the protein and the DNA molecules, in this manner of Δ
H calculation. Yet, there are introns in the eukaryotic genome and the sizes of the proteins are larger and much more varied in the eukaryotes. So, introns increase the corresponding DNA sizes to a great extent and in a variable manner. The presence of introns is implemented accordingly in the Δ
H calculations, by using a multiplication factor
A for the
Hdna part. This multiplication factor represents the non-coding/coding regions ratio in the gene. Eventually, equalization of the information amounts of the protein and DNA molecules is started to be observed at around diverse length ranges of the proteins and the corresponding DNA molecules, as the multiplication factor
A changes.
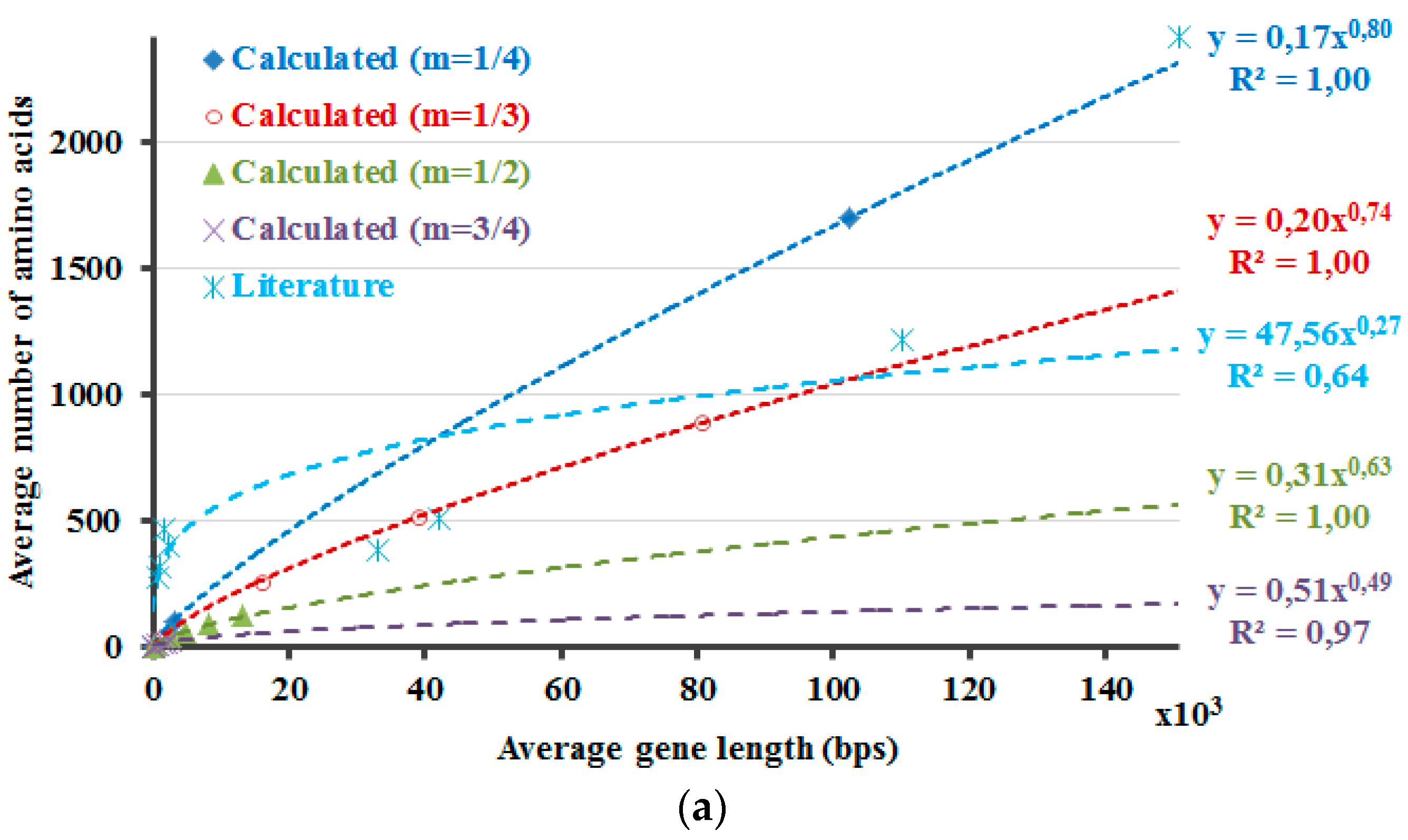
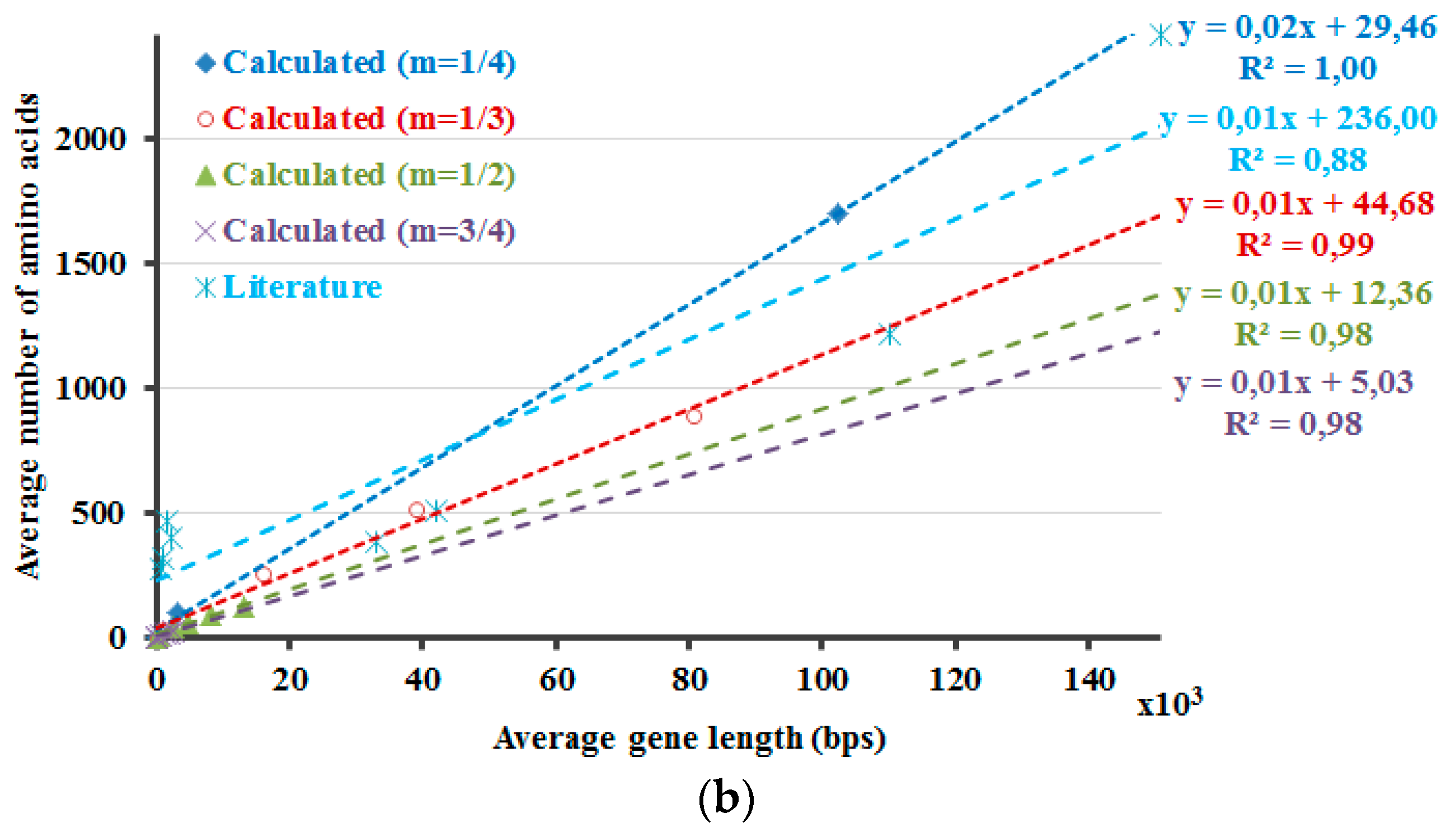
The protein lengths that reveal the minimum non-negative Δ
H values that are calculated by Equation (5) are compared with the average protein and gene sizes in the literature (
Figure 2). Accordingly, this method envisages the requirement of introns and the higher dissimilarity in the lengths of the proteins in the eukaryotic genome due to the occurrence of introns. If 5% of the DNA is composed of exons, with the rest part introns, the non-coding DNA, Δ
H will attain the minimum non-negative value in case of a protein with 266 amino acids (
Figure 1, purple curve,
Hp′
—20 ×
Hdna). The number 266 is about the point where the graph crosses the x-axis. If ~3.3% of the DNA is comprised of exons, the differences of the calculated information amounts would be zero or close to zero at about 900 amino acids (
Figure 2,
m = 1/3). These results are in line with the amplification of the length discrepancies in the eukaryotic proteins. Based on the same observations, it can be expected that the prokaryotic proteins would be smaller and less varied in length. Moreover, the values in the literature are correlated well with the calculated results when different sizes of the proteins in the human genome are included, as described in the last part of the
methods section (
Figure 2). This can also be interpreted in such a manner that the relation between the model that is proposed here and the literature-base data is noticeable when the variation of the intron amounts in different genes of the human genome and the respective proteins’ lengths are considered and included in the calculations. This relation is revealed better when 1/3 is rather used as the value of the exponential
m in the calculations.
In
Figure 2, the DNA and the protein lengths that reveal the minimum non-negative values as the result of the Δ
H calculations are shown. First f all, the Δ
H calculations are required for the determination of these values. These Δ
H calculations are performed by using the
Hp′ and by accounting for the presence of introns. So, in case of the
Hdna calculations part, the
Hdna is multiplied with distinct values of the multiplication factor
A. For each distinct value of the exponential
m that is used in the calculations of
Hp′, separate
Hdna calculations are also performed with dissimilar values of the multiplication factor A. Δ
H′s are determined for all combinations of those calculations with distinct values of the exponential
m and the multiplication factor
A. Then, the minimum non-negative values of the respective Δ
H calculations are determined to plot the graphs in
Figure 2. Further, these values are compared with the average protein and gene sizes in the literature (
Figure 2). At a first glance, the results that are presented in
Figure 2 seem to indicate that the current approach requires further work to be improved and/or to be validated. This is because the literature values cannot be fitted with the linear or exponential models as good as the calculation results. However, this does not necessarily deteriorate the reliability of the calculations at this point. This is because the selection of the literature is critical in studies such as this one, regardless of the toughness of accessing the literature values, which are sometimes contradictory even among themselves. Therefore, the biological data that is presented here should better be considered as just a rough comparison rather than validation tool of the model, the presented-calculations. Actually, this is the major reason why the model for the equalization of the information amounts of the protein and the DNA was not derived initially simply through the literature-based data. Yet, the literature-based data is still actual, real, observed values, regardless of the properness of the data that is selected for comparison. So, the literature-based data is informative as well and one should consider, think about the possible reasons of the fact that the literature-based data is fitted better with a linear function. As a result, the prospected future work involves such considerations along with working on the improvement of the literature data set, on the model itself and also even on the development of a new and better parameter in Equation (4).
4. Summary and Conclusions
Information amounts of the proteins gradually decays as the protein length increases if only the residue variations are accounted for while comparing the information amounts of the proteins and the corresponding lengths of the genes that encode for the same sizes of the proteins. Here, it is assumed that there is no loss (and actually also no gain) of information amount, in bits, during the translation of a protein from its encoding gene, although the information in the genes that is eliminated during protein synthesis due to being untranslated is not known to be residing in the proteins. However, the assumption of this work is that the information amounts of the protein and the gene that would be coding the same length of a protein are equal. Accordingly, the aim here is to suggest a possible model to realize the condition of the equalization of the information amounts of the proteins and the corresponding sizes of the DNA molecules. With this purpose, to compare the information amounts, Δ
H values are calculated through taking the difference of the information amounts of the proteins, (
Hp or
Hp′), and the information amounts of the DNA (
Hdna).
Hp is obtained by calculating the information amount in the protein by accounting only for the residue variation through the entire length of a protein. Likewise,
Hdna is obtained by calculating the information amount in a gene, by accounting also only for the residue variation through the entire length of the gene. So, the Δ
H value that would be calculated by taking the difference of the
Hp and
Hdna reveal a constant decay with increase in the protein size, as mentioned in the beginning.
Hp is multiplied with a protein-length dependent parameter to end up in
Hp′, as the actual information content of the proteins. This prevented the decay in the information content of the proteins. However, these Δ
H calculations reveal that the protein and the corresponding DNA lengths that are estimated by this means would be present in certain ranges, which would not be representing the immense variation in life, or it would be representing maybe only one or a few species, or only certain protein and DNA sizes. This problem is resolved by itself upon considering the contribution of the untranslated regions in the genes that encode for the proteins. These regions are the introns and they increase and vary the corresponding lengths of the DNA molecules that encode for the proteins. So, the presence of different amounts of introns enable the observation of different, increasing sizes of the proteins, together with the corresponding DNA lengths that have equal information amounts with those proteins. This is in line with the notion that the protein sizes are elevated with the genomes of different organisms and the presence of introns. In this sense, this work can be considered to be presenting a novel means of evaluation of the protein length conservation and variation in species. New techniques can provide further insight to the existing discussions on the evolutionary forces that are shaping the protein sizes and distributions in the modern organisms. In the future, further implications of this work will be explored together with our recent study on the equalization of the information amounts in the messenger RNA molecules and the proteins [
18]. The literature data set is also aimed to be improved, together with the model itself and the parameter that is implemented in the
Hp calculation.

{kind=link}
{kind=link}
{kind=link}


